Low-Temperature Steam Reforming of Natural Gas after LPG-Enrichment with MFI Membranes


 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
2.2. Catalyst Characterization
2.3. Membrane Separation
2.4. Catalytic Steam Reforming Tests
3. Results
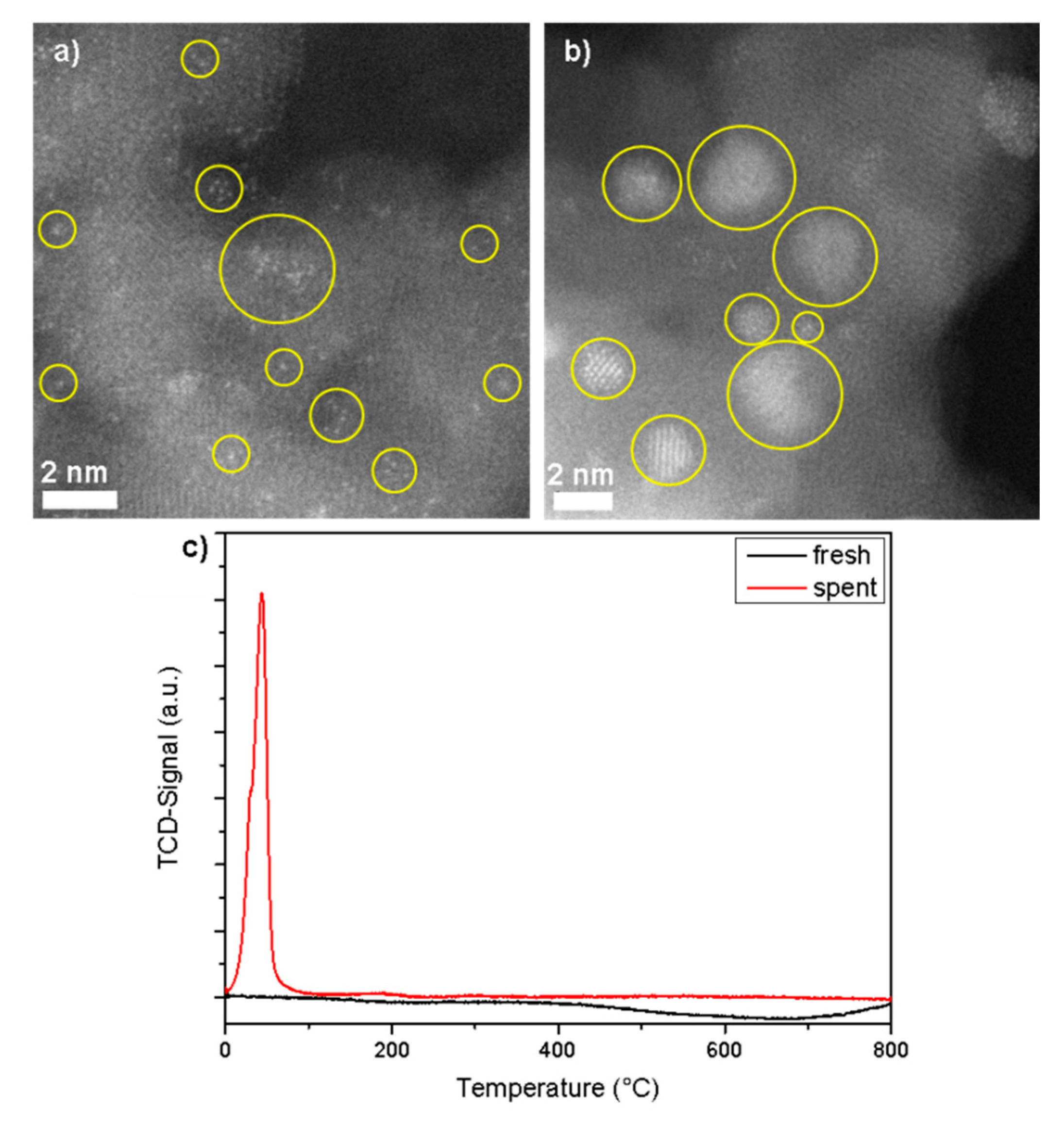
3.1. Rh1/Al2O3 Catalyst
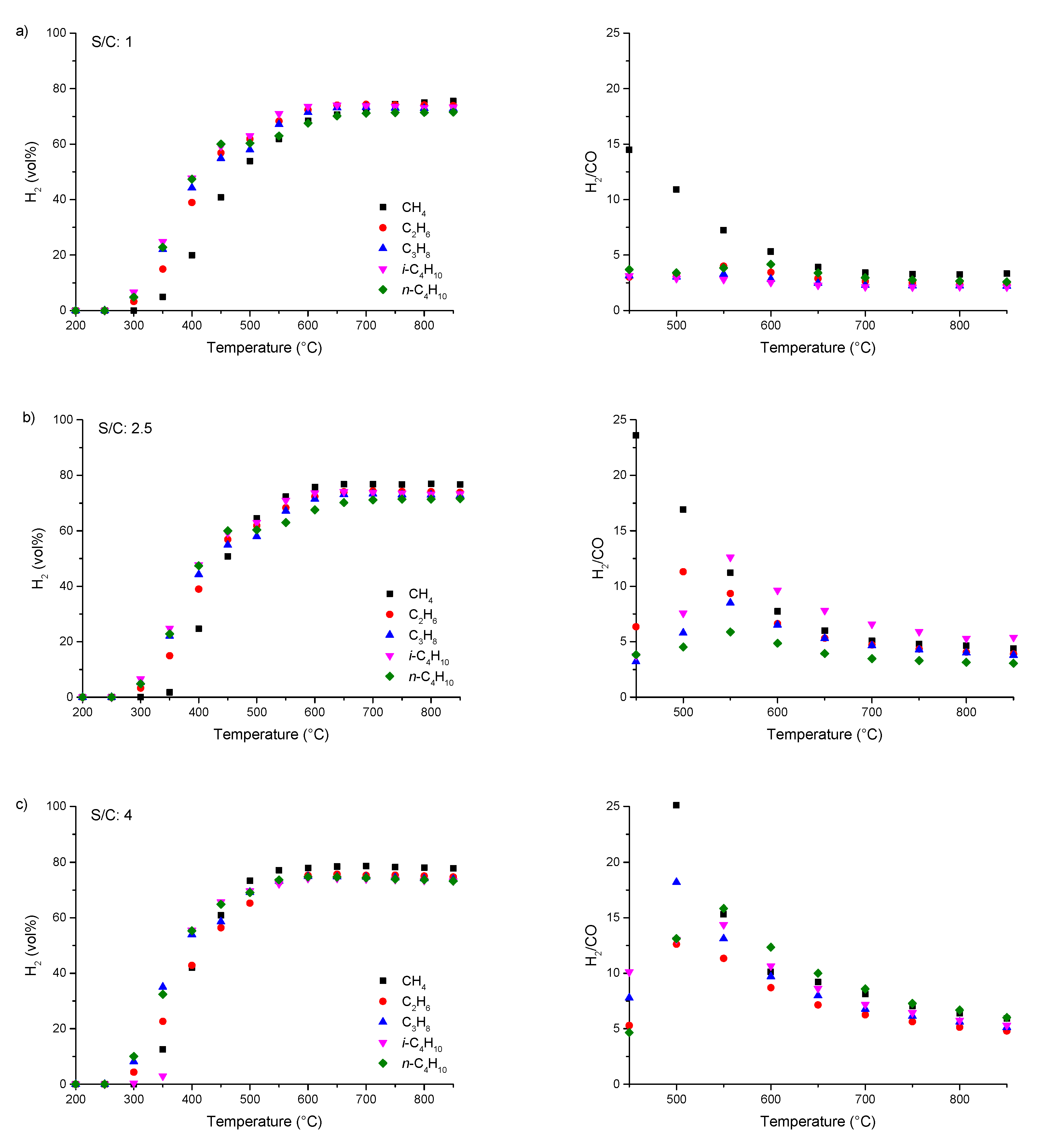
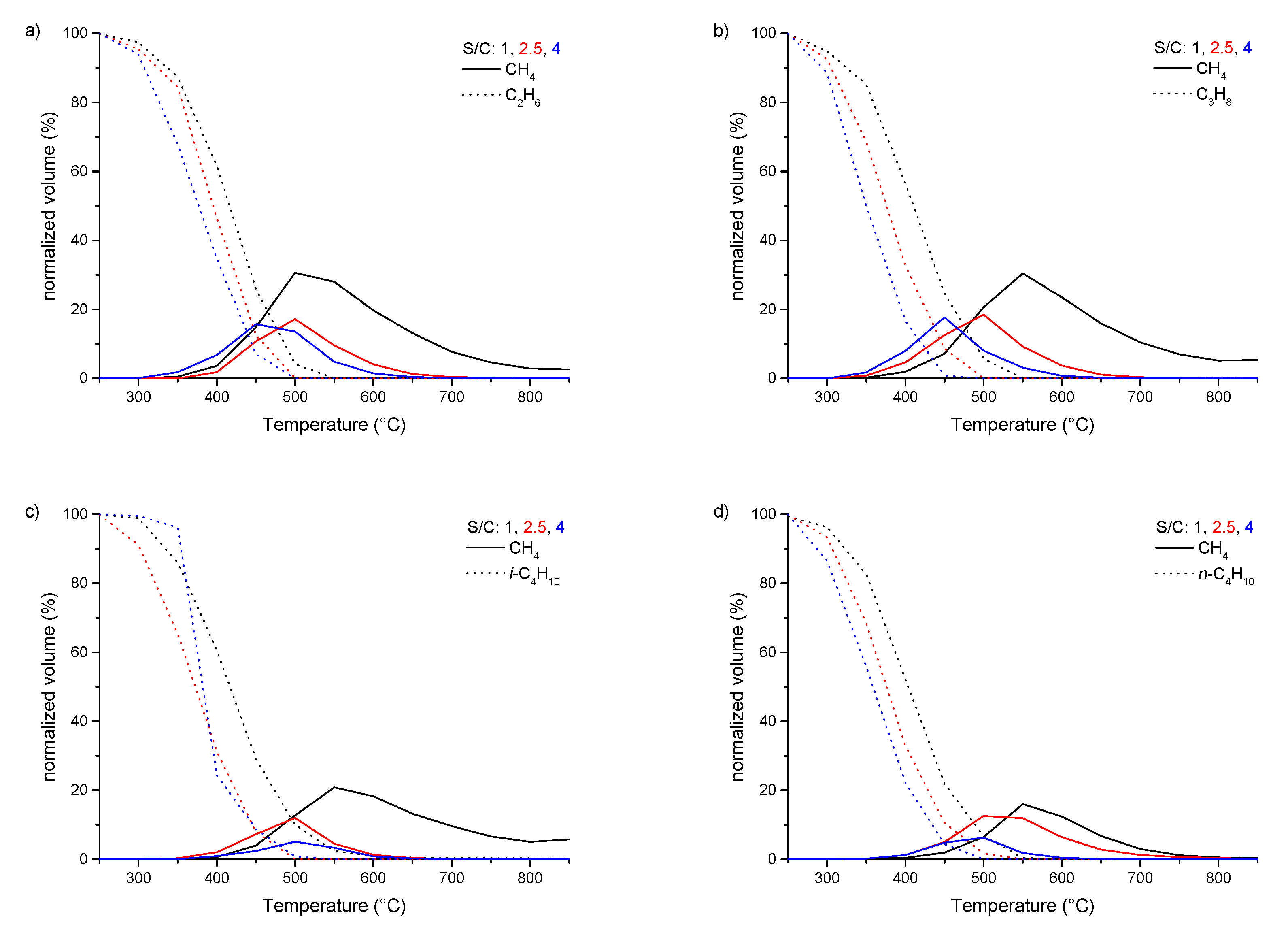
3.2. Steam Reforming of pure C1–4 Alkanes and Mixtures of C2–4 with Methane over Rh1/Al2O3
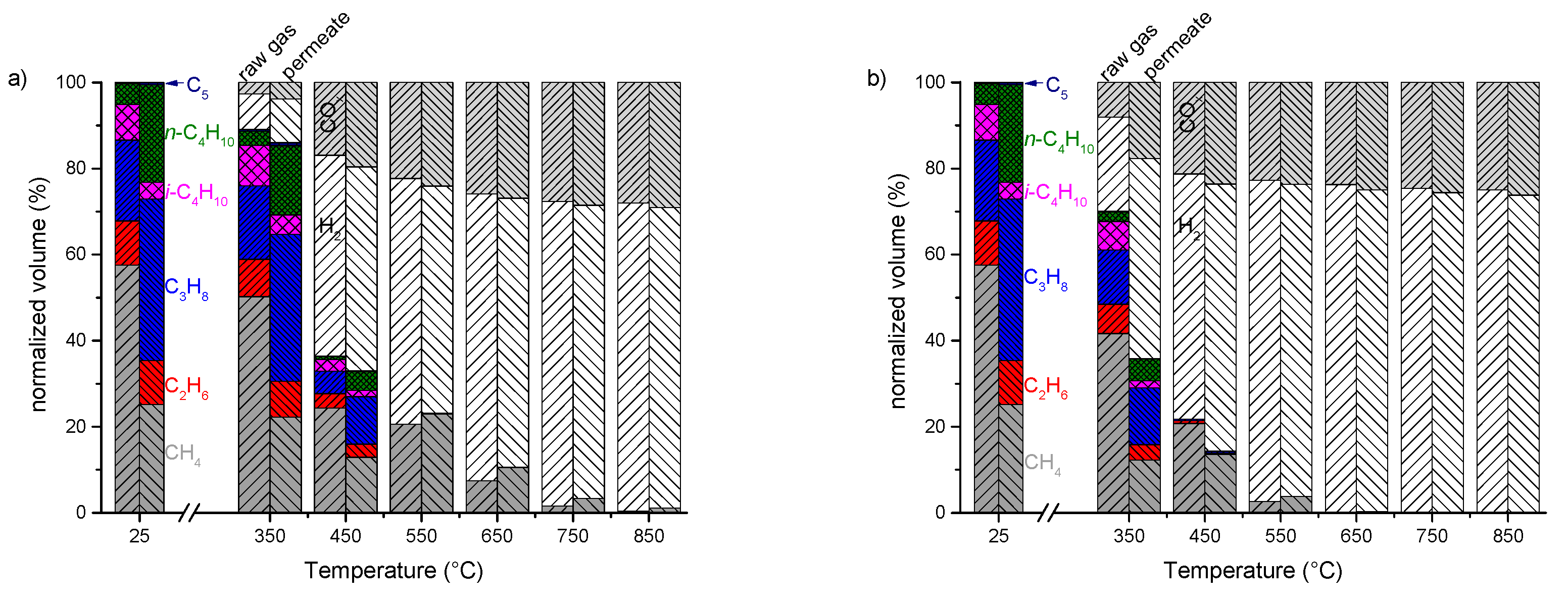
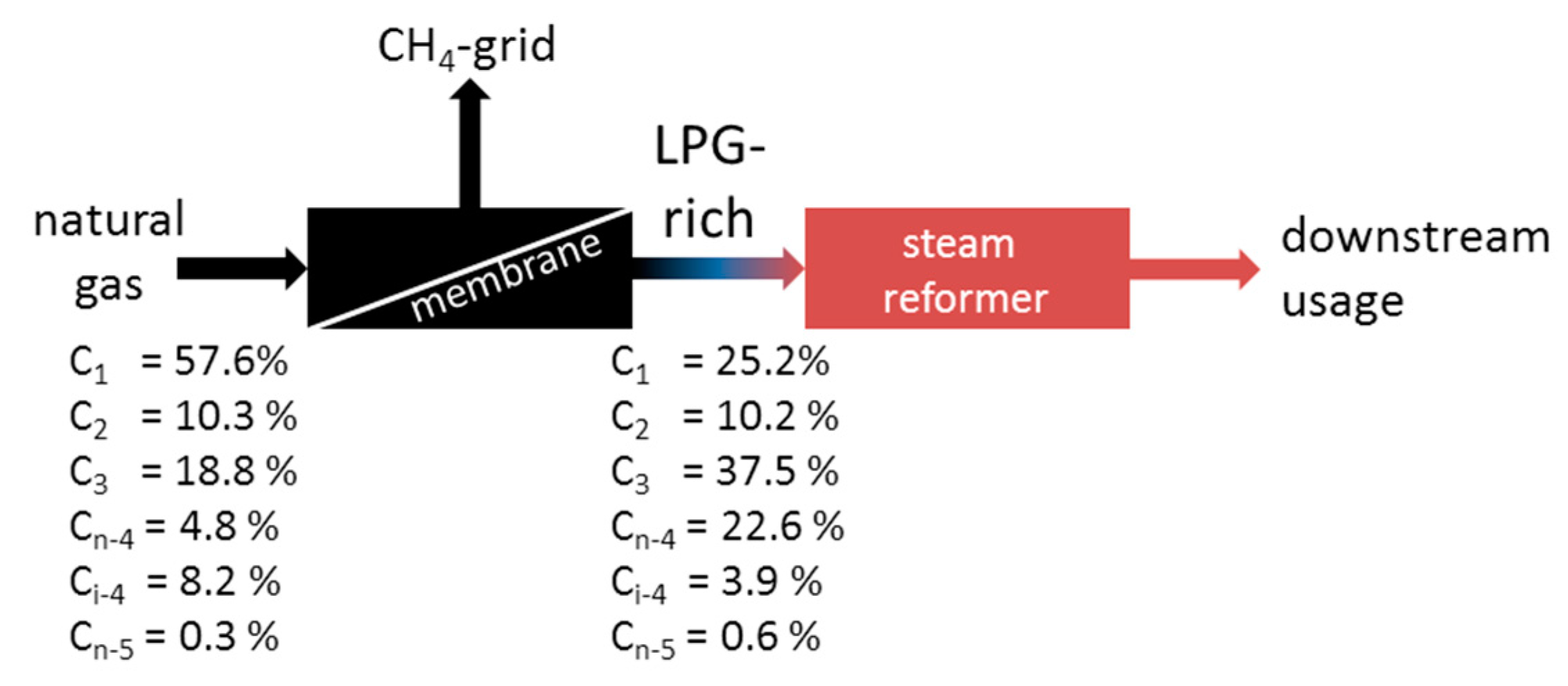
3.3. Enrichment of LPG from Natural Gas Using MFI-Membranes
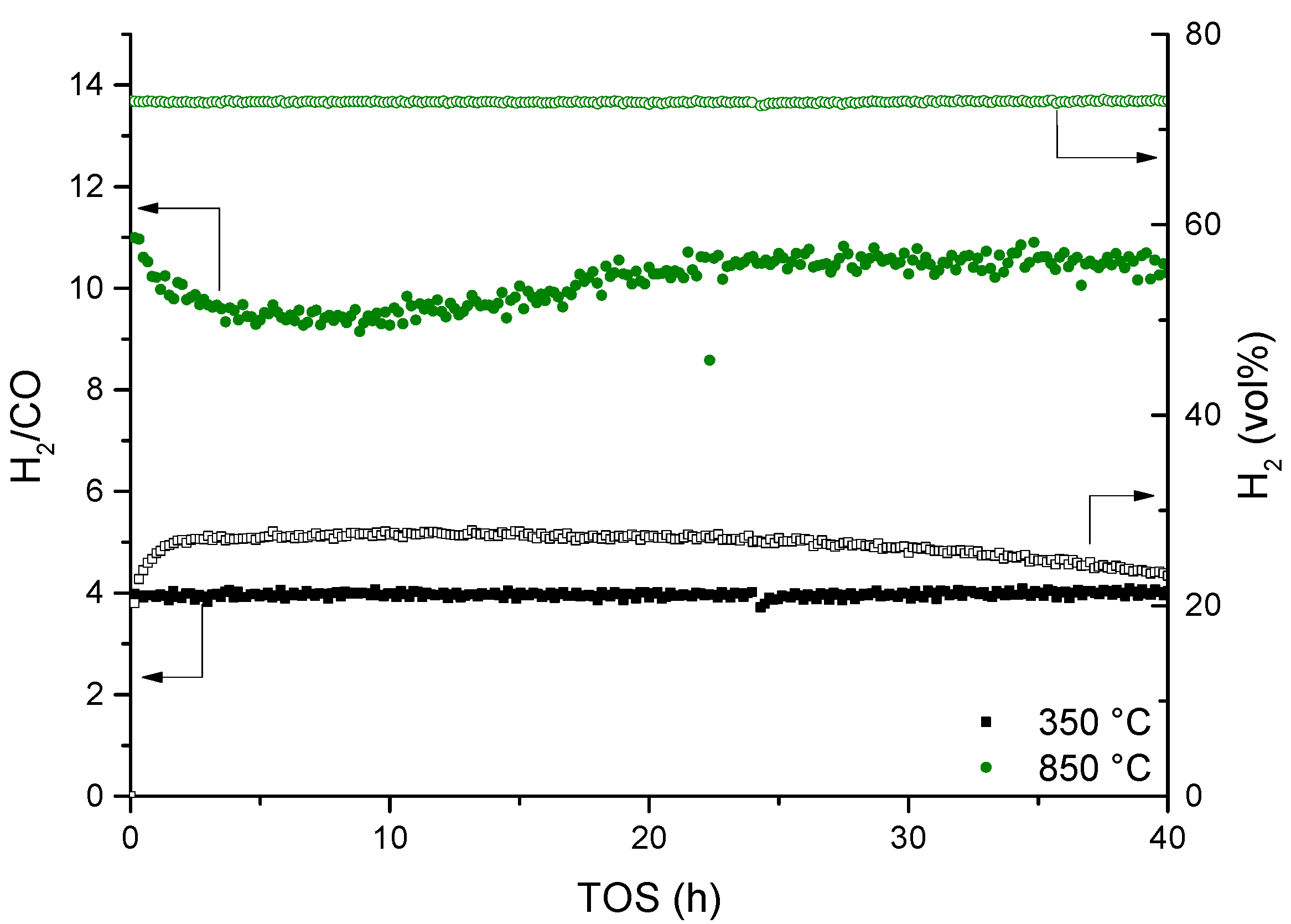
3.4. Steam Reforming of Real Natural Gas Mixtures over Rh1/Al2O3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- McFarland, E. Unconventional Chemistry for Unconventional Natural Gas. Science 2012, 338, 340–342. [Google Scholar] [CrossRef]
- Tang, P.; Zhu, Q.; Wu, Z.; Ma, D. Methane activation: The past and future. Energy Environ. Sci. 2014, 7, 2580–2591. [Google Scholar] [CrossRef]
- Horn, R.; Schlögl, R. Methane Activation by Heterogeneous Catalysis. Catal. Lett. 2015, 145, 23–39. [Google Scholar] [CrossRef]
- Olivos-Suarez, A.I.; Szécsényi, À.; Hensen, E.J.M.; Ruiz-Martinez, J.; Pidko, E.A.; Gascon, J. Strategies for the Direct Catalytic Valorization of Methane Using Heterogeneous Catalysis: Challenges and Opportunities. ACS Catal. 2016, 6, 2965–2981. [Google Scholar] [CrossRef]
- Schwach, P.; Pan, X.; Bao, X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef]
- Kondratenko, E.V.; Peppel, T.; Seeburg, D.; Kondratenko, V.A.; Kalevaru, N.; Martin, A.; Wohlrab, S. Methane conversion into different hydrocarbons or oxygenates: Current status and future perspectives in catalyst development and reactor operation. Catal. Sci. Technol. 2017, 7, 366–381. [Google Scholar] [CrossRef]
- Ross, J.R.H.; van Keulen, A.N.J.; Hegarty, M.E.S.; Seshan, K. The catalytic conversion of natural gas to useful products. Catal. Today 1996, 30, 193–199. [Google Scholar] [CrossRef]
- Lunsford, J.H. Catalytic conversion of methane to more useful chemicals and fuels: A challenge for the 21st century. Catal. Today 2000, 63, 165–174. [Google Scholar] [CrossRef]
- Aasberg-Petersen, K.; Dybkjær, I.; Ovesen, C.V.; Schjødt, N.C.; Sehested, J.; Thomsen, S.G. Natural gas to synthesis gas—Catalysts and catalytic processes. J. Nat. Gas Sci. Eng. 2011, 3, 423–459. [Google Scholar] [CrossRef]
- Baliban, R.C.; Elia, J.A.; Weekman, V.; Floudas, C.A. Process synthesis of hybrid coal, biomass, and natural gas to liquids via Fischer–Tropsch synthesis, ZSM-5 catalytic conversion, methanol synthesis, methanol-to-gasoline, and methanol-to-olefins/distillate technologies. Comput. Chem. Eng. 2012, 47, 29–56. [Google Scholar] [CrossRef]
- Wender, I. Reactions of synthesis gas. Fuel Process. Technol. 1996, 48, 189–297. [Google Scholar] [CrossRef]
- Song, X.P.; Guo, Z.C. Technologies for direct production of flexible H2/CO synthesis gas. Energy Convers. Manag. 2006, 47, 560–569. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R. New aspects of syngas production and use. Catal. Today 2000, 63, 159–164. [Google Scholar] [CrossRef]
- Joensen, F.; Rostrup-Nielsen, J.R. Conversion of hydrocarbons and alcohols for fuel cells. J. Power Sources 2002, 105, 195–201. [Google Scholar] [CrossRef]
- Avcı, A.K.; Trimmb, D.L.; Aksoylu, A.E.; Önsan, Z.I. Hydrogen production by steam reforming of n-butane over supported Ni and Pt-Ni catalysts. Appl. Catal. A 2004, 258, 235–240. [Google Scholar] [CrossRef]
- Hotz, N.; Stutz, M.J.; Loher, S.; Stark, W.J.; Poulikakos, D. Syngas production from butane using a flame-made Rh/Ce0.5Zr0.5O2 catalyst. Appl. Catal. B 2007, 73, 336–344. [Google Scholar] [CrossRef]
- Von Rickenbach, J.; Nabavi, M.; Zinovik, I.; Hotz, N.; Poulikakos, D. A detailed surface reaction model for syngas production from butane over Rhodium catalyst. Int. J. Hydrogen Energy 2011, 36, 12238–12248. [Google Scholar] [CrossRef]
- Pasel, J.; Wohlrab, S.; Kreft, S.; Rotov, M.; Löhken, K.; Peters, R.; Stolten, D. Routes for deactivation of different autothermal reforming catalysts. J. Power Sources 2016, 325, 51–63. [Google Scholar] [CrossRef]
- Auprêtre, F.; Descorme, C.; Duprez, D. Bio-ethanol catalytic steam reforming over supported metal catalysts. Catal. Commun. 2002, 3, 263–267. [Google Scholar] [CrossRef]
- Schädel, B.T.; Duisberg, M.; Deutschmann, O. Steam reforming of methane, ethane, propane, butane, and natural gas over a rhodium-based catalyst. Catal. Today 2009, 142, 42–51. [Google Scholar] [CrossRef]
- Karakaya, C.; Maier, L.; Deutschmann, O. Surface Reaction Kinetics of the Oxidation and Reforming of CH4 over Rh/Al2O3 Catalysts. Int. J. Chem. Kinet. 2016, 48, 144–160. [Google Scholar] [CrossRef]
- Angeli, S.D.; Monteleone, G.; Giaconia, A.; Lemonidou, A.A. State-of-the-art catalysts for CH4 steam reforming at low temperature. Int. J. Hydrogen Energy 2014, 39, 1979–1997. [Google Scholar] [CrossRef]
- Supat, K.; Chavadej, S.; Lobban, L.L.; Mallinson, R.G. Combined Steam Reforming and Partial Oxidation of Methane to Synthesis Gas under Electrical Discharge. Ind. Eng. Chem. Res. 2003, 42, 1654–1661. [Google Scholar] [CrossRef]
- Bakker, W.J.W.; Kapteijn, F.; Poppe, J.; Moulijn, J.A. Permeation characteristics of a metal-supported silicalite-1 zeolite membrane. J. Membr. Sci. 1996, 117, 57–78. [Google Scholar] [CrossRef]
- Krishna, R.; Vandenbroeke, L.J.P. The Maxwell-Stefan Description of Mass-Transport across Zeolite Membranes. Chem. Eng. J. Biochem. Eng. J. 1995, 57, 155–162. [Google Scholar] [CrossRef]
- Vroon, Z.A.E.P.; Keizer, K.; Gilde, M.J.; Verweij, H.; Burggraaf, A.J. Transport properties of alkanes through ceramic thin zeolite MFI membranes. J. Membr. Sci. 1996, 113, 293–300. [Google Scholar] [CrossRef]
- Keizer, K.; Burggraaf, A.J.; Vroon, Z.A.E.P.; Verweij, H. Two component permeation through thin zeolite MFI membranes. J. Membr. Sci. 1998, 147, 159–172. [Google Scholar] [CrossRef]
- Van de Graaf, J.M.; Kapteijn, F.; Moulijn, J.A. Methodological and operational aspects of permeation measurements on silicalite-1 membranes. J. Membr. Sci. 1998, 144, 87–104. [Google Scholar] [CrossRef]
- Gump, C.J.; Lin, X.; Falconer, J.L.; Noble, R.D. Experimental configuration and adsorption effects on the permeation of C4 isomers through ZSM-5 zeolite membranes. J. Membr. Sci. 2000, 173, 35–52. [Google Scholar] [CrossRef]
- Arruebo, M.; Coronas, J.; Menendez, M.; Santamaria, J. Separation of hydrocarbons from natural gas using silicalite membranes. Sep. Purif. Technol. 2001, 25, 275–286. [Google Scholar] [CrossRef]
- Wohlrab, S.; Meyer, T.; Stöhr, M.; Hecker, C.; Lubenau, U.; Oßmann, A. On the performance of customized MFI membranes for the separation of n-butane from methane. J. Membr. Sci. 2011, 369, 96–104. [Google Scholar] [CrossRef]
- Dragomirova, R.; Stohr, M.; Hecker, C.; Lubenau, U.; Paschek, D.; Wohlrab, S. Desorption-controlled separation of natural gas alkanes by zeolite membranes. RSC Adv. 2014, 4, 59831–59834. [Google Scholar] [CrossRef]
- Neubauer, K.; Dragomirova, R.; Stöhr, M.; Mothes, R.; Lubenau, U.; Paschek, D.; Wohlrab, S. Combination of membrane separation and gas condensation for advanced natural gas conditioning. J. Membr. Sci. 2014, 453, 100–107. [Google Scholar] [CrossRef]
- Dragomirova, R.; Jorabchi, M.N.; Paschek, D.; Wohlrab, S. Operational Criteria for the Separation of Alkanes by Zeolite Membranes. Chem. Ing. Tech. 2017, 89, 926–934. [Google Scholar] [CrossRef]
- Angeli, S.D.; Pilitsis, F.G.; Lemonidou, A.A. Methane steam reforming at low temperature: Effect of light alkanes’ presence on coke formation. Catal. Today 2015, 242, 119–128. [Google Scholar] [CrossRef]
- Guo, X.; Fang, G.; Li, G.; Ma, H.; Fan, H.; Yu, L.; Ma, C.; Wu, X.; Deng, D.; Wei, M.; et al. Direct, Nonoxidative Conversion of Methane to Ethylene, Aromatics, and Hydrogen. Science 2014, 344, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhang, S.; Tang, Y.; Li, Y.; Nguyen, L.; Li, Y.; Shan, J.; Xiao, D.; Gagne, R.; Frenkel, A.I.; et al. Low-Temperature Transformation of Methane to Methanol on Pd1O4 Single Sites Anchored on the Internal Surface of Microporous Silicate. Angew. Chem. 2016, 128, 13639–13643. [Google Scholar] [CrossRef]
- Vanelderen, P.; Vancauwenbergh, J.; Tsai, M.-L.; Hadt, R.G.; Solomon, E.I.; Schoonheydt, R.A.; Sels, B.F. Spectroscopy and Redox Chemistry of Copper in Mordenite. Chem. Phys. Chem. 2014, 15, 91–99. [Google Scholar] [CrossRef]
- Grundner, S.; Markovits, M.A.C.; Li, G.; Tromp, M.; Pidko, E.A.; Hensen, E.J.M.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J.A. Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol. Nat. Commun. 2015, 6, 7546. [Google Scholar] [CrossRef]
- Grundner, S.; Luo, W.; Sanchez-Sanchez, M.; Lercher, J.A. Synthesis of single-site copper catalysts for methane partial oxidation. Chem. Commun. 2016, 52, 2553–2556. [Google Scholar] [CrossRef]
- Kulkarni, A.R.; Zhao, Z.-J.; Siahrostami, S.; Nørskov, J.K.; Studt, F. Monocopper Active Site for Partial Methane Oxidation in Cu-Exchanged 8MR Zeolites. ACS Catal. 2016, 6, 6531–6536. [Google Scholar] [CrossRef]
- Wallis, P.; Schonborn, E.; Kalevaru, V.N.; Martin, A.; Wohlrab, S. Enhanced formaldehyde selectivity in catalytic methane oxidation by vanadia on Ti-doped SBA-15. RSC Adv. 2015, 5, 69509–69513. [Google Scholar] [CrossRef]
- Wallis, P.; Wohlrab, S.; Kalevaru, V.N.; Frank, M.; Martin, A. Impact of support pore structure and morphology on catalyst performance of VOx/SBA-15 for selective methane oxidation. Catal. Today 2016, 278, 120–126. [Google Scholar] [CrossRef]
- Dang, T.T.H.; Seeburg, D.; Radnik, J.; Kreyenschulte, C.; Atia, H.; Vu, T.T.H.; Wohlrab, S. Influence of V-sources on the catalytic performance of VMCM-41 in the selective oxidation of methane to formaldehyde. Catal. Commun. 2018, 103, 56–59. [Google Scholar] [CrossRef]
- Duarte, R.B.; Krumeich, F.; van Bokhoven, J.A. Structure, Activity, and Stability of Atomically Dispersed Rh in Methane Steam Reforming. ACS Catal. 2014, 4, 1279–1286. [Google Scholar] [CrossRef]
- Lian, J.; Ma, J.; Duan, X.; Kim, T.; Li, H.; Zheng, W. One-step ionothermal synthesis of g-Al2O3 mesoporous nanoflakes at low temperature. Chem. Commun. 2010, 46, 2650–2652. [Google Scholar] [CrossRef]
- Fargeot, D.; Mercurio, D.; Dauger, A. Structural characterization of alumina metastable phases in plasma sprayed deposits. Mater. Chem. Phys. 1990, 24, 299–314. [Google Scholar] [CrossRef]
- Husson, E.; Repelin, Y. Structural studies of transition aluminas. Theta alumina. Eur. J. Solid State Inorg. Chem. 1996, 33, 1223–1231. [Google Scholar]
- Weng, W.Z.; Pei, X.Q.; Li, H.M.; Luo, C.R.; Liu, Y.; Lin, H.Q.; Huang, C.J.; Wan, H.L. Effects of calcination temperatures on the catalytic performance of Rh/Al2O3 for methane partial oxidation to synthesis gas. Catal. Today 2006, 117, 53–61. [Google Scholar] [CrossRef]
- Yao, H.C.; Japar, S.; Shelef, M. Surface interactions in the system RhAl2O3. J. Catal. 1977, 50, 407–418. [Google Scholar] [CrossRef]
- Guan, H.L.; Lin, J.; Qiao, B.T.; Miao, S.; Wang, A.Q.; Wang, X.D.; Zhang, T. Enhanced performance of Rh1/TiO2 catalyst without methanation in water-gas shift reaction. AIChE J. 2017, 63, 2081–2088. [Google Scholar] [CrossRef]
- Graf, P.O.; Mojet, B.L.; van Ommen, J.G.; Lefferts, L. Comparative study of steam reforming of methane, ethane and ethylene on Pt, Rh and Pd supported on yttrium-stabilized zirconia. Appl. Catal. A 2007, 332, 310–317. [Google Scholar] [CrossRef]
- Igarashi, A.; Ohtaka, T.; Motoki, S. Low-temperature steam reforming of n-butane over Rh and Ru catalysts supported on ZrO2. Catal. Lett. 1992, 13, 189–194. [Google Scholar] [CrossRef]
- Alcheikhhamdon, Y.; Hoorfar, M. Natural gas quality enhancement: A review of the conventional treatment processes, and the industrial challenges facing emerging technologies. J. Nat. Gas Sci. Eng. 2016, 34, 689–701. [Google Scholar] [CrossRef]
- Dragomirova, R.; Wohlrab, S. Zeolite Membranes in Catalysis—From Separate Units to Particle Coatings. Catalysts 2015, 5, 2161–2222. [Google Scholar] [CrossRef]
- Krishna, R.; Paschek, D. Separation of hydrocarbon mixtures using zeolite membranes: A modelling approach combining molecular simulations with the Maxwell–Stefan theory. Sep. Purif. Technol. 2000, 21, 111–136. [Google Scholar] [CrossRef]
- Hansen, T.W.; DeLaRiva, A.T.; Challa, S.R.; Datye, A.K. Sintering of Catalytic Nanoparticles: Particle Migration or Ostwald Ripening? Acc. Chem. Res. 2013, 46, 1720–1730. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | BET | ICP | ||
|---|---|---|---|---|
| SBET (m2/g) | Vt (mL/g) | D (nm) | Rh (mol%) | |
| Al2O3 | 181 | 0.413 | 9.1 | - |
| Rh1/Al2O3 | 169 | 0.406 | 9.6 | 1.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seeburg, D.; Liu, D.; Dragomirova, R.; Atia, H.; Pohl, M.-M.; Amani, H.; Georgi, G.; Kreft, S.; Wohlrab, S. Low-Temperature Steam Reforming of Natural Gas after LPG-Enrichment with MFI Membranes. Processes 2018, 6, 263. https://doi.org/10.3390/pr6120263
Seeburg D, Liu D, Dragomirova R, Atia H, Pohl M-M, Amani H, Georgi G, Kreft S, Wohlrab S. Low-Temperature Steam Reforming of Natural Gas after LPG-Enrichment with MFI Membranes. Processes. 2018; 6(12):263. https://doi.org/10.3390/pr6120263
Chicago/Turabian StyleSeeburg, Dominik, Dongjing Liu, Radostina Dragomirova, Hanan Atia, Marga-Martina Pohl, Hadis Amani, Gabriele Georgi, Stefanie Kreft, and Sebastian Wohlrab. 2018. "Low-Temperature Steam Reforming of Natural Gas after LPG-Enrichment with MFI Membranes" Processes 6, no. 12: 263. https://doi.org/10.3390/pr6120263
APA StyleSeeburg, D., Liu, D., Dragomirova, R., Atia, H., Pohl, M.-M., Amani, H., Georgi, G., Kreft, S., & Wohlrab, S. (2018). Low-Temperature Steam Reforming of Natural Gas after LPG-Enrichment with MFI Membranes. Processes, 6(12), 263. https://doi.org/10.3390/pr6120263