On the Thermal Self-Initiation Reaction of n-Butyl Acrylate in Free-Radical Polymerization


Abstract
:1. Introduction
2. Materials and Methods
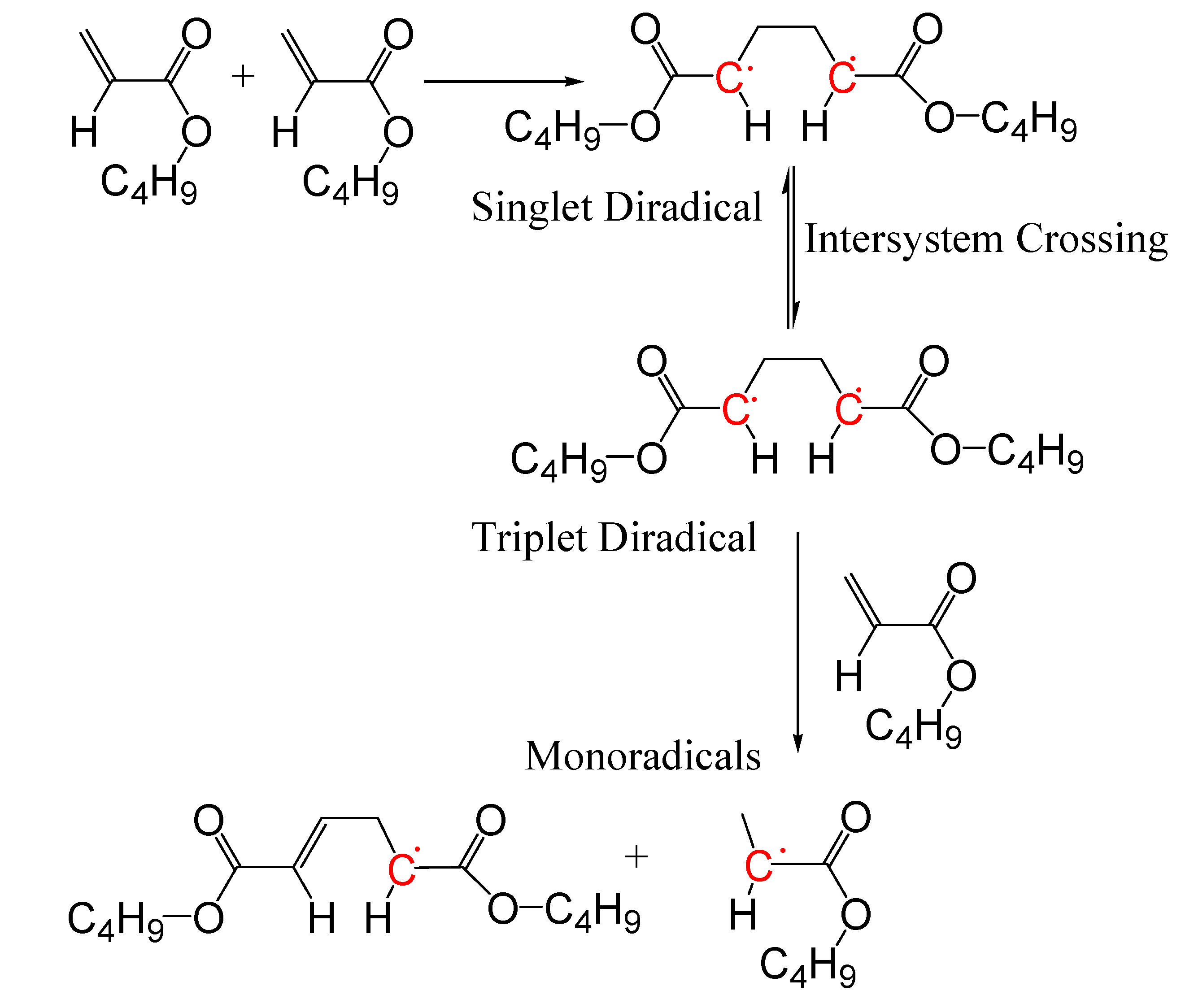
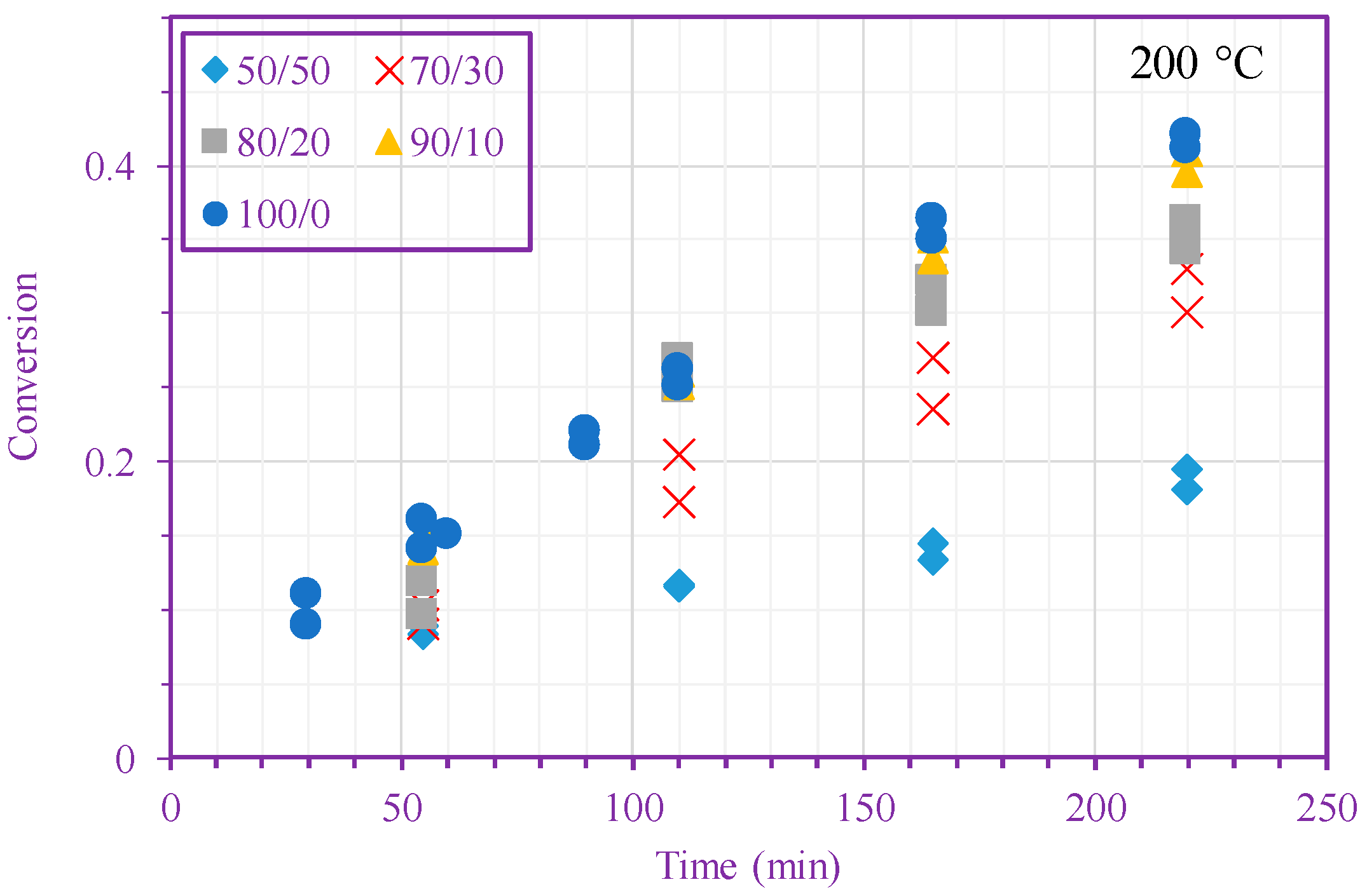
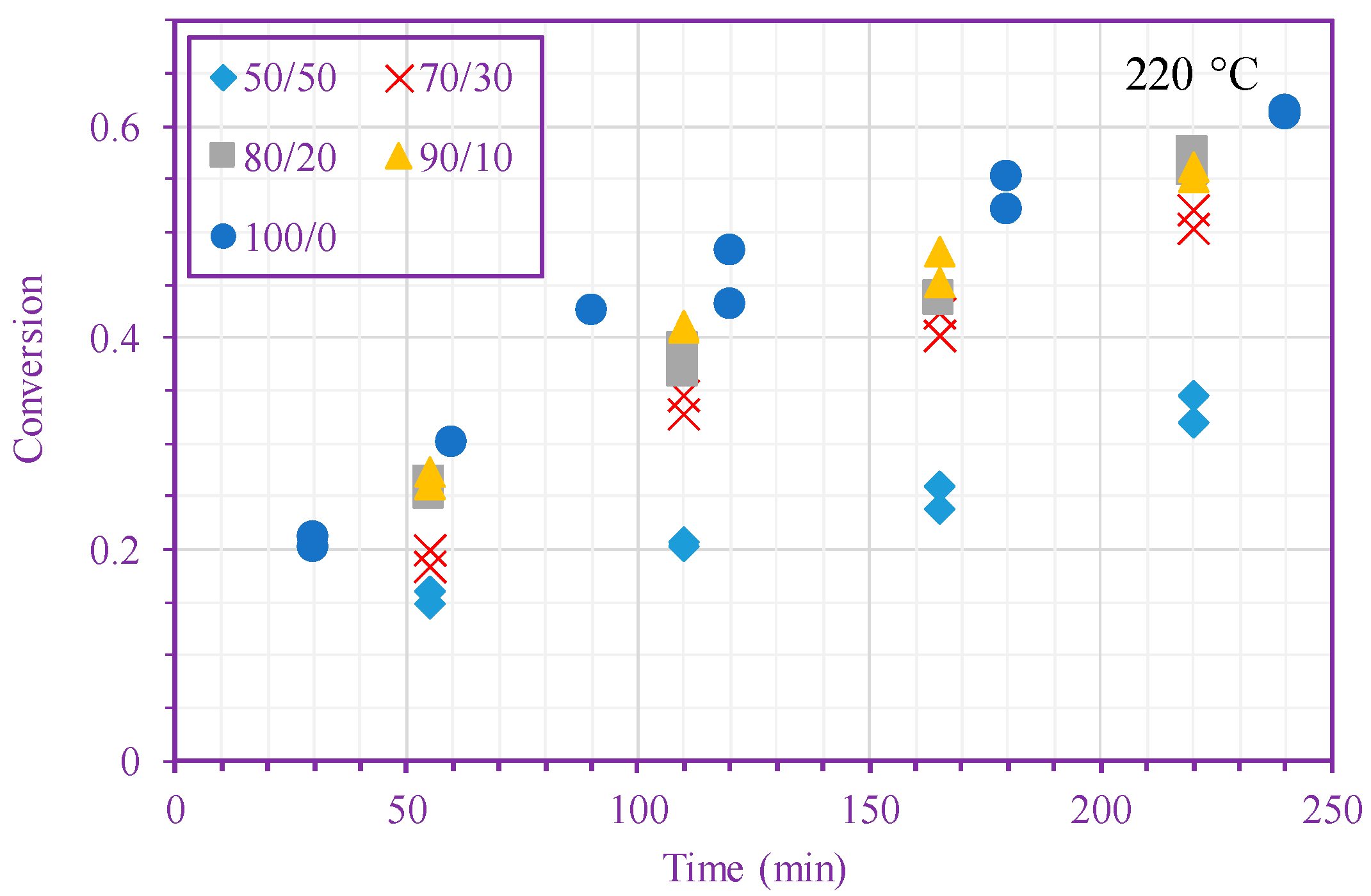
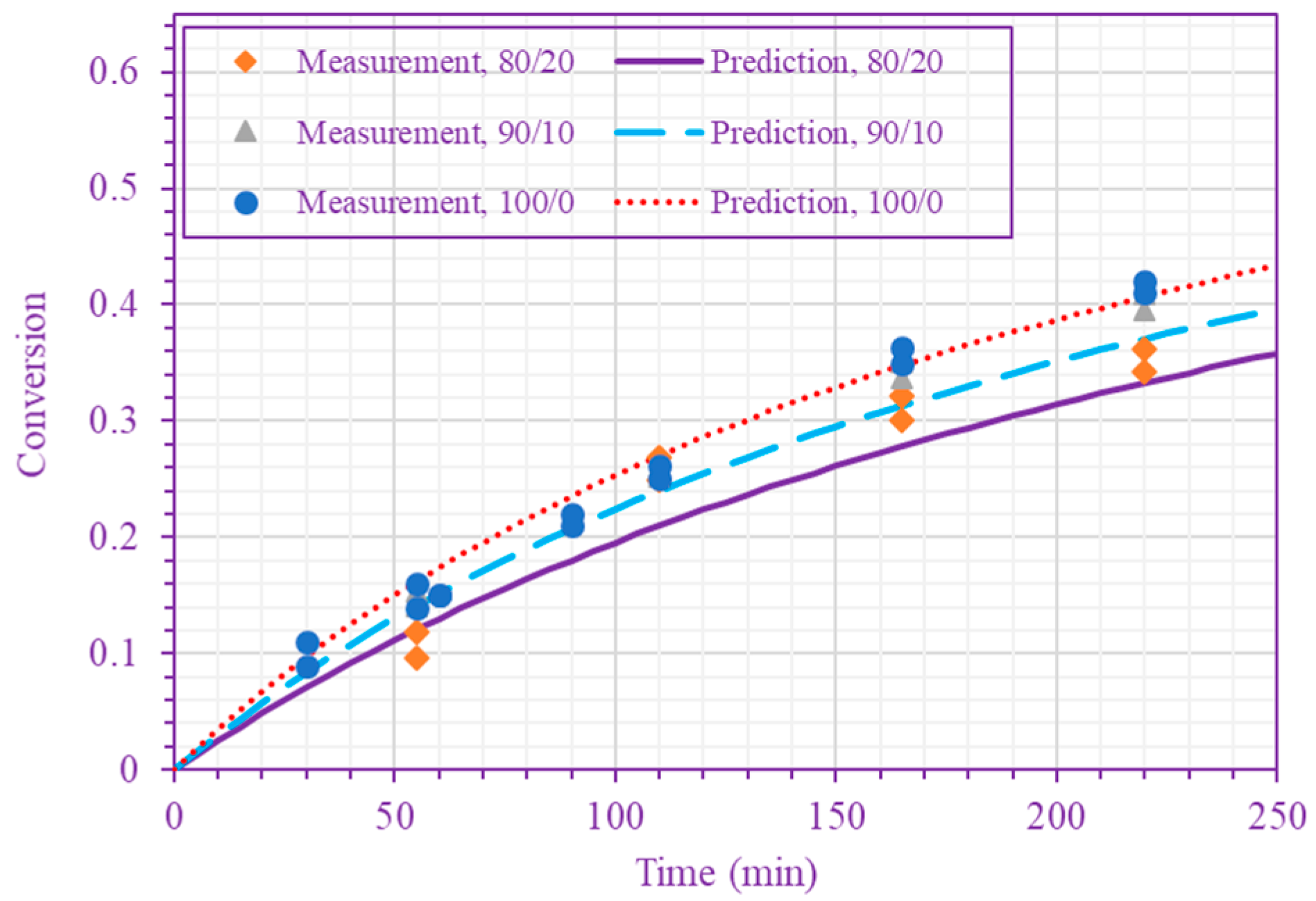
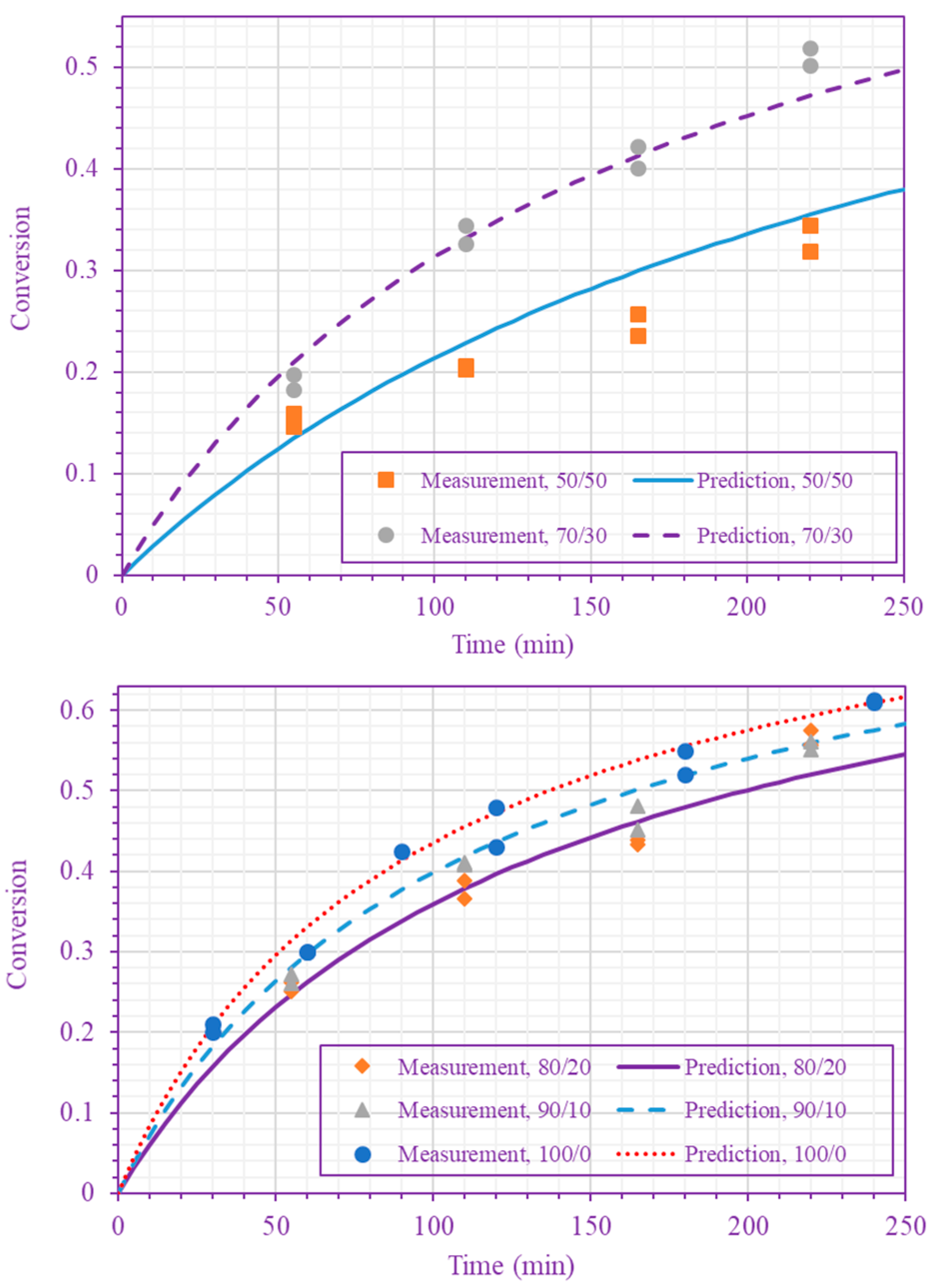
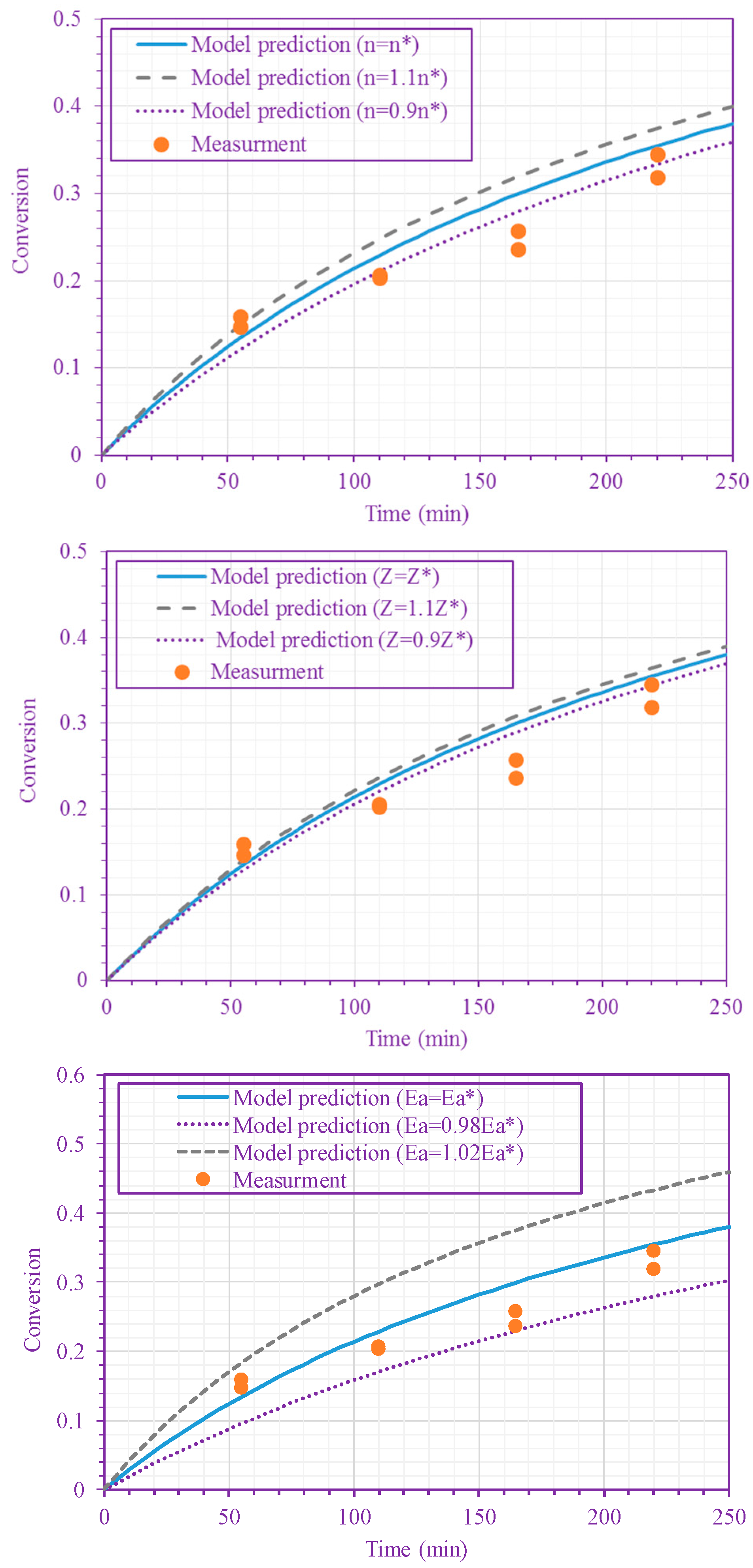
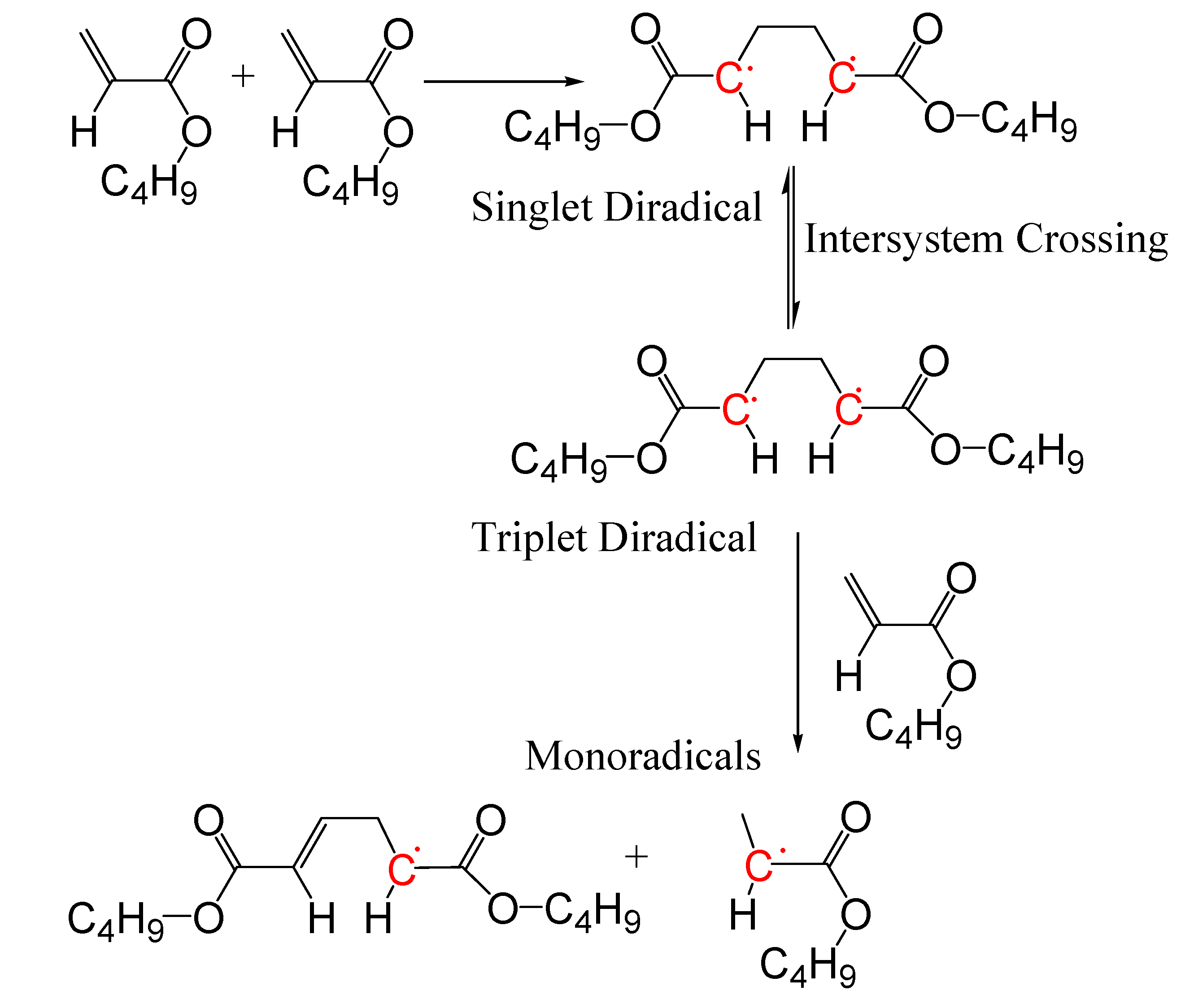
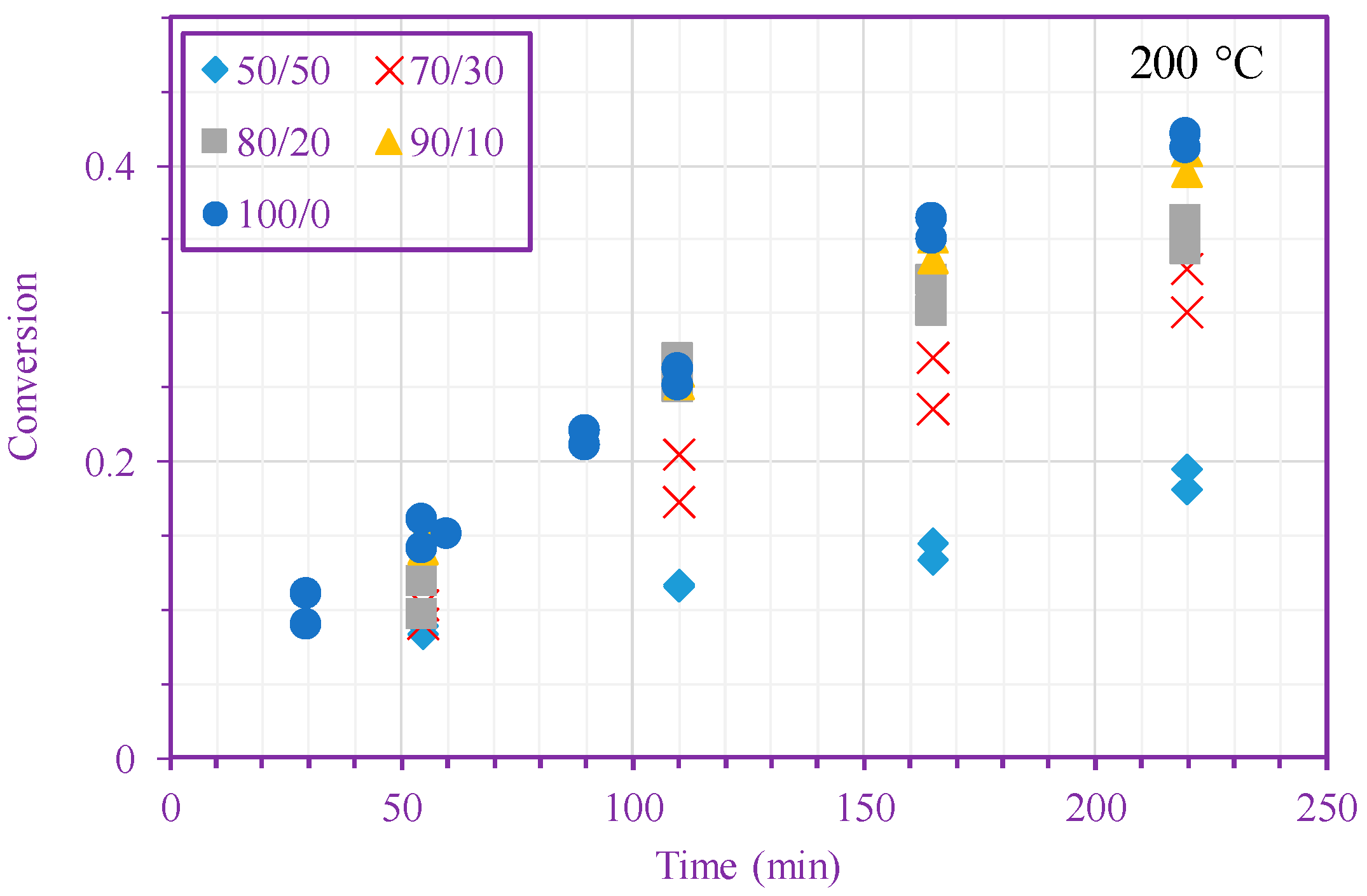
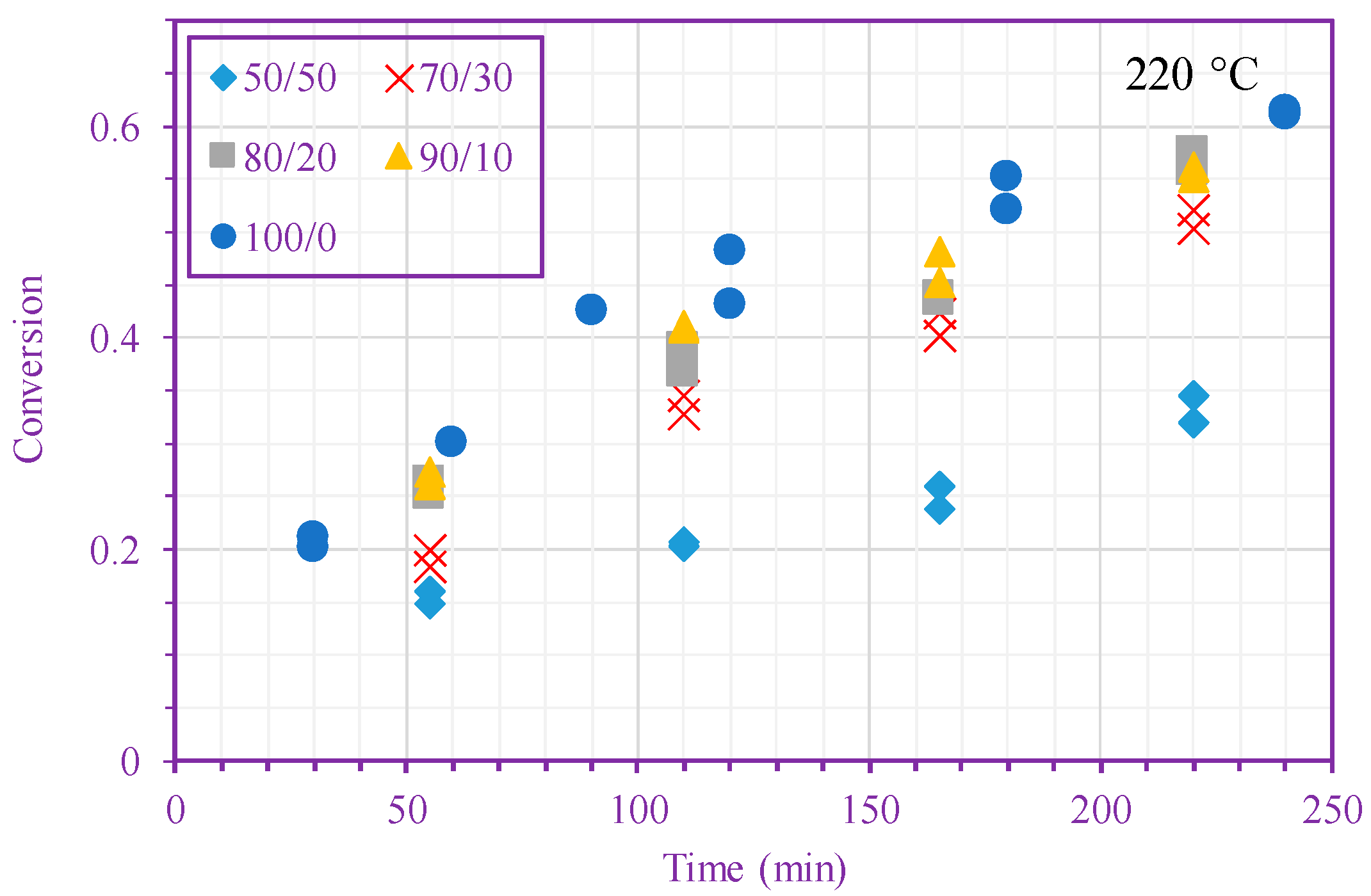
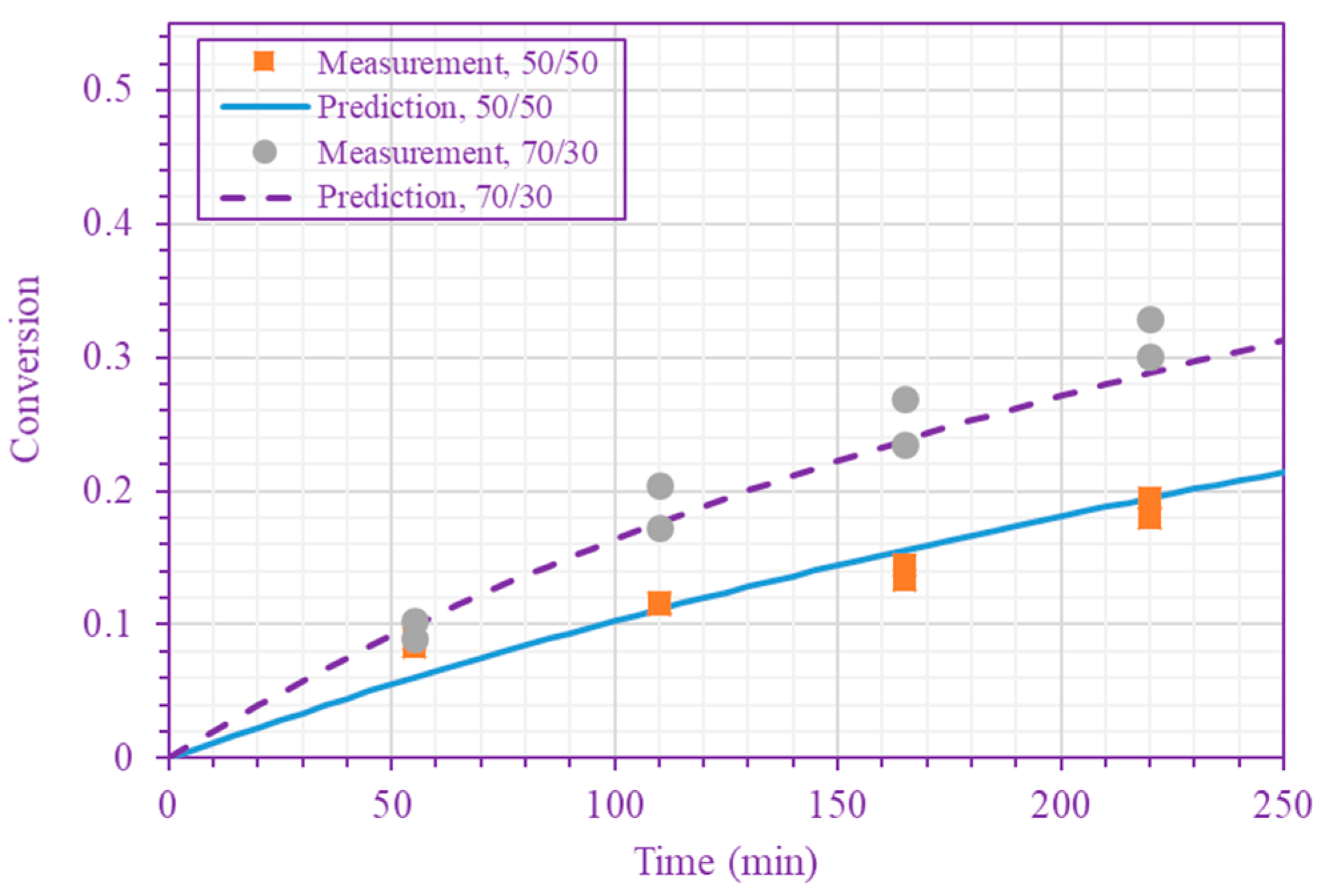
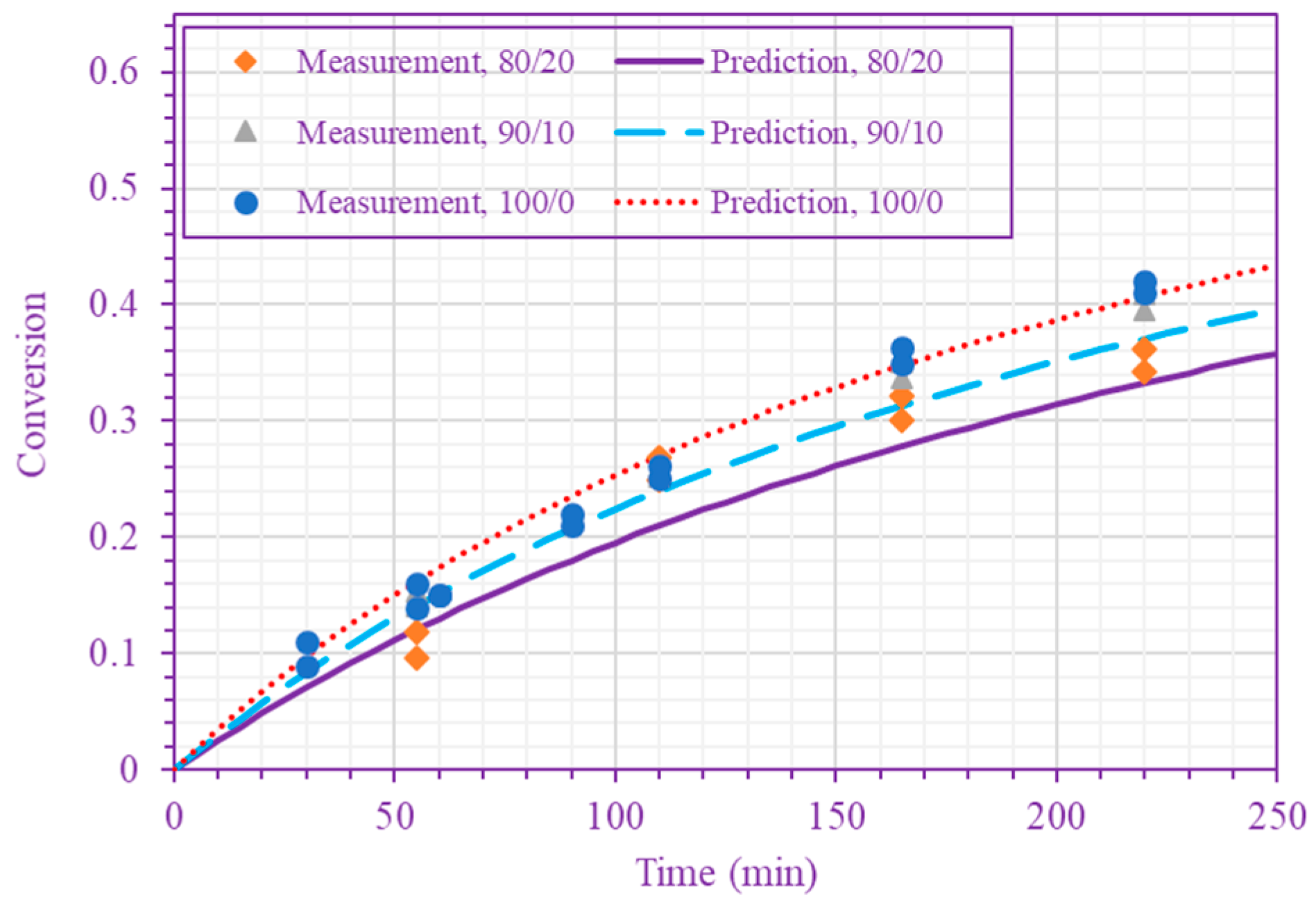
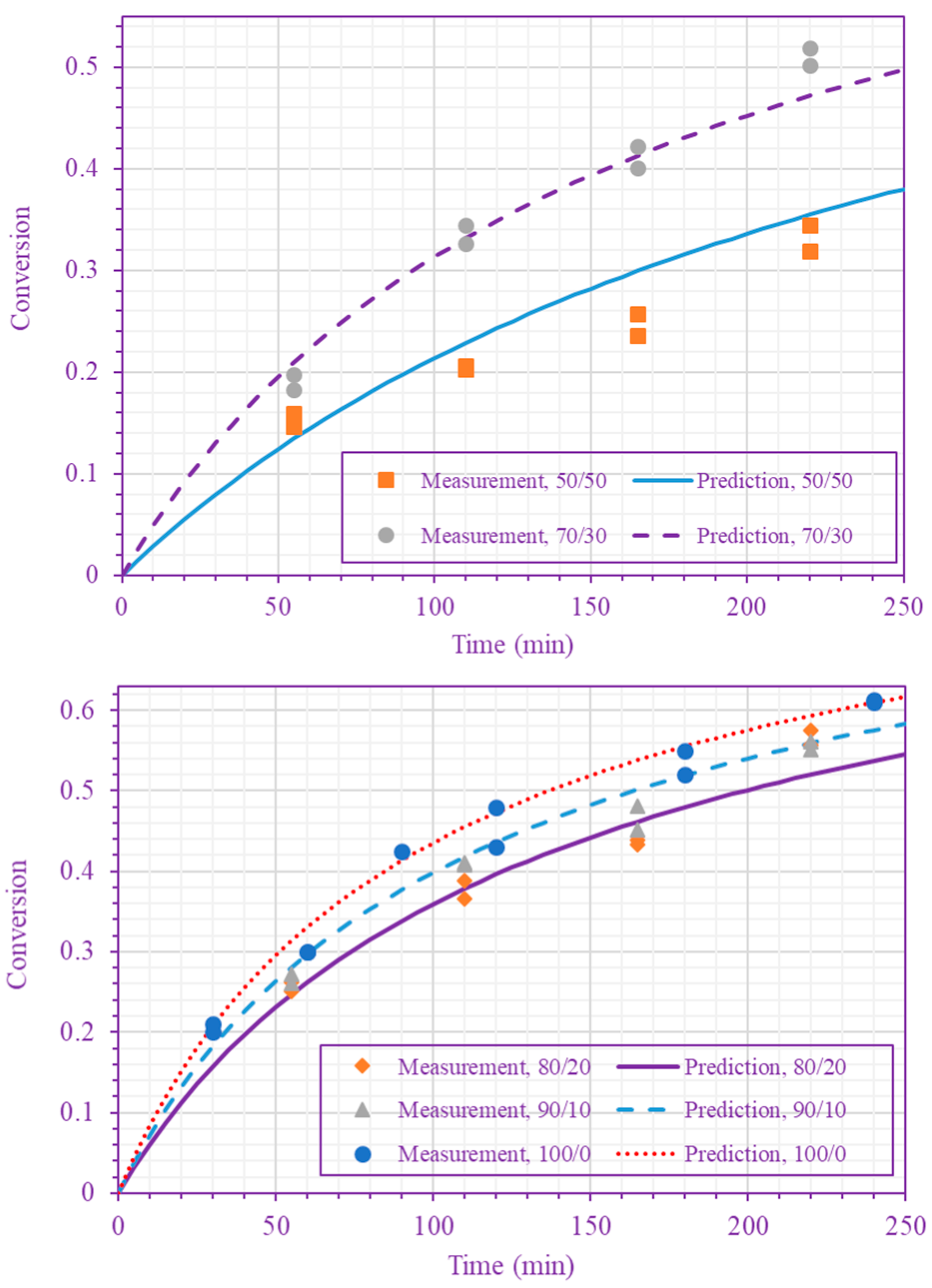
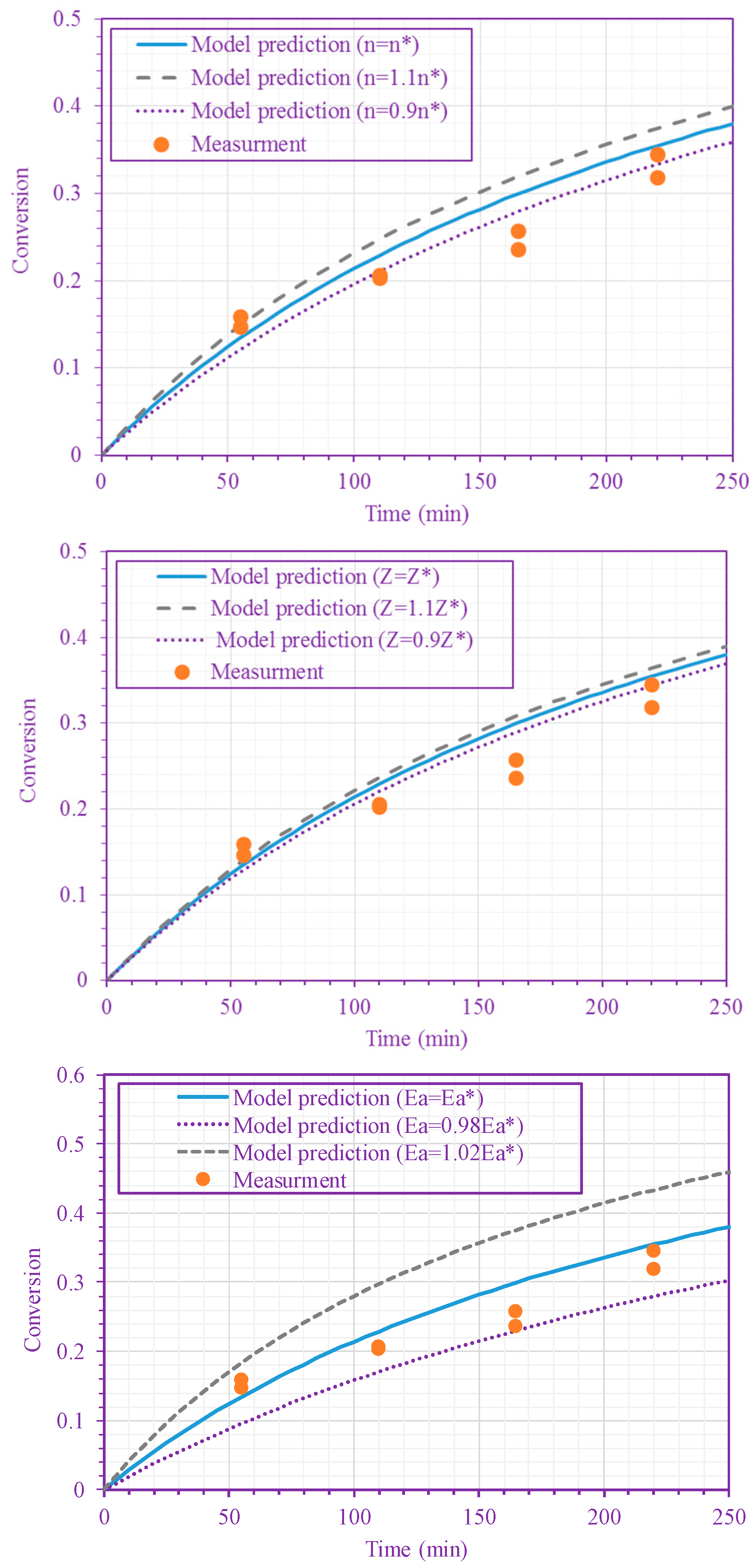
3. Results and Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
|
References
- Adelnia, H.; Gavgani, J.N.; Riazi, H.; Bidsorkhi, H.C. Transition behavior, su characteristics and film formation of functionalized poly (methyl methacrylate-co-butyl acrylate) particles. Prog. Org. Coat. 2014, 77, 1826–1833. [Google Scholar] [CrossRef]
- Adelnia, H.; Riazi, H.; Saadat, Y.; Hosseinzadeh, S. Synthesis of monodisperse anionic submicron polystyrene particles by stabilizer-free dispersion polymerization in alcoholic media. Colloid Polym. Sci. 2013, 291, 1741–1748. [Google Scholar] [CrossRef]
- Yasuda, H.; Wang, C. Plasma polymerization investigated by the substrate temperature dependence. J. Polym. Sci. A1 1985, 23, 87–106. [Google Scholar] [CrossRef]
- Price, G.J.; Norris, D.J.; West, P.J. Polymerization of methyl methacrylate initiated by ultrasound. Macromolecules 1992, 25, 6447–6454. [Google Scholar] [CrossRef]
- Decker, C. The use of UV irradiation in polymerization. Polym. Int. 1998, 45, 133–141. [Google Scholar] [CrossRef]
- Marans, N.S. Ionizing Irradiation Polymerization of Trioxane with Copolymerizable Stabilizing Comonomers. Google Patents. U.S. Patent 3,366,561, 30 January 1968. [Google Scholar]
- Riazi, H.; Mohammadi, N.; Mohammadi, H. Emulsion copolymerization of methyl methacrylate/butyl acrylate/iodine system to monosize rubbery nanoparticles containing iodine and triiodide mixture. Ind. Eng. Chem. Res. 2013, 52, 2449–2456. [Google Scholar] [CrossRef]
- Riazi, H.; Arabi Shamsabadi, A.; Grady, M.; Rappe, A.; Soroush, M. Experimental and Macroscopic Mechanistic Modeling Studies of the Methyl Acrylate Self Initiation Reaction; AIChE Annual Meeting: Minneapolis, MN, USA, 2017. [Google Scholar]
- Riazi, H.; Arabi Shamsabadi, A.; Grady, M.; Rappe, A.M.; Soroush, M. Experimental and Theoretical Study of the Self-Initiation Reaction of Methyl Acrylate in Free-Radical Polymerization. Ind. Eng. Chem. Res. 2017. [Google Scholar] [CrossRef]
- Alsharaeh, E.H.; Ibrahim, Y.M.; El-Shall, M.S. Direct evidence for the gas phase thermal polymerization of styrene. Determination of the initiation mechanism and structures of the early oligomers by ion mobility. J. Am. Chem. Soc. 2005, 127, 6164–6165. [Google Scholar] [PubMed]
- Pan, G.; Sudol, E.D.; Dimonie, V.L.; El-Aasser, M.S. Thermal self-initiation of styrene in the presence of TEMPO radicals: Bulk and miniemulsion. J. Polym. Sci. A1 2004, 42, 4921–4932. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Davis, T.P. Handbook of Radical Polymerization; John Wiley & Sons: Hoboken, NJ, USA, 2003; p. 878. [Google Scholar]
- Vana, P.; Barner-Kowollik, C.; Davis, T.P.; Matyjaszewski, K. Radical Polymerization; Encyclopedia of Polymer Science and Technology; John Wiely & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Moad, G.; Solomon, D.H. The Chemistry of Radical Polymerization, 2rd ed.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 108–109. [Google Scholar]
- Wang, W.; Hutchinson, R.A. Free-Radical Acrylic Polymerization Kinetics at Elevated Temperatures. Chem. Eng. Technol. 2010, 33, 1745–1753. [Google Scholar] [CrossRef]
- Srinivasan, S.; Kalfas, G.; Petkovska, V.I.; Bruni, C.; Grady, M.C.; Soroush, M. Experimental study of the spontaneous thermal homopolymerization of methyl and n-butyl acrylate. J. Appl. Polym. Sci. 2010, 118, 1898–1909. [Google Scholar] [CrossRef]
- Liu, S.; Srinivasan, S.; Grady, M.C.; Soroush, M.; Rappe, A.M. Computational study of cyclohexanone-monomer co-initiation mechanism in thermal homo-polymerization of methyl acrylate and methyl methacrylate. J. Phys. Chem. A 2012, 116, 5337–5348. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Yagci, Y. Handbook of Radical Vinyl Polymerization; CRC Press: Boca Raton, FL, USA, 1998; p. 48. [Google Scholar]
- Mattamal, G.J. US FDA perspective on the regulations of medical-grade polymers: Cyanoacrylate polymer medical device tissue adhesives. Exp. Rev. Med. Devices 2008, 5, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Frommelt, H. Polymers for medical applications. Macromol. Symp. 1987, 12, 281–301. [Google Scholar] [CrossRef]
- Jin, T.; Zhang, H. Biodegradable polylactic acid polymer with nisin for use in antimicrobial food packaging. J. Food Sci. 2008, 73, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulou, M.; Beyazit, S.; Nestora, S.; Haupt, K.; Bui, B.T.S. Initiator-free synthesis of molecularly imprinted polymers by polymerization of self-initiated monomers. Polymer 2015, 66, 43–51. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, Y.; Chen, L.; Fu, Z.; Shi, Y. Thermal Self-Initiation in Stable Free-Radical Polymerization of Styrene. Polym. J. 2009, 41, 954–960. [Google Scholar] [CrossRef]
- Peck, A.N.; Hutchinson, R.A. Secondary reactions in the high-temperature free radical polymerization of butyl acrylate. Macromolecules 2004, 37, 5944–5951. [Google Scholar] [CrossRef]
- Yu, X.; Pfaendtner, J.; Broadbelt, L.J. Ab initio study of acrylate polymerization reactions: Methyl methacrylate and methyl acrylate propagation. J. Phys. Chem. A 2008, 112, 6772–6782. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, N.; Liu, S.; Srinivasan, S.; Grady, M.C.; Rappe, A.M.; Soroush, M. Theoretical study of intermolecular chain transfer to polymer reactions of alkyl acrylates. Ind. Eng. Chem. Res. 2015, 54, 4148–4165. [Google Scholar] [CrossRef]
- Hui, C.M.; Dang, A.; Chen, B.; Yan, J.; Konkolewicz, D.; He, H.; Ferebee, R.; Bockstaller, M.R.; Matyjaszewski, K. Effect of thermal self-initiation on the synthesis, composition, and properties of particle brush materials. Macromolecules 2014, 47, 5501–5508. [Google Scholar] [CrossRef]
- Shalati, M.D.; Scott, R.M. Thermal polymerization of dimethylaminoethyl methacrylate. Macromolecules 1975, 8, 127–130. [Google Scholar] [CrossRef]
- Zorn, A.M.; Junkers, T.; Barner-Kowollik, C. Synthesis of a Macromonomer Library from High-Temperature Acrylate Polymerization. Macromol. Rapid Commun. 2009, 30, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Junkers, T.; Bennet, F.; Koo, S.P.; Barner-Kowollik, C. Self-directed formation of uniform unsaturated macromolecules from acrylate monomers at high temperatures. J. Polym. Sci. Pol. Chem. 2008, 46, 3433–3437. [Google Scholar] [CrossRef]
- Beckert, F.; Rostas, A.M.; Thomann, R.; Weber, S.; Schleicher, E.; Friedrich, C.; Mulhaupt, R. Self-initiated free radical grafting of styrene homo-and copolymers onto functionalized graphene. Macromolecules 2013, 46, 5488–5496. [Google Scholar] [CrossRef]
- Soroush, M.; Grady, M.C.; Kalfas, G.A. Free-radical polymerization at higher temperatures: Systems impacts of secondary reactions. Comput. Chem. Eng. 2008, 32, 2155–2167. [Google Scholar] [CrossRef]
- Meyer, T.; Keurentjes, J. Handbook of Polymer Reaction Engineering; Wiley-VCH: Weinheim, Germany, 2005; p. 169. [Google Scholar]
- Zhang, C.; Wang, X.; Liu, L.; Wang, Y.; Peng, X. Modeling the spontaneous initiation of the polymerization of methyl methacrylate. J. Mol. Model. 2008, 14, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Lee, M.W.; Grady, M.C.; Soroush, M.; Rappe, A.M. Self-initiation mechanism in spontaneous thermal polymerization of ethyl and n-butyl acrylate: A theoretical study. J. Phys. Chem. A 2010, 114, 7975–7983. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Srinivasan, S.; Tao, J.; Grady, M.C.; Soroush, M.; Rappe, A.M. Modeling spin-forbidden monomer self-initiation reactions in spontaneous free-radical polymerization of acrylates and methacrylates. J. Phys. Chem. A 2014, 118, 9310–9318. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Lee, M.W.; Grady, M.C.; Soroush, M.; Rappe, A.M. Computational study of the self-initiation mechanism in thermal polymerization of methyl acrylate. J. Phys. Chem. A 2009, 113, 10787–10794. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Srinivasan, S.; Grady, M.C.; Soroush, M.; Rappe, A.M. Backbiting and β-scission reactions in free-radical polymerization of methyl acrylate. Int. J. Quantum Chem. 2014, 114, 345–360. [Google Scholar] [CrossRef]
- Shamsabadi, A.A.; Moghadam, N.; Srinivasan, S.; Corcoran, P.; Grady, M.C.; Rappe, A.M.; Soroush, M. Study of n-Butyl Acrylate Self-Initiation Reaction Experimentally and via Macroscopic Mechanistic Modeling. Processes 2016, 4, 15. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H.; Grulke, E.A.; Abe, A.; Bloch, D.R. Polymer Handbook; Wiley: New York, NY, USA, 1989; p. II/120. [Google Scholar]
- Moghadam, N.; Srinivasan, S.; Grady, M.C.; Rappe, A.M.; Soroush, M. Theoretical study of chain transfer to solvent reactions of alkyl acrylates. J. Phys. Chem. A 2014, 118, 5474–5487. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riazi, H.; Shamsabadi, A.A.; Corcoran, P.; Grady, M.C.; Rappe, A.M.; Soroush, M. On the Thermal Self-Initiation Reaction of n-Butyl Acrylate in Free-Radical Polymerization. Processes 2018, 6, 3. https://doi.org/10.3390/pr6010003
Riazi H, Shamsabadi AA, Corcoran P, Grady MC, Rappe AM, Soroush M. On the Thermal Self-Initiation Reaction of n-Butyl Acrylate in Free-Radical Polymerization. Processes. 2018; 6(1):3. https://doi.org/10.3390/pr6010003
Chicago/Turabian StyleRiazi, Hossein, Ahmad Arabi Shamsabadi, Patrick Corcoran, Michael C. Grady, Andrew M. Rappe, and Masoud Soroush. 2018. "On the Thermal Self-Initiation Reaction of n-Butyl Acrylate in Free-Radical Polymerization" Processes 6, no. 1: 3. https://doi.org/10.3390/pr6010003
APA StyleRiazi, H., Shamsabadi, A. A., Corcoran, P., Grady, M. C., Rappe, A. M., & Soroush, M. (2018). On the Thermal Self-Initiation Reaction of n-Butyl Acrylate in Free-Radical Polymerization. Processes, 6(1), 3. https://doi.org/10.3390/pr6010003