
Mining Biosynthetic Gene Clusters of Bacillus subtilis MGE 2012 Using Whole Genome Sequencing



Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Assembly and Annotation
2.2. Phylogenetic Analysis
2.3. Prediction of Biosynthetic Gene Cluster
3. Results
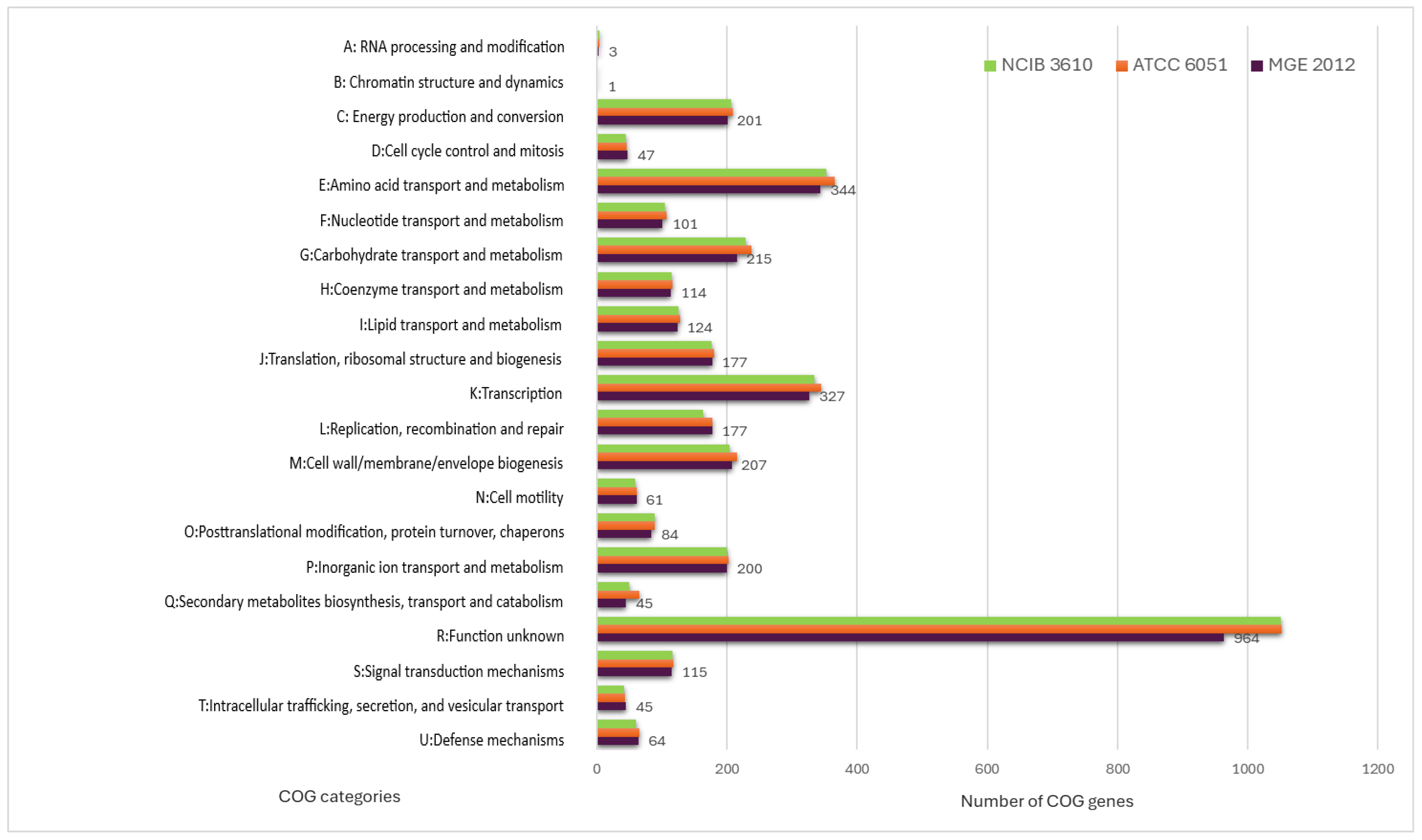
3.1. Genome Annotation
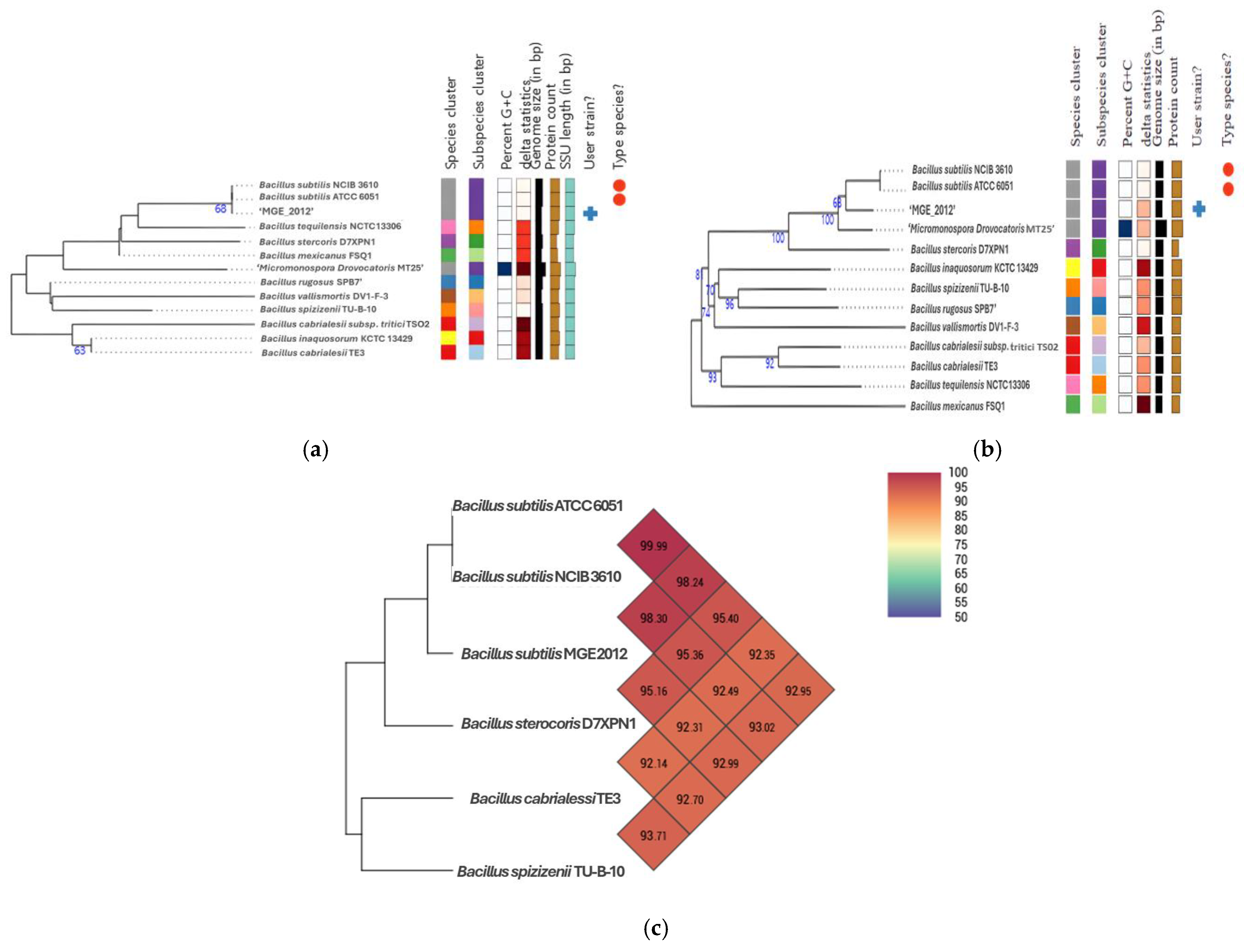
3.2. Phylogenetic Relationship of B. subtilis MGE2012
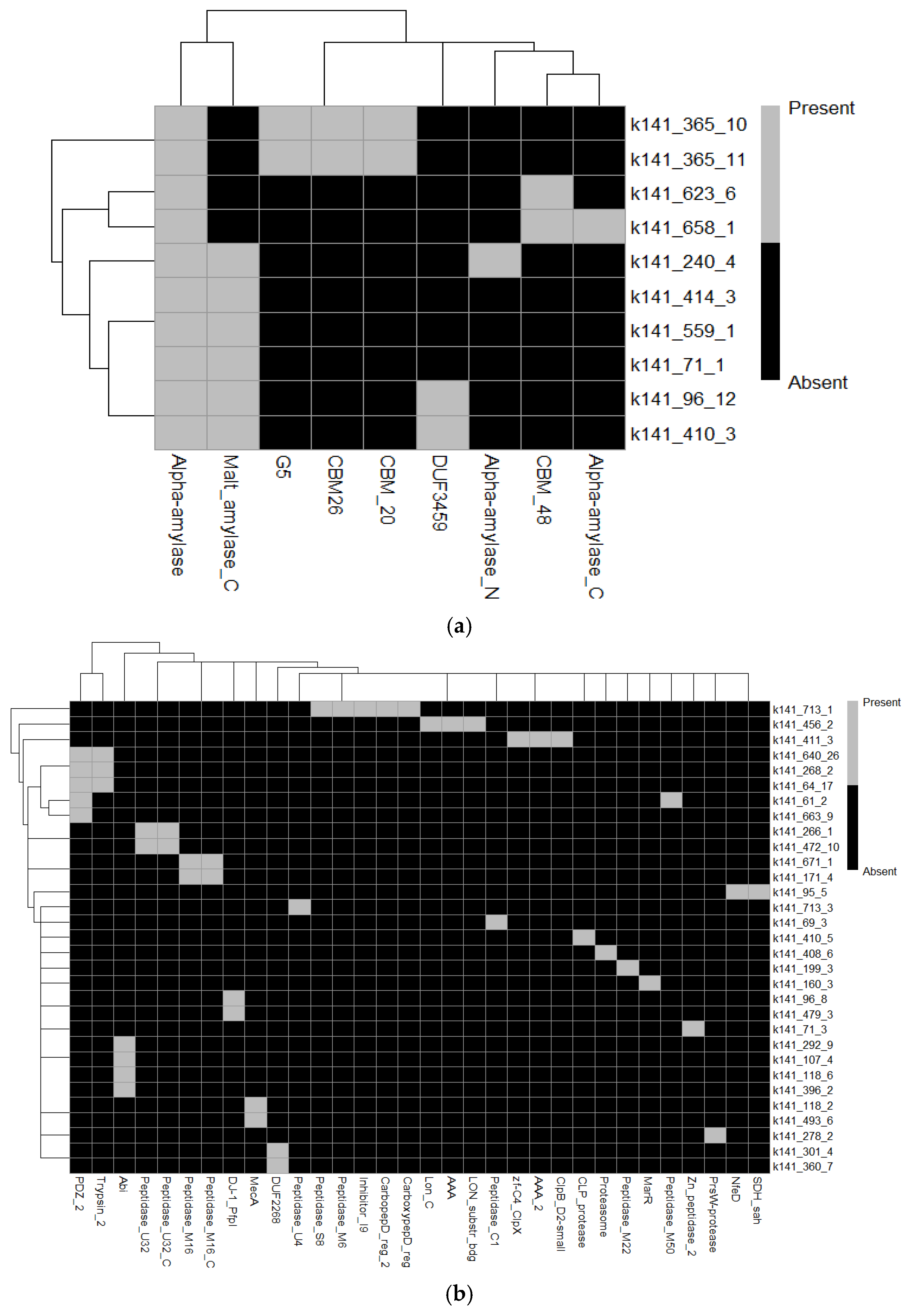
3.3. Genomic Identification and Functional Annotation of Amylases and Proteases
3.4. Biosynthetic Gene Clusters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Iqbal, S.; Begum, F.; Rabaan, A.A.; Aljeldah, M.; Al Shammari, B.R.; Alawfi, A.; Alshengeti, A.; Sulaiman, T.; Khan, A. Classification and Multifaceted Potential of Secondary Metabolites Produced by Bacillus subtilis Group: A Comprehensive Review. Molecules 2023, 28, 927. [Google Scholar] [CrossRef] [PubMed]
- Medeot, D.; Sannazzaro, A.; Estrella, M.J.; Torres Tejerizo, G.; Contreras-Moreira, B.; Pistorio, M.; Jofré, E. Unraveling the Genome of Bacillus Velezensis MEP218, a Strain Producing Fengycin Homologs with Broad Antibacterial Activity: Comprehensive Comparative Genome Analysis. Sci. Rep. 2023, 13, 22168. [Google Scholar] [CrossRef]
- Fira, D.; Dimkić, I.; Berić, T.; Lozo, J.; Stanković, S. Biological Control of Plant Pathogens by Bacillus Species. J. Biotechnol. 2018, 285, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Penha, R.O.; Vandenberghe, L.P.S.; Faulds, C.; Soccol, V.T.; Soccol, C.R. Bacillus Lipopeptides as Powerful Pest Control Agents for a More Sustainable and Healthy Agriculture: Recent Studies and Innovations. Planta 2020, 251, 70. [Google Scholar] [CrossRef]
- Chen, I.; Christie, P.J.; Dubnau, D. The Ins and Outs of DNA Transfer in Bacteria. Science 2005, 310, 1456–1460. [Google Scholar] [CrossRef] [PubMed]
- Benoit, I.; van den Esker, M.H.; Patyshakuliyeva, A.; Mattern, D.J.; Blei, F.; Zhou, M.; Dijksterhuis, J.; Brakhage, A.A.; Kuipers, O.P.; de Vries, R.P.; et al. Bacillus subtilis Attachment to Aspergillus niger Hyphae Results in Mutually Altered Metabolism. Environ. Microbiol. 2015, 17, 2099–2113. [Google Scholar] [CrossRef]
- Jones, S.E.; Paynich, M.L.; Kearns, D.B.; Knight, K.L. Protection from Intestinal Inflammation by Bacterial Exopolysaccharides. J. Immunol. 2014, 192, 4813–4820. [Google Scholar] [CrossRef]
- Kearns, D.B.; Losick, R. Swarming Motility in Undomesticated Bacillus subtilis. Mol. Microbiol. 2003, 49, 581–590. [Google Scholar] [CrossRef]
- Tjalsma, H.; Antelmann, H.; Jongbloed, J.D.H.; Braun, P.G.; Darmon, E.; Dorenbos, R.; Dubois, J.-Y.F.; Westers, H.; Zanen, G.; Quax, W.J.; et al. Proteomics of Protein Secretion by Bacillus subtilis: Separating the “Secrets” of the Secretome. Microbiol. Mol. Biol. Rev. 2004, 68, 207–233. [Google Scholar] [CrossRef]
- Beauregard, P.B.; Chai, Y.; Vlamakis, H.; Losick, R.; Kolter, R. Bacillus subtilis Biofilm Induction by Plant Polysaccharides. Proc. Natl. Acad. Sci. USA 2013, 110, E1621–E1630. [Google Scholar] [CrossRef]
- Kovács, Á.T. Bacillus subtilis. Trends Microbiol. 2019, 27, 724–725. [Google Scholar] [CrossRef]
- Lee, J.-H.; Moon, G.-S. Development of Functional Material by Using Bacillus subtilis Harboring α-Amylase and Protease Enzyme Activity. Curr. Top. Lact. Acid Bact. Probiotics 2023, 9, 81–85. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations Using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. EggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A Database Tandem for Fast and Reliable Genome-Based Classification and Nomenclature of Prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Öker, M.G. Genome Sequence-Based Species Delimitation with Confidence Intervals and Improved Distance Functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast Genome and Metagenome Distance Estimation Using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and Rapid Annotation of Ribosomal RNA Genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Chalita, M.; Kim, Y.O.; Park, S.; Oh, H.S.; Cho, J.H.; Moon, J.; Baek, N.; Moon, C.; Lee, K.; Yang, J.; et al. EzBioCloud: A Genome-Driven Database and Platform for Microbiome Identification and Discovery. Int. J. Syst. Evol. Microbiol. 2024, 74, 6421. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.N.; et al. AntiSMASH 7.0: New and Improved Predictions for Detection, Regulation, Chemical Structures and Visualisation. Nucleic Acids Res. 2023, 51, W46–W50. [Google Scholar] [CrossRef]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef]
- Farris, J.S. Estimating Phylogenetic Trees from Distance Matrices. Am. Nat. 1972, 106, 645–668. [Google Scholar] [CrossRef]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: An Improved Algorithm and Software for Calculating Average Nucleotide Identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Foster, K.R.; Bell, T. Competition, Not Cooperation, Dominates Interactions among Culturable Microbial Species. Curr. Biol. 2012, 22, 1845–1850. [Google Scholar] [CrossRef]
- Straight, P.D.; Willey, J.M.; Kolter, R. Interactions between Streptomyces coelicolor and Bacillus subtilis: Role of Surfactants in Raising Aerial Structures. J. Bacteriol. 2006, 188, 4918–4925. [Google Scholar] [CrossRef] [PubMed]
- Romero, D.; Traxler, M.F.; López, D.; Kolter, R. Antibiotics as Signal Molecules. Chem. Rev. 2011, 111, 5492–5505. [Google Scholar] [CrossRef]
- Linares, J.F.; Gustafsson, I.; Baquero, F.; Martinez, J.L. Antibiotics as Intermicrobiol Signaling Agents Instead of Weapons. Proc. Natl. Acad. Sci. USA 2006, 103, 19484–19489. [Google Scholar] [CrossRef]
- Vaz Jauri, P.; Bakker, M.G.; Salomon, C.E.; Kinkel, L.L. Subinhibitory Antibiotic Concentrations Mediate Nutrient Use and Competition among Soil Streptomyces. PLoS ONE 2013, 8, e81064. [Google Scholar] [CrossRef]
- Fan, B.; Blom, J.; Klenk, H.P.; Borriss, R. Bacillus amyloliquefaciens, Bacillus velezensis, and Bacillus siamensis Form an “Operational Group B. amyloliquefaciens” within the B. subtilis Species Complex. Front. Microbiol. 2017, 8, 22. [Google Scholar] [CrossRef]
- Ongena, M.; Jacques, P. Bacillus Lipopeptides: Versatile Weapons for Plant Disease Biocontrol. Trends Microbiol. 2008, 16, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, K.J.; Bleich, R.M.; Santa Maria, K.C.; Allen, S.E.; Farag, S.; Shank, E.A.; Bowers, A.A. Large-Scale Bioinformatics Analysis of Bacillus Genomes Uncovers Conserved Roles of Natural Products in Bacterial Physiology. mSystems 2017, 2, e00040-17. [Google Scholar] [CrossRef]
- Harwood, C.R.; Mouillon, J.M.; Pohl, S.; Arnau, J. Secondary Metabolite Production and the Safety of Industrially Important Members of the Bacillus subtilis Group. FEMS Microbiol. Rev. 2018, 42, 721–738. [Google Scholar] [CrossRef] [PubMed]
- Zha, W.; Rubin-Pitel, S.B.; Zhao, H. Characterization of the Substrate Specificity of PhlD, a Type III Polyketide Synthase from Pseudomonas fluorescens. J. Biol. Chem. 2006, 281, 32036–32047. [Google Scholar] [CrossRef]
- Kiesewalter, H.T.; Lozano-Andrade, C.N.; Wibowo, M.; Strube, M.L.; Maróti, G.; Snyder, D.; Jørgensen, T.S.; Larsen, T.O.; Cooper, V.S.; Weber, T.; et al. Genomic and Chemical Diversity of Bacillus subtilis Secondary Metabolites against Plant Pathogenic Fungi. mSystems 2021, 6, e00770-20. [Google Scholar] [CrossRef]
- Grau, R.R.; De Oña, P.; Kunert, M.; Leñini, C.; Gallegos-Monterrosa, R.; Mhatre, E.; Vileta, D.; Donato, V.; Hölscher, T.; Boland, W.; et al. A Duo of Potassium-Responsive Histidine Kinases Govern the Multicellular Destiny of Bacillus subtilis. mBio 2015, 6, e00581. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, J.D.; Jumarie, C.; Cooper, D.G.; Laprade, R. Ionic Channels Induced by Surfactin in Planar Lipid Bilayer Membranes. BBA Biomembr. 1991, 1064, 13–23. [Google Scholar] [CrossRef]
- Heerklotz, H.; Seelig, J. Leakage and Lysis of Lipid Membranes Induced by the Lipopeptide Surfactin. Eur. Biophys. J. 2007, 36, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Sabaté, D.C.; Audisio, M.C. Inhibitory Activity of Surfactin, Produced by Different Bacillus subtilis Subsp. subtilis Strains, against Listeria monocytogenes Sensitive and Bacteriocin-Resistant Strains. Microbiol. Res. 2013, 168, 125–129. [Google Scholar] [CrossRef]
- Loiseau, C.; Schlusselhuber, M.; Bigot, R.; Bertaux, J.; Berjeaud, J.M.; Verdon, J. Surfactin from Bacillus subtilis Displays an Unexpected Anti-Legionella Activity. Appl. Microbiol. Biotechnol. 2015, 99, 5083–5093. [Google Scholar] [CrossRef]
- Gao, L.; Han, J.; Liu, H.; Qu, X.; Lu, Z.; Bie, X. Plipastatin and Surfactin Coproduction by Bacillus subtilis PB2-L and Their Effects on Microorganisms. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 2017, 110, 1007–1018. [Google Scholar] [CrossRef]
- Arjes, H.A.; Vo, L.; Dunn, C.M.; Willis, L.; DeRosa, C.A.; Fraser, C.L.; Kearns, D.B.; Huang, K.C. Biosurfactant-Mediated Membrane Depolarization Maintains Viability during Oxygen Depletion in Bacillus subtilis. Curr. Biol. 2020, 30, 1011–1022.e6. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wen, J.; Zhao, X.; Ding, J.; Qi, G. Surfactin: A Quorum-Sensing Signal Molecule to Relieve CCR in Bacillus amyloliquefaciens. Front. Microbiol. 2020, 11, 631. [Google Scholar] [CrossRef]
- Steinke, K.; Mohite, O.S.; Weber, T.; Kovács, Á.T. Phylogenetic Distribution of Secondary Metabolites in the Bacillus subtilis Species Complex. mSystems 2021, 6, e00057-21. [Google Scholar] [CrossRef]
- Umezawa, H.; Aoyagi, T.; Nishikiori, T.; Okuyama, A.; Yamagishi, Y.; Hamada, M.; Takeuchi, T. Plipastatins: New Inhibitors of Phospholipase A2, Produced by Bacillus cereus BMG302-FF67 I. Taxonomy, Production, Isolation and Preliminary Characterization. J. Antibiot. 1986, 39, 737–744. [Google Scholar] [CrossRef]
- Deleu, M.; Paquot, M.; Nylander, T. Fengycin Interaction with Lipid Monolayers at the Air-Aqueous Interface—Implications for the Effect of Fengycin on Biological Membranes. J. Colloid Interface Sci. 2005, 283, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Romero, D.; De Vicente, A.; Rakotoaly, R.H.; Dufour, S.E.; Veening, J.W.; Arrebola, E.; Cazorla, F.M.; Kuipers, O.P.; Paquot, M.; Pérez-García, A. The Iturin and Fengycin Families of Lipopeptides Are Key Factors in Antagonism of Bacillus subtilis toward Podosphaera fusca. Mol. Plant Microbe Interact. 2007, 20, 430–440. [Google Scholar] [CrossRef]
- Alvarez, F.; Castro, M.; Príncipe, A.; Borioli, G.; Fischer, S.; Mori, G.; Jofré, E. The Plant-Associated Bacillus amyloliquefaciens Strains MEP218 and ARP23 Capable of Producing the Cyclic Lipopeptides Iturin or Surfactin and Fengycin Are Effective in Biocontrol of Sclerotinia Stem Rot Disease. J. Appl. Microbiol. 2012, 112, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Falardeau, J.; Wise, C.; Novitsky, L.; Avis, T.J. Ecological and Mechanistic Insights Into the Direct and Indirect Antimicrobial Properties of Bacillus subtilis Lipopeptides on Plant Pathogens. J. Chem. Ecol. 2013, 39, 869–878. [Google Scholar] [CrossRef]
- Roy, A.; Mahata, D.; Paul, D.; Korpole, S.; Franco, O.L.; Mandal, S.M. Purification, Biochemical Characterization and Self-Assembled Structure of a Fengycin-like Antifungal Peptide from Bacillus thuringiensis Strain SM. Front. Microbiol. 2013, 4, 332. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Bie, X.; Lu, Z.; Lv, F.; Tao, Y.; Qu, X. Effects of Fengycin from Bacillus subtilis FmbJ on Apoptosis and Necrosis in Rhizopus Stolonifer. J. Microbiol. 2014, 52, 675–680. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, C. Fengycins, Cyclic Lipopeptides from Marine Bacillus subtilis Strains, Kill the Plant-Pathogenic Fungus Magnaporthe grisea by Inducing Reactive Oxygen Species Production and Chromatin Condensation. Appl. Environ. Microbiol. 2018, 84, e00445-18. [Google Scholar] [CrossRef]
- Mayerl, F.; Fisher, S.; Pirnik, D.; Aklonis, C.; Dean, L.; Meyers, E.; Fernandes, P. Bacillaene, a Novel Inhibitor of Procaryotic Protein Synthesis Produced by Bacillus subtilis: Production, Taxonomy, Isolation, Physico-Chemical Characterization and Biological Activity. J. Antibiot. 1995, 48, 997–1003. [Google Scholar] [CrossRef]
- Müller, S.; Strack, S.N.; Hoefler, B.C.; Straight, P.D.; Kearns, D.B.; Kirby, J.R. Bacillaene and Sporulation Protect Bacillus subtilis from Predation by Myxococcus Xanthus. Appl. Environ. Microbiol. 2014, 80, 5603–5610. [Google Scholar] [CrossRef]
- May, J.J.; Wendrich, T.M.; Marahiel, M.A. The Dhb Operon of Bacillus subtilis Encodes the Biosynthetic Template for the Catecholic Siderophore 2,3-Dihydroxybenzoate-Glycine-Threonine Trimeric Ester Bacillibactin. J. Biol. Chem. 2001, 276, 7209–7217. [Google Scholar] [CrossRef]
- Stein, T.; Düsterhus, S.; Stroh, A.; Entian, K.D. Subtilosin Production by Two Bacillus subtilis Subspecies and Variance of the Sbo-Alb Cluster. Appl. Environ. Microbiol. 2004, 70, 2349–2353. [Google Scholar] [CrossRef] [PubMed]
- Kawulka, K.; Sprules, T.; McKay, R.T.; Mercier, P.; Diaper, C.M.; Zuber, P.; Vederas, J.C. Structure of Subtilosin A, an Antimicrobial Peptide from Bacillus subtilis with Unusual Posttranslational Modifications Linking Cysteine Sulfurs to α-Carbons of Phenylalanine and Threonine. J. Am. Chem. Soc. 2003, 125, 4726–4727. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.M.; Zheng, G.; Zuber, P. Dual Control of Sbo-Alb Operon Expression by the Spo0 and ResDE Systems of Signal Transduction under Anaerobic Conditions in Bacillus subtilis. J. Bacteriol. 2000, 182, 3274–3277. [Google Scholar] [CrossRef]
- Chen, X.H.; Scholz, R.; Borriss, M.; Junge, H.; Mögel, G.; Kunz, S.; Borriss, R. Difficidin and Bacilysin Produced by Plant-Associated Bacillus amyloliquefaciens Are Efficient in Controlling Fire Blight Disease. J. Biotechnol. 2009, 140, 38–44. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Type | From | To | Most Similar Known Cluster | Similarity |
|---|---|---|---|---|---|
| 48.1 | RiPP-like | 832 | 9194 | - | - |
| 59.1 | NRPS | 1 | 4553 | - | - |
| 137.1 | T3PKS | 1 | 4380 | - | - |
| 152.1 | NRPS | 1 | 10,195 | bacillibactin | 42% |
| 170.1 | other | 1 | 25,018 | bacilysin | 100% |
| 218.1 | betalactone | 1 | 3916 | fengycin | 33% |
| 224.1 | NRPS-like | 1 | 3118 | plipastatin | 23% |
| 237.1 | NRPS | 1 | 18,801 | surfactin | 30% |
| 254.1 | NRPS | 1 | 6562 | plipastatin | 38% |
| 287.1 | terpene | 1 | 4320 | ||
| 320.1 | NRPS | 1 | 3789 | surfactin | 8% |
| 357.1 | sactipeptide | 1 | 18,018 | subtilosin A | 87% |
| 467.1 | NRPS | 1 | 7694 | plipastatin | 38% |
| 492.1 | Terpene | 2012 | 14,459 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Bashir, H.H.; Hwang, J.; Moon, G.-S. Mining Biosynthetic Gene Clusters of Bacillus subtilis MGE 2012 Using Whole Genome Sequencing. Processes 2025, 13, 1503. https://doi.org/10.3390/pr13051503
Kim J, Bashir HH, Hwang J, Moon G-S. Mining Biosynthetic Gene Clusters of Bacillus subtilis MGE 2012 Using Whole Genome Sequencing. Processes. 2025; 13(5):1503. https://doi.org/10.3390/pr13051503
Chicago/Turabian StyleKim, Jiyoun, Hafiza Hira Bashir, Joon Hwang, and Gi-Seong Moon. 2025. "Mining Biosynthetic Gene Clusters of Bacillus subtilis MGE 2012 Using Whole Genome Sequencing" Processes 13, no. 5: 1503. https://doi.org/10.3390/pr13051503
APA StyleKim, J., Bashir, H. H., Hwang, J., & Moon, G.-S. (2025). Mining Biosynthetic Gene Clusters of Bacillus subtilis MGE 2012 Using Whole Genome Sequencing. Processes, 13(5), 1503. https://doi.org/10.3390/pr13051503