Abstract
Polygalacturonic acid (PGA), derived from the natural plant polysaccharide, pectin, has been suggested as a biomaterial for implantable medical devices and tissue engineering; particularly in the field of bone implant materials. As a negatively charged polysaccharide, PGA can be considered similar to hyaluronic acid, a component of the extracellular matrix (ECM). PGA-based biomaterials may therefore exhibit favorable biocompatibility with surface chemistry mimicking the natural ECM. In this study, we synthesized semi-interpenetrating polymer networks (SIPNs) incorporating PGA, and conducted physical characterization and in vitro biocompatibility studies. Biocompatibility testing revealed the SIPNs to be cytocompatible, with the PGA component conferring some resistance to the adherence of the macrophage cell line RAW264.7. In addition, SIPNs did not support the fusion of primary murine macrophages into foreign body giant cells (FBGCs). Macrophage adherence and FBGC formation on implanted biomaterial surfaces are important events in the progression of a foreign body response. Our in vitro studies suggest that PGA-based materials may offer desirable biocompatibility profiles, holding promise for future clinical applications.
1. Introduction
Biomaterials for implantable medical devices are designed to exist in contact with tissues of the human body without eliciting harm. Traditionally, the term “biocompatible” with reference to biomaterials was viewed on the basis of the material being non-irritant, non-toxic, non-thrombogenic, non-carcinogenic, and so on []. Essentially, the material was required to be “bio-inert”. There has since been a re-evaluation of this position and the definition of truly biocompatible materials has somewhat shifted. Rather than simply being “tolerated” by the body, recent thinking has placed an emphasis on biomaterial performance in terms of bio-integration, tissue regeneration, or the desired modulation of biological processes. This introduces the concept of a dynamic host/material interaction within the microenvironment of the implant. An ideal biomaterial would be able to reproduce the complex functionality of the natural extracellular matrix (ECM), providing an environment for the desired adhesion of an appropriate cell type, and for the desired cellular function [,].
Ratner has put forward a definition of biocompatibility as “the ability of a material to locally trigger and guide non-fibrotic wound healing, reconstruction and tissue integration”, whereas previous thinking on biocompatibility would be best viewed as “biotolerability”, and this defined by Ratner as “the ability of a material to reside in the body for long periods of time with only low degrees of inflammatory reaction” [].
A major factor in determining the biocompatibility (or in fact the biotolerability) of an implanted biomaterial is its propensity to evoke an immunological and inflammatory reaction known as the foreign body response (FBR). The FBR describes the dynamic cascade of molecular and cellular events occurring subsequent to material implantation. Inflammatory cells, in particular macrophages, migrate to the peri-implant area, adhere to the biomaterial surface, and fuse to form Foreign Body Giant Cells (FBGCs) [,]. It is the presence of these large, multinucleated FBGCs that is the hallmark of the FBR []. FBGCs express an armory of potent enzymes and reactive oxygen species (ROS) that are released at the biomaterial/tissue interface during a process described as “frustrated phagocytosis”, and can result in considerable damage to both the structural and functional integrity of the device [,]. A notable example is clinical device failure of cardiac pacemakers due to damage of their polyurethane-coated pacing leads [,]. Adherent macrophages and FBGCs were found to be responsible for the in vivo cracking, with macrophage-derived enzymes facilitating polymer degradation [,,]. Chemokines, such as interleukin-1 (IL-1) are also expressed, attracting fibroblasts to the implant site and culminating in the laying down of a fibrous, avascular capsule surrounding the device and isolating it from neighboring tissues [].
Towards the development of truly biocompatible materials, much focus has been placed on elucidating the biomolecular events that orchestrate the FBR in order to identify a “druggable” target, or in the design of biomaterials with the capability to modulate this host response. For example, the inhibition of Rac1, a GTPase important in cell motility, attenuates FBGC formation [], while incorporating anti-inflammatory or anti-fibrotic drugs into a the biomaterial, or as a coating, has also been investigated [,,]. Materials-based approaches have included the development of nano-patterned material surfaces which can prevent the process of macrophage fusion [,]. Zwitterionic hydrogels have also been reported to attenuate the FBR in vivo [].
In addition to improvements in biocompatibility, an aspect of material design must also be afforded to the intended application and site of implantation. A notable example lies with biomaterials for orthopedic application, where strength, osseointegration and/or osteogensis, are desirable for the appropriate functioning of the material and longevity of the implanted device. A failure of the biomaterial to osseointegrate can result in poor performance due to aseptic loosening of a total joint replacement or marginal bone loss around oral implants; the latter suggested as a result of a provoked foreign body reaction [,]. For orthopedic devices, metallic materials (including stainless steel and titanium alloys) have been widely used due to their physical strength, while functional polymeric coatings, such as hydrogels, have been investigated as a means to improve biocompatibility, prevent infection, or improve osseointegration [,,,].
Naturally occurring polysaccharides have shown immense potential in the design of biomaterials [,,,], and materials derived from the plant polysaccharide, pectin (polygalacturonic acid), are gaining momentum [,,,,,,,]. Pectin is a heterosaccharide found in the plant cell wall, and consists mainly of D-galacturonic acid residues in α-(1–4) chains. It is a water-soluble polyanionic polysaccharide and plays an important role in the formation of ionic interactions with proteins via calcium ions. Pectins are widely used in the food industry as thickeners in jams and jellies, and commercially are obtained from apple pomace, a waste product of the fruit juice industry making it an inexpensive, abundant, and renewable resource. As a biomaterial, pectin has shown immunoregulatory, antibacterial, anti-inflammatory, anti-tumor, and antioxidant activities []. For bone tissue engineering, pectin is able to trigger the growth and differentiation of osteoblasts and provide an appropriate microenvironment for the regeneration of mineralized bone. Preosteoblastic cells immobilized inside pectin-based materials are able to differentiate into the osteoblastic phenotype and will synthesize new ECM collagen fibers, resulting in complex and natural new bone tissue []. In addition, fully differentiated human osteoblasts can adhere and proliferate on the surface of pectin-based scaffolds []. The ability of pectin to influence the behavior of osteoblasts is likely due to its glycomolecule structure. Glycomolecules naturally exist in bone tissue, for example, biglycan which is found in the bone ECM, and which is needed for osteoblast differentiation [,]. Such properties of pectin-derived molecules could be exploited in the field of orthopedic or dental biomaterials to obtain adequate osseotintegration of implants, the surface of which must be compatible with biological phases of osteoblastic development (proliferation and differentiation), yet not elicit an inflammatory or immune reaction, namely the FBR. Kokkonen et al. have developed pectin coatings for titanium-based orthopedic and dental implants, and have confirmed that pectin-coated titanium implants are well tolerated in a rat implant model. Interestingly, histological examination failed to observe any FBGCs at the site of implantation, although mononuclear macrophages and a collagenous capsule were observed at the implant-tissue interface []. As FBGC are the hallmark of the FBR, their absence on the surface of the pectin-coated implant holds promise in the development of biomaterials with improved biocompatibility profiles for a wide range of applications. In addition, pectin has been investigated as a carrier for oral drug delivery due to its ability to remain intact throughout the gastrointestinal tract until degraded by colonic microflora [,]. Interpenetrating polymer networks incorporating pectin have recently been investigated for controlled/triggered release drug delivery applications [,].
In this study, we report on the fabrication and in vitro characterization of a novel hydrogel incorporating the anionic polygalacturonic acid (PGA), the major polysaccharide component of pectin, with the aim of developing materials or coatings with improved biocompatibility for implantable medical devices. The PGA-incorporated hydrogel developed is a semi-interpenetrating polymer network (SIPN), defined by IUPAC as “a polymer comprising one or more networks and one or more linear or branched polymer(s) characterized by the penetration on a molecular scale of at least one of the networks by at least some of the linear or branched macromolecules” []. The monomers (Hydroxyethyl)methacrylate (HEMA, neutral) and [2-(Methacryloyloxy)ethyl]trimethylammonium chloride (METAC, cationic) are mixed with the linear PGA polysaccharide prior to the initiation of polymerization. Once polymerized, the PGA becomes entrapped within the poly (HEMA-co-METAC) network. Following physical characterization of the material, in vitro cytotoxicity, protein adsorption, and biocompatibility studies (macrophage adherence and fusion assays) are reported.
2. Materials and Methods
2.1. SIPN Preparation
SIPN hydrogels were synthesized by the in situ polymerization of HEMA (Sigma Aldrich, Saint Louis, MO, USA) and METAC monomers following mixing with an aqueous solution of PGA sodium salt (Supplementary Figure S1). PGA sodium salt was obtained from Sigma Aldrich, Saint Louis, MO, USA (product number: P3850, batch number: SLBG0917V, assay by titration ≥ 75% as reported on manufacturer’s Certificate of Analysis). Then, 2,2′-Azobis (2-methylpropionitrile) (AIBN, Sigma Aldrich, Saint Louis, MO, USA) was used as the thermal initiator at a concentration of 0.1% w/w, and materials were prepared with a final concentration of 1% w/w PGA and 2% w/w PGA. A control material, p(HEMA-co-METAC), was prepared by substituting the aqueous PGA solution with an equal volume of deionized water prior to polymerization. Monomer/PGA mixtures were poured into a mold of 1 mm thickness (Supplementary Figure S2A) and polymerized at 60 °C for 1 h, 70 °C for 16 h, and finally, 85 °C for 1 h. (Pectin has good heat stability and is routinely used in the food industry at temperatures exceeding 85 °C) The resultant SIPN hydrogel films (Supplementary Figure S2B) were washed three times for at least 24 h in deionized water in order to remove unreacted monomers, and this was confirmed by UV analysis of the spent water washes. Discs were cut from the hydrated hydrogel using a No. 6 cork borer, and stored in deionized water until required for analysis.
2.2. Swelling Studies
Swelling studies were conducted in both deionized water and phosphate buffered saline (PBS). Water uptake was analyzed by gravimetric measurements using an analytical balance. Hydrogel discs were fully air-dried until a constant dry weight was measured and recorded. Discs were then immersed in deionized water or PBS at 37 °C, and removed at regular intervals (15–30 min) for kinetic studies, or after 24 h to determine equilibrium swelling. Prior to weighing, samples were blotted with filter paper to remove excess surface water. Equilibrium water content (EWC) was determined using Equation (1), where ωs represents the swollen polymer weight, and ωd represents the dried polymer weight.
2.3. Scanning Electron Microscopy
Semi-IPNs in their hydrated state were analyzed directly by cryo-SEM on a HITACHI model TM3030 Tabletop Microscope (Hitachi High-Tech Europe, Maidenhead, UK ) with a TM-300 Deben Coolstage (Deben UK Ltd., Bury St. Edmunds, UK ) and high-sensitivity four-segment semiconductor BSE detector. Samples were analyzed at 15 kV and 2000× magnification. Cryo-SEM prevents water loss from the sample under vacuum, negating the need to dry the sample prior to analysis.
2.4. Differential Scanning Calorimetry
The glass transition temperature (Tg) was determined from DSC thermograms generated using a DSC Q100 calorimeter (TA Instruments, Wilmslow, UK ). Hydrogels were oven-dried for 24 h. Samples were then fragmented, weight recorded, and placed within an aluminum pan with lid. A pre-scan was conducted from 20 °C up to 100 °C to eliminate sample moisture. The second scan was run at 20–160 °C with an incremental increase in temperature of 10 °C min−1 under nitrogen atmosphere (30 mL min−1). An empty aluminum pan and lid were employed as a reference. STARe software v13.00a (Mettler Toledo, Leicester, UK) was used to analyze the Tg for each material.
2.5. Mechanical Testing
Swollen test specimens (n ≥ 4) were cut into dog-bone-shaped samples using a RayRan profile cutter. Using digital Vernier calipers, samples were measured at a total length of 30 mm, a gauge length of 10 mm, and an overall thickness of 1 mm. Tensile strength analysis was performed using a Stable Micro Systems TA.XT plus Texture Analyzer (Mason Technology, Belfast, UK) with a 30 kg load-cell. Samples were extended at a constant strain rate of 0.50 mm/s until fracture. Generation of Stress–Strain diagrams allowed for the calculation of the parameters including Young’s modulus (E), Ultimate Tensile Strength (UTS), and Maximum Elongation (emax) of each hydrogel.
2.6. Contact Angle Analysis
Contact angle analysis was performed using the captive bubble method. Hydrogel discs were immersed in a bath of deionized water and air bubbles dispensed from a hooked needle. The contact angle was measured between the air bubble and the surface of the hydrogel using the FTÅ 200 apparatus (First Ten Ångstroms, Portsmouth, VA, USA). At least three measurements were carried out on three replicate samples, and the mean contact angle determined for each hydrogel.
2.7. Protein Adsorption
Hydrogel discs (in triplicate) were placed in a 10% w/v solution of bovine serum albumin (BSA) (Sigma Aldrich, Saint Louis, MO, USA), 10% w/v vitronectin (PeproTech, Rocky Hill, NJ, USA), or 10% w/v fibrinogen from bovine serum (Sigma Aldrich, Saint Louis, MO, USA) in PBS and incubated for 2 h with gentle agitation. Adsorbed protein was removed from the samples by sonication (30 min) in 1% w/v SDS in PBS (1 mL), and the protein content of the resulting solution was analyzed using a Micro BCA Assay (Thermo Fisher Scientific, Waltham, MA, USA), with BSA as a standard. An 8-point calibration curve was generated to determine protein concentration.
2.8. Cytotoxicity Assays
Cytotoxicity assays were carried out in accordance with ISO 10993-5 Biological Evaluation of Medical Devices, Part 5: Tests for in vitro Cytotoxicity [].
2.8.1. Direct Contact
Murine L-929 fibroblasts (ATCC CCL-1) were cultured under standard cell culture conditions (37 °C, 5% CO2) in MEM supplemented with 10% horse serum and penicillin/streptomycin. All experiments were conducted below passage number 15. Cells were passaged using trypsin-EDTA and standard cell culture protocols. Then, 6 × 105 cells were seeded into 6 well plates and allowed to recover for 24 h, after which the 2 mL of media was aspirated and replaced with 0.8 mL complete growth medium. After sterilization by autoclaving, hydrogel samples (in triplicate) were placed directly onto the cell monolayers and incubated under cell culture conditions for 24 h. Experimental controls consisted of poly (HEMA-co-EGDMA) (negative control), and poly (HEMA-co-EGDMA) impregnated with benzalkonium chloride (BAK, positive control). Control materials were also tested in triplicate. After 24 h incubation, cell monolayers were stained using the vital stain, trypan blue, and cell morphology examined under an inverted light microscope. Materials were graded for cytotoxic potential as per reactivity grades defined by ISO 10993-5.
2.8.2. Tests on Extracts
Extracts of SIPNs and corresponding positive and negative controls (as per direct contact test detailed above), were prepared by placing samples individually in 5 mL of complete growth medium within a sterile 15 mL conical tube and incubating for 24 h at 37 °C. Then, 5 × 104 L-929 fibroblasts were seeded into the wells of a 96-well plate, and allowed to recover overnight after which time the media was aspirated and replaced with 200 μL of material extract. Monolayers were incubated for a further 24 h before cell viability was determined by an MTT assay.
2.9. Macrophage Adherence
RAW 264.7 (ATCC TIB-71) cells were cultured in DMEM supplemented with 10% endotoxin-free FBS, and 1% penicillin-streptomycin. Cells were passaged using a cell scraper to dislodge adherent cells, as per ATCC directions for this cell line. All experiments were conducted below passage number 20. Macrophage adherence to samples was assessed by seeding 1 × 104 RAW 264.7 cells directly onto sterilized hydrogel discs, placed in individual wells of a 24-well plate. Macrophages were allowed to adhere to the materials for 72 h. Materials were removed from the 24-well plate, washed gently in PBS, and fixed in 4% paraformaldehyde for 20 min. Nuceli were stained with DAPI before analysis using fluorescence microscopy. The experiment was carried out in triplicate, with five random fields of vision (FOV) captured per experimental replicate, and the numbers of nuclei per FOV counted.
2.10. Macrophage Fusion
Expansion of murine monocytes from the bone marrow of wild type C57BL/6 mice was performed as per standard protocols []. C57BL/6 femurs from which the bone marrow was removed were kindly donated by Dr. Rebecca Ingram, Centre for Infection and Immunity, Queen’s University Belfast. Following primary macrophage (MΦ) differentiation, cells were suspended in IMDM with 20% FBS and penicillin/streptomycin at a density of 3.5 × 105 cells/mL, and 1 mL of this cell suspension was seeded onto the semi-IPN hydrogels, placed in individual wells of a 24-well plate. Recombinant murine IL-4 (PeproTech) and murine GM-CSF (PeproTech) were added to each well (both at a concentration of 10 ng/mL) to induce fusion and FBGC formation, as per the previously described methodology [,]. Untreated polystyrene served as a positive fusion control. After 5 days, samples were stained with Giemsa/May–Grünwald (polystyrene) or DAPI and rhodamine-phalloidin (SIPN hydrogels) and examined by light or fluorescence microscopy, respectively. Samples were examined in at least triplicate, and five random FOV were captured per sample. The percentage fusion was determined by counting the number of nuclei per FBGC, and expressing this as a percentage of the total nuclei per FOV. Multinucleate cells containing three or more nuclei were classed as FBGCs.
2.11. Statistical Analysis
All statistical analysis was conducted using GraphPad Prism, version 10.4.1. One-way ANOVA with Tukey’s Multiple Comparison Test was used to test the significance of contact angle analysis, protein adsorption, and macrophage adherence. Kruskal–Wallis with Dunn’s Multiple Comparison Test was used to analyze the mechanical testing data. In all analyses, the significance level was set at α = 0.05.
3. Results
3.1. Swelling Studies
Figure 1 displays the swelling behavior of p(HEMA-co-METAC), p(HEMA-co-METAC)/1%PGA, and p(HEMA-co-METAC)/2%PGA in deionised H2O and PBS. Equilibrium swelling in both PBS and deionised water was achieved after 120 min. When soaked in dH2O, all samples including the control material showed a higher EWC than those samples immersed in PBS. This is likely due to cationic groups being present in the METAC component of the material self-repelling and causing molecular chain expansion when in dH2O. On incorporation of PGA, a slight decrease in EWC for dH2O immersed samples was observed, although not statistically significant. It is expected that the electrostatic interactions between the oppositely charged carboxyl groups in PGA (anionic) and the nitrogen-bound methyl groups in METAC (cationic) also would counteract swelling.
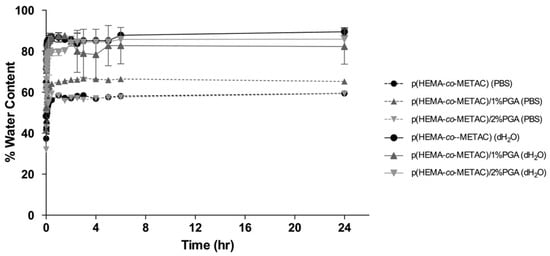
Figure 1.
Kinetic swelling and equilibrium water content (EWC) of PGA-incorporated SIPNs and the control material, p(HEMA-co-METAC). Swelling studies were carried out in both deionized water and PBS.
When soaked in PBS, all materials showed a reduced EWC compared to that observed for dH2O, although no significant difference was observed between samples. The lower EWC observed with PBS may be explained by an increase in ionic strength altering the polyelectrolyte conformation, becoming increasingly coiled and tightly wound, as opposed to their looser, extended form when imbibed in lower ionic strength solutions [].
3.2. Scanning Electron Microscopy
SEM analysis is a useful tool for characterizing semi-IPNs because it shows arrangements, voids, and microscopic structure of the material []. Figure 2 shows representative SEM images of p(HEMA-co-METAC)/1% PGA, p(HEMA-co-METAC)/2% PGA, and p(HEMA-co-METAC) control hydrogel films obtained to determine surface characteristics. Both control and PGA-incorporated hydrogels have an irregular, porous, sponge-like structure, with pits and perforations of micrometer scale, ranging from 0.28 to 9.46 μm in diameter.

Figure 2.
SEM images of (A) p(HEMA-co-METAC), (B) p(HEMA-co-METAC)/1% PGA, and (C) p(HEMA-co-METAC)/2% PGA. Scale bar represents 10 μm.
3.3. Differential Scanning Calorimetry
Tg varied subject to sample composition, with a slight upward shift in Tg with increasing PGA content (Table 1). The single Tg value obtained for all samples is indicative of a homogeneous mixture with no phase separation of the individual SIPN components in any of the fabricated materials. Representative thermograms can be found in Supplementary Figure S3.

Table 1.
Glass transition temperatures, as determined by DSC, of p(HEMA-co-METAC) and PGA-incorporated SIPNs. Values reported are means ± standard deviations of three experimental replicates.
3.4. Mechanical Testing
The results of the mechanical testing are shown in Table 2. The SIPN incorporating 2% PGA displays an increase in all mechanical properties: ultimate tensile strength (UTS), maximum elongation (emax), and Young’s modulus (E). The SIPN incorporating 1% PGA did not show a corresponding increase in UTS or emax, but did display an increase in E, when compared to p(HEMA-co-METAC).
Table 2.
Mechanical analysis of p(HEMA-co-METAC) and PGA-incorporated SIPNs. Values reported are means ± standard deviations of three experimental replicates.
3.5. Contact Angle Analysis
In order to evaluate hydrogel wettability, contact angle measurements of hydrogels under water were obtained using the captive bubble method. Incorporation of PGA into the polymeric material increased the contact angle, as shown in Table 3, although this increase was only significant for p(HEMA-co-METAC)/1%PGA, with a contact angle of 47.306° ± 5.667 (p < 0.001). In this study, we have adopted the following definitions: “super-hydrophobic” (with a contact angle measurement of a > 110°), “hydrophobic”, “intermediate hydrophilic” (30° < a ≤ 90°), “hydrophilic”, and “superhydrophilic” (0 ≤ a ≤ 30°) []. Our materials fell into the class of “intermediate hydrophilic” materials.
Table 3.
Contact angle measurements of p(HEMA-co-METAC) and PGA-incorporated SIPNs. Values reported are means ± standard deviations of three experimental replicates.
3.6. Protein Adsorption
Of the three proteins investigated in this study (BSA, vitronectin, and fibrinogen), BSA displayed a greater affinity for each of the hydrogel SIPNs incorporating PGA, and also for the p(HEMA-co-METAC) control material (Figure 3). All protein solutions used in this assay were prepared at identical concentrations (10% w/v); therefore, the approximately 4-fold greater BSA adsorption, when compared with the adsorption of vitronectin and fibronectin, is likely due to a stronger interaction between BSA and the hydrogels tested.
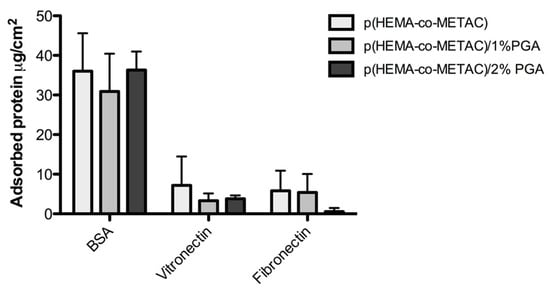
Figure 3.
Adsorption of bovine serum albumin (BSA), vitronectin, and fibrinogen to p(HEMA-co-METAC) and PGA-incorporated SIPNs. Error bars represent standard deviations.
3.7. Cytotoxicity Assays
The cytotoxic potential of the PGA-incorporated SIPNs was tested in accordance with ISO 10993-5 “Biological evaluation of medical devices—Part 5: Tests for in vitro cytotoxicity”. Both direct contact and indirect tests on material extracts were conducted in the present study.
3.7.1. Direct Contact
Direct contact testing utilizes a monolayer of cells onto which is directly placed a sample of the biomaterial which is incubated for 24 h, after which time cell viability is assessed using the vital stain, trypan blue. Reactivity is assessed in accordance with a graded scale provided by ISO 10993-5. In order to be deemed cytocompatible, material must achieve an numerical grade of 2 or less. A Grade 2 reactivity is considered “mild” with any cellular reactivity limited to the zone directly under the biomaterial specimen. We observed slight reactivity (Grade 1) in monolayers exposed to the test samples with minimal morphological changes or trypan blue staining. Some cell detachment was noted directly under the specimen; however, this was deemed to be a result of cellular trauma upon specimen removal. As exposure of L929 to SIPNs incorporating PGA did not display cytotoxicity with cells remaining attached, elongated, and acceptably confluent when compared with controls, we deem this material to be cytocompatible under the conditions of the assay. A summary of reactivity grades for each material is provided in Table 4.
Table 4.
Direct contact cytotoxicity testing, with reactivity graded as per ISO 10993-5. Positive and negative experimental controls were p(HEMA) imbibed with benzalkonium chloride (BAK) and p(HEMA), respectively.
3.7.2. Tests on Extracts
The viability of the L929 fibroblast cells was determined by the reduction in MTT to formazan via metabolism by mitochondria dehydrogenases. Figure 4 displays the percentage viability, relative to the blank (L929 monolayer without extract) of the negative control p(HEMA-co-EGDMA) (a material which reproducibly displays negligible cytotoxic potential), positive control p(HEMA-co-EGDMA) impregnated with benzalkonium chloride (a material which provides a reproducible cytotoxic effect), the material control p(HEMA-co-METAC), and the PGA incorporated SIPNs. ISO 10993-5 states that if cell viability is reduced to <70% of the blank, the material is deemed to have a cytotoxic potential. Our results show that the material control p(HEMA-co-METAC) and each SIPN incorporating PGA, at both 1% and 2% composition, display no discernable cytotoxic potential. The percentage viability of the material control and SIPNs incorporating PGA at 1% and 2% were 74.55%, 71.88%, and 79.14% viability, respectively.
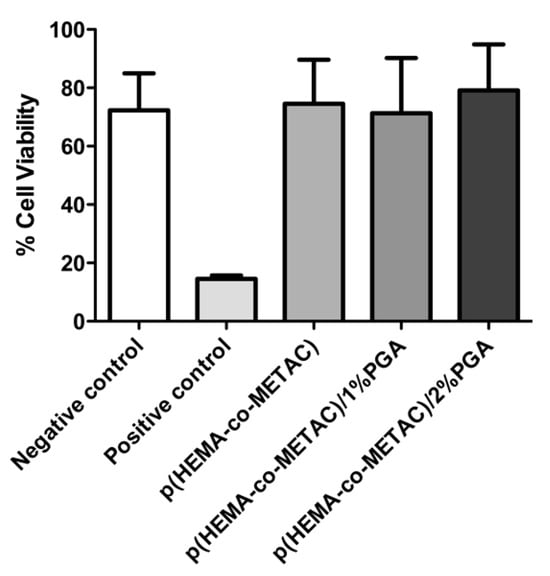
Figure 4.
Cell viability, as determined by MTT assay, of PGA-incorporated SIPNs and corresponding experimental controls. Viability is expressed as a percentage of that observed for a L929 monolayer, unexposed to sample extracts. Error bars represent standard deviations.
3.8. Macrophage Adherence
After 72 h incubation with material samples, the number of adherent RAW264.7 cells on each material was determined through DAPI staining and fluorescence microscopy. The numbers of adherence MΦ per square centimeter for p(HEMA-co-METAC) (control material), p(HEMA-co-METAC)/1% PGA, and p(HEMA-co-METAC)/2% PGA was 192.2 ± 92.81 cells/cm2, 72.72 ± 83.08 cells/cm2, and 136.90 ± 67.31 cells/cm2, respectively (Figure 5). There was a significant reduction in the number of adherent MΦ on p(HEMA-co-METAC)/1% compared to the material control without PGA (p < 0.001); however, there was no significant reduction seen with p(HEMA-co-METAC)/2%. A difference in MΦ adherence was seen between the two PGA incorporated SIPNs, with significantly more cells adhered to the material incorporating 2% PGA after 72 h incubation (p < 0.05).
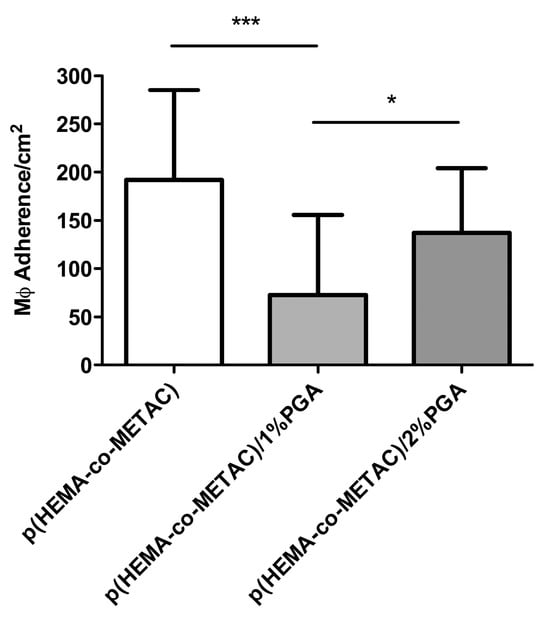
Figure 5.
Number of adherent macrophage (RAW264.7) cells on p(HEMA-co-METAC) and PGA-incorporated SIPNs 72 h after seeding. Error bars represent standard deviations. *** represents significance of p ≤ 0.001, and * represents significance of p ≤ 0.05.
3.9. Macrophage Fusion
After seeding on test materials and polystyrene (PS) controls, primary murine MΦ were incubated in the presence of 10 ng/mL GM-CSF and 10 ng/mL IL-4 in order to promote fusion and FBGC formation. After 3 days, cell fusion and FBGC formation was clearly evident in the PS control (PS is known to facilitate in vitro FBGC formation), as shown in Figure 6B. This FBGC formation was not observed on either p(HEMA-co-METAC), p(HEMA-co-METAC)/1% PGA, or p(HEMA-co-METAC)/2% PGA where MΦ were adherent, but remained as mononuclear cells. Cell morphology was rounded with little indication of cell spreading. F-actin is localized around the parameter of the cells, with no podosomes or lamellipodia evident in any FOV recorded (Figure 6C–E).
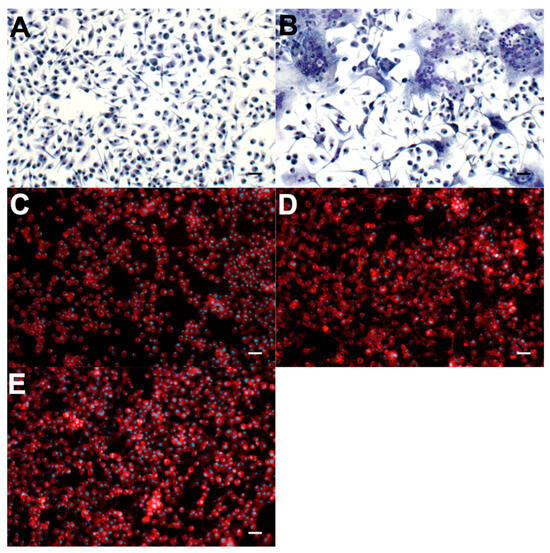
Figure 6.
Primary murine MΦ grown in fusogenic conditions on control materials and PGA-incorporated SIPNs. (A) Negative experimental control (no IL-4) and (B) positive control (with IL-4). Both of these experimental control materials are non-treated sterile polystyrene and are stained with Giemsa/May Grünwald. (C) p(HEMA-co-METAC), (D) p(HEMA-co-METAC)/1% PGA, and (E) p(HEMA-co-METAC)/2% PGA are stained with DAPI (blue, nuclei) and rhodamine phalloidin (red, F-actin). Scale bars represent 10 μm.
4. Discussion
Materials incorporating a natural or cellular fraction aspire to be a functionally superior biomaterial alternative when considering the requirements for integration in tissue engineering and implantable medical devices []. Recently, polysaccharide-incorporated polyelectrolyte gels and multi-layer systems have generated a lot of interest for biomedical and pharmaceutical applications such as drug delivery, medical device coatings, biomimesis, and tissue engineering. This is mostly due to the fact that, in general, polysaccharides display many favorable characteristics such as biocompatibility, bioresorbability, negligible cytotoxicity, anti-inflammatory properties, and can strengthen the polymer’s macromolecular structure [,,,,].
This study details the synthesis of a novel PGA-impregnated polyelectrolyte-modified SIPN, poly(HEMA-co-METAC)/PGA. It was hoped that the polyelectrolyte system would display desirable physical characteristics to support its application as a biomaterial, in addition to performing well in in vitro biocompatibility studies. PGA can be considered similar in structure and charge to hyaluronic acid [], and therefore, we aim to mimic the natural extracellular matrix architecture and thus, enhance biocompatibility. Herein, we have characterized and assessed physical characteristics and in vitro biological responses to our synthesized materials.
In order to evaluate hydrogel wettability, contact angle measurements were obtained using the captive bubble method. Pectins are hydrophilic linear polysaccharides, however, as PGA concentrations increase within our SIPN, water content of the hydrogels decrease, correlating with a marginal increase in contact angle. Within the SIPN, HEMA is co-polymerized with METAC, offering cationic characteristics to the hydrogel component of the SIPN. It is likely that electrostatic interactions between the cationic METAC and anionic PGA reduce chain motility and contribute to a tighter polymer network, hence the reduction in water content observed for the PGA-incorporated materials. It is possible that this tighter polymer network and the electrostatic interactions between METAC and PGA may restrict the reorientation of the polymer side chain to adjacent water molecules. In the case of p(HEMA), the α-methyl group is hydrophobic, whereas the hydroxyethyl is hydrophilic. Restriction of p(HEMA) chain mobility, through electrostatic interaction of the copolymerized METAC component and incorporated PGA, may prevent the tendency of the hydroxyethyl moiety aligning with the surrounding aqueous phase, and so the contact angle of the material is increased. These electrostatic interactions may also explain the reduced swelling observed for each material when placed in phosphate buffered saline, compared to the greater swelling propensity in deionized water. The electrolytes present in PBS will interact with the charged moieties within the polymer, and may effectively function as an ionic cross-linker, hence reducing the degree of swelling. Additionally, the electrostatic interactions between the PGA and METAC components may explain the overall increase in mechanical properties (UTS, emax, and E) displayed in particular by p(HEMA-co-METAC)/2%, due to tighter interactions within the polymer network effectively reinforcing the material. The Young’s modulus of our prepared materials lies within the same values exhibited by soft mammalian tissue. Most mammalian organs have elastic moduli between 0.1 and 10 kPa, for example, lung tissue has an elastic modulus of 5–6 kPa, and kidney tissue 4 kPa []. The SIPNs therefore display appropriate physical characteristics to mimic the physical properties of physiological tissues, and it would be anticipated that this would contribute positively to the materials’ biocompatibility profile.
Protein adsorption resulting in a “conditioning layer” on the surface of the implanted biomaterial is the first stage in the tissue reaction towards the foreign material. Inflammatory cells are not thought to recognize the surface chemistry of the biomaterial itself, rather the adsorbed proteins, thus initiating a cascade of events that culminate in the foreign body response []. While hydrophilic materials are often considered relatively resistant to the adsorption of proteins, recent studies have shown that proteins do in fact interact and adsorb to hydrophilic materials []. Albumin, an abundant plasma protein, will flood the interstitial space upon implantation, and has been considered by some to be a “passivating” protein when adsorbed or coated onto a biomaterial [], possibly attenuating a post-implantation inflammatory response.
We have observed albumin absorption in the range of 30 to 45 μg/cm2 across all of the tested materials, including control materials which do not incorporate PGA. This is likely due to electrostatic interactions between the net negative charge of the albumin protein and the positive charge of the METAC component of the hydrogel. The inclusion of PGA within the material displayed no influence on albumin adsorption, explained by the relatively small proportion of the negatively charged PGA within the SIPNs, in comparison to the METAC component. With albumin being a potentially passivating protein, its readiness to adsorb to our developed materials may not be detrimental to the overall biocompatibility profile.
Several other proteins are known to adsorb to implanted biomaterials, and have been studied in terms of their influence on biocompatibility and propensity to elicit the FBR. Adsorption and denaturation of fibrinogen is crucial to the procession of the FBR, with the protein appearing to play a role in the accumulation of phagocytes (including macrophages) on the implant surface. This has been demonstrated in hypofibrinogenemic mice wherein no inflammatory response is mounted against an implanted material, unless that material is pre-coated with fibrinogen, and is believed to be due to interactions between the phagocyte integrin Mac-1 (CD11b/CD18) and a short sequence on the fibrinogen D domain []. Likewise, vitronectin is striking in its support of significant macrophage adhesion and FBGC formation [,]. Our materials have demonstrated less adsorption per unit area of the potentially detrimental vitronectin and fibrinogen, when compared to albumin adsorption. Additionally, the SIPNs containing PGA appeared to adsorb less vitronectin and fibrinogen, although this observation was not determined to be statistically significant. While these in vitro observations highlight promise, the in vivo situation is much more complex and dynamic. The Vroman effect describes how adsorbed proteins may be competitively displaced and replaced over time [], therefore the in vivo pattern of protein adsorption may vary dramatically from in vitro observations.
In determining the biological acceptability of any material intended for implantation, it is necessary to conduct cytotoxicity testing since the chemical and/or physical nature of the material may exhibit a lack of cytocompatibility, thereby precluding use as a biomaterial. Here, we have used in vitro cell culture assays described in ISO 10993-5 [], in which the outcome is measured by observing changes in cellular morphology, as well as cellular toxicity levels assessed in viable cell populations. Both the direct contact assay and the material extracts comply with the requirements of this International Standard; hence, we have demonstrated that these materials are cytocompatible and have potential for biomaterial applications, subject to further in vivo biocompatibility testing. These promising in vitro results are in agreement with cytotoxicity testing of PGA-based materials reported in the recent literature [].
Since macrophage adherence to an implanted biomaterial is fundamental to the early stage FBR, we investigated the ability of a macrophage cell line (RAW 264.7) to adhere to the surface of our SIPNs and corresponding control material under in vitro conditions. Macrophages bind to foreign materials through a mechanism involving integrin-driven interactions that mediate adhesion to various ligands present on the absorbed protein layer [,]. Cellular adherence therefore reflects distinct adsorption profiles which are characteristic of the underlying surface properties []. Results displayed in Figure 5 show that incorporation of PGA resulted in a significant decrease in macrophage cell adhesion to the polymer surface, when compared with the p(HEMA-co-METAC) control. Considering the role of integrins in cellular adhesion, this is likely due to the particular adsorption of serum proteins on PGA-incorporated materials. Several reports exist in the literature detailing the effects of pectin (from which PGA is derived) on cellular adhesion. Enzymatically modified pectin has been reported to decrease cell adhesion, spreading and proliferation [], and has been shown to have a preventative effect on mice with colitis, proposed due to a decrease in neutrophil and macrophage infiltration and adhesion as well as anti-oxidant affects []. If these in vitro observations are replicated in vivo, the developed SIPNs incorporating PGA may show promise in terms of their overall biocompatibility through a dampening of the post-implantation inflammatory response mediated by macrophages.
In addition to initial macrophage adherence, we have investigated the ability of macrophages to fuse and differentiate into FBGCs on the material surfaces. Monocyte/macrophage membrane fusion occurs in the formation of FBGCs, although the exact sequence of events, and the exact functional contribution to inflammation is not yet fully understood []. Adhesion proteins, membrane lipid rafts, and actin rearrangement are all thought to be critical in the process []. It has been proposed that fusion occurs in three programmed steps: acquiring the ability to fusion, migration and attachment to approximating membranes, and finally fusion and sharing of cellular components []. As with initial cellular attachment and adhesion, the binding of integrins to foreign materials is known to cause a change in movement, gene transcription, proliferation, and alteration of the cellular cytoskeleton []. The expression profile of FBGCs includes the following integrins: αmβ2, αxβ2, α5β1, αvβ1, and α2β1, indicating a strong role for subunits β2 and β1 []. Fibrinogen and vitronectin are recognised by the β2-associated pathway [], so again the adsorbed protein layer is likely to play an instrumental role in the fusion of macrophages at the biomaterial surface. In Figure 6, it is illustrated that p(HEMA-co-METAC), p(HEMA-co-METAC)/1% PGA, and p(HEMA-co-METAC)/2% PGA do not support the fusion of primary murine MΦ, nor their differentiation to FBGCs. On each of these materials, we observed cells to be adherent, but with little spreading. Rhodamine-phalloidin staining revealed F-actin to be localized at the perimeter of the cells, with no evidence of the typical ring-like podosome structures seen in spreading cells (Figure 6C–E). In stark contrast, FBGC formation proceeds unimpeded when MΦ obtained from the same isolation and expansion were plated under fusogenic conditions on non-treated polystyrene (Figure 6A,B), a material which has been widely demonstrated to support macrophage fusion [,,,,,]. These results highlight that the developed PGA-incorporated SIPNs possess desirable properties in terms of in vitro biocompatibility, and future work is planned to extend this work to in vivo implantation models.
5. Conclusions
The present study describes the synthesis, physical characterization, and in vitro biocompatibility profile of SIPN hydrogels incorporating the natural plant-derived polysaccharide, polygalacturonic acid. The results detailed above demonstrate the potential of these SIPNs as biomaterials, since both physical and biological characterization proved to support this intended end-application. Given the recent attention received by pectin/polygalacturonic acid in the field of bone tissue regeneration, we suggest that our developed SIPNs may ultimately be applied as coatings on orthopedic or dental implants, conferring a biocompatible surface and affording osseointegration without a heightened foreign body response. Future work will study these materials in the context of the cellular activity involved in bone remodeling (osteoblasts and osteoclasts), and we will further characterize biocompatibility using in vivo implant models.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/pr13051502/s1, Figure S1. Schematic diagram illustrating the in-situ fabrication of poly(HEMA-co-METAC) semi-interpenetrating polymer networks. Figure S2. (A) Monomer/PGA mixture were added to a mould of 1 mm thickness, comprising two glass sheets (100 × 100 mm), which contained a loop of silicone tubing between a sheet of silicone coated release liner, and clamped together. (B) The resultant hydrogel film after polymerization. Figure S3. Representative thermograms of poly(HEMA-co-METAC) semi-IPNs, and poly(HEMA-co-METAC) control material.
Author Contributions
Conceptualization, A.N.O., C.P.M. and L.C.; methodology, A.N.O. and L.C.; software, data curation, A.N.O. and L.C.; writing—original draft preparation, A.N.O. and L.C.; writing—review and editing, L.C.; supervision, project administration, funding acquisition, L.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, [L.C.], upon request.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ECM | Extracellular Matrix |
| FBR | Foreign Body Response |
| FBGC | Foreign Body Giant Cell |
| ROS | Reactive Oxygen Species |
| IL-1 | Interleukin-1 |
| HEMA | (Hydroxyethyl)methacrylate |
| METAC | [2-(Methacryloyloxy)ethyl]trimethylammonium chloride |
| PGA | Polygalacturonic acid |
| SIPN | Semi-interpenetrating polymer network |
| AIBN | 2,2′-Azobis(2-methylpropionitrile) |
| SEM | Scanning Electron Microscopy |
| DSC | Differential Scanning Calorimetry |
| PBS | Phosphate Buffered Saline |
| DAPI | 4′,6-diamidino-2-phenylindole |
| FOV | Fields of Vision |
| BAK | Benzalkonium Chloride |
| MTT | 4′,6-diamidino-2-phenylindole |
References
- Williams, D.F. On the mechanisms of biocompatibility. Biomaterials 2008, 29, 2941–2953. [Google Scholar] [CrossRef] [PubMed]
- Bryers, J.D.; Giachelli, C.M.; Ratner, B.D. Engineering biomaterials to integrate and heal: The biocompatibility paradigm shifts. Biotechnol. Bioeng. 2012, 109, 1898–1911. [Google Scholar] [CrossRef]
- Anderson, J.M.; Rodriguez, A.; Chang, D.T. Foreign body reaction to biomaterials. Semin. Immunol. 2008, 20, 86–100. [Google Scholar] [CrossRef]
- Brodbeck, W.G.; Anderson, J.M. Giant cell formation and function. Curr. Opin. Hematol. 2009, 16, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Jay, S.M.; Skokos, E.A.; Zeng, J.; Knox, K.; Kyriakides, T.R. Macrophage fusion leading to foreign body giant cell formation persists under phagocytic stimulation by microspheres in vitro and in vivo in mouse models. J. Biomed. Mater. Res. Part A 2010, 93A, 189–199. [Google Scholar] [CrossRef]
- Henson, P.M. Immunologic release of constituents from neutrophil leukocytes. 1. Role of antibody and complement on nonphagocytosable surfaces or phagocytosable particles. J. Immunol. 1971, 107, 1535–1546. [Google Scholar] [CrossRef]
- Yoko, S.; Kobayashi, Y.; Iiri, T.; Kitazawa, H.; Okabe, M.; Kobayashi, H.; Okazaki, E.; Aizawa, Y. Pacing lead-induced granuloma in the atrium: A foreign body reaction to polyurethane. Case Rep. Cardiol. 2013, 2013, 396595. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, M.J.; Wilkoff, B.; Anderson, J.M.; Hiltner, A. Biodegradation of polyether polyurethane inner insulation in bipolar pacemaker leads. J. Biomed. Mater. Res. 2001, 58, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Labow, R.S.; Meek, E.; Matheson, L.A.; Santerre, J.P. Human macrophage-mediated biodegradation of polyurethanes: Assessment of candidate enzyme activities. Biomaterials 2002, 23, 3969–3975. [Google Scholar] [CrossRef]
- Miller, K.M.; Anderson, J.M. In vitro stimulation of fibroblast activity by factors generated from human monocytes activated by biomedical polymers. J. Biomed. Mater. Res. 1989, 23, 911–930. [Google Scholar] [CrossRef]
- Jay, S.M.; Skokos, E.; Laiwalla, F.; Krady, M.M.; Kyriakides, T.R. Foreign body giant cell formation is preceded by lamellipodia formation and can be attenuated by inhibition of Rac1 activation. Am. J. Pathol. 2007, 171, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Harmon, K.A.; Lane, B.A.; Boone, R.E.; Afshari, A.; Berdel, H.O.; Yost, M.J.; Goodwin, R.L.; Friedman, H.I.; Eberth, J.F. Therapeutic Engineered Hydrogel Coatings Attenuate the Foreign Body Response in Submuscular Implants. Ann. Plast. Surg. 2018, 80, S410–S417. [Google Scholar] [CrossRef]
- Love, R.J.; Jones, K.S. Biomaterials, fibrosis, and the use of drug delivery systems in future antifibrotic strategies. Crit. Rev. Biomed. Eng. 2009, 37, 259–281. [Google Scholar] [CrossRef] [PubMed]
- Rujitanaroj, P.O.; Jao, B.; Yang, J.; Wang, F.; Anderson, J.M.; Wang, J.; Chew, S.Y. Controlling fibrous capsule formation through long-term down-regulation of collagen type I (COL1A1) expression by nanofiber-mediated siRNA gene silencing. Acta Biomater. 2013, 9, 4513–4524. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, J.; Kinser, E.R.; Stalter, M.A.; Duncan-Lewis, C.; Balestrini, J.L.; Sawyer, A.J.; Schroers, J.; Kyriakides, T.R. Engineering Cellular Response Using Nanopatterned Bulk Metallic Glass. ACS Nano 2014, 8, 4366–4375. [Google Scholar] [CrossRef]
- Padmanabhan, J.; Kyriakides, T.R. Nanomaterials, Inflammation, and Tissue Engineering. Wiley Interdiscip. Rev.-Nanomed. Nanobiotechnology 2015, 7, 355–370. [Google Scholar] [CrossRef]
- Zhang, L.; Cao, Z.; Bai, T.; Carr, L.; Ella-Menye, J.R.; Irvin, C.; Ratner, B.D.; Jiang, S. Zwitterionic hydrogels implanted in mice resist the foreign-body reaction. Nat. Biotechnol. 2013, 31, 553–556. [Google Scholar] [CrossRef]
- Albrektsson, T.; Dahlin, C.; Jemt, T.; Sennerby, L.; Turri, A.; Wennerberg, A. Is marginal bone loss around oral implants the result of a provoked foreign body reaction? Clin. Implant Dent. Relat. Res. 2014, 16, 155–165. [Google Scholar] [CrossRef]
- Abu-Amer, Y.; Darwech, I.; Clohisy, J.C. Aseptic loosening of total joint replacements: Mechanisms underlying osteolysis and potential therapies. Arthritis Res. Ther. 2007, 9, S6. [Google Scholar] [CrossRef]
- Simsek, G.M.; Barthes, J.; Muller, C.; McGuinness, G.B.; Vrana, N.E.; Yapici, G.G. PVA/gelatin-based hydrogel coating of nickel-titanium alloy for improved tissue-implant interface. Appl. Phys. A 2021, 127, 387. [Google Scholar] [CrossRef]
- Leng, J.; He, Y.; Yuan, Z.; Tao, B.; Li, K.; Lin, C.; Xu, K.; Chen, M.; Dai, L.; Li, X.; et al. Enzymatically-degradable hydrogel coatings on titanium for bacterial infection inhibition and enhanced soft tissue compatibility via a self-adaptive strategy. Bioact. Mater. 2021, 6, 4670–4685. [Google Scholar] [CrossRef] [PubMed]
- Saveleva, M.; Vladescu, A.; Cotrut, C.; Van Der Meeren, L.; Surmeneva, M.; Surmenev, R.; Parakhonskiy, B.; Skirtach, A.G. The effect of hybrid coatings based on hydrogel, biopolymer and inorganic components on the corrosion behavior of titanium bone implants. J. Mater. Chem. B 2019, 7, 6778–6788. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Bodón, J.; Andrade del Olmo, J.; Alonso, J.M.; Moreno-Benítez, I.; Vilas-Vilela, J.L.; Pérez-Álvarez, L. Bioactive Coatings on Titanium: A Review on Hydroxylation, Self-Assembled Monolayers (SAMs) and Surface Modification Strategies. Polymers 2022, 14, 165. [Google Scholar] [CrossRef]
- Rinaudo, M. Main properties and current applications of some polysaccharides as biomaterials. Polym. Int. 2008, 57, 397–430. [Google Scholar] [CrossRef]
- Shelke, N.B.; James, R.; Laurencin, C.T.; Kumbar, S.G. Polysaccharide biomaterials for drug delivery and regenerative engineering. Polym. Adv. Technol. 2014, 25, 448–460. [Google Scholar] [CrossRef]
- Sun, J.; Tan, H. Alginate-Based Biomaterials for Regenerative Medicine Applications. Materials 2013, 6, 1285–1309. [Google Scholar] [CrossRef]
- Sood, A.; Gupta, A.; Agrawal, G. Recent advances in polysaccharides based biomaterials for drug delivery and tissue engineering applications. Carbohydr. Polym. Technol. Appl. 2021, 2, 100067. [Google Scholar] [CrossRef]
- Coimbra, P.; Ferreira, P.; de Sousa, H.C.; Batista, P.; Rodrigues, M.A.; Correia, I.J.; Gil, M.H. Preparation and chemical and biological characterization of a pectin/chitosan polyelectrolyte complex scaffold for possible bone tissue engineering applications. Int. J. Biol. Macromol. 2011, 48, 112–118. [Google Scholar] [CrossRef]
- Gurzawska, K.; Svava, R.; Yihua, Y.; Haugshoj, K.B.; Dirscherl, K.; Levery, S.B.; Byg, I.; Damager, I.; Nielsen, M.W.; Jorgensen, B.; et al. Osteoblastic response to pectin nanocoating on titanium surfaces. Mater. Sci. Eng. C Mater. Biol. Appl. 2014, 43, 117–125. [Google Scholar] [CrossRef]
- Kokkonen, H.; Cassinelli, C.; Verhoef, R.; Morra, M.; Schols, H.A.; Tuukkanen, J. Differentiation of osteoblasts on pectin-coated titanium. Biomacromolecules 2008, 9, 2369–2376. [Google Scholar] [CrossRef]
- Kokkonen, H.; Verhoef, R.; Kauppinen, K.; Muhonen, V.; Jorgensen, B.; Damager, I.; Schols, H.A.; Morra, M.; Ulvskov, P.; Tuukkanen, J. Affecting osteoblastic responses with in vivo engineered potato pectin fragments. J. Biomed. Mater. Res. Part A 2012, 100, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, H.E.; Ilvesaro, J.M.; Morra, M.; Schols, H.A.; Tuukkanen, J. Effect of modified pectin molecules on the growth of bone cells. Biomacromolecules 2007, 8, 509–515. [Google Scholar] [CrossRef]
- Munarin, F.; Guerreiro, S.G.; Grellier, M.A.; Tanzi, M.C.; Barbosa, M.A.; Petrini, P.; Granja, P.L. Pectin-based injectable biomaterials for bone tissue engineering. Biomacromolecules 2011, 12, 568–577. [Google Scholar] [CrossRef]
- Gurzawska, K.; Dirscherl, K.; Jørgensen, B.; Berglundh, T.; Jørgensen, N.R.; Gotfredsen, K. Pectin nanocoating of titanium implant surfaces—An experimental study in rabbits. Clin. Oral Implant. Res. 2017, 28, 298–307. [Google Scholar] [CrossRef]
- Sultana, N. Biological Properties and Biomedical Applications of Pectin and Pectin-Based Composites: A Review. Molecules 2023, 28, 7974. [Google Scholar] [CrossRef]
- Chen, X.D.; Bian, X.P.; Teslovich, T.M.; Stephan, D.A.; Young, M.F. Dissection of the sets of genes that control the behavior of biglycan-deficient pre-osteoblasts using oligonucleotide microarrays. Bone 2005, 37, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.D.; Fisher, L.W.; Robey, P.G.; Young, M.F. The small leucine-rich proteoglycan biglycan modulates BMP-4-induced osteoblast differentiation. Faseb J. 2004, 18, 948–958. [Google Scholar] [CrossRef]
- Kokkonen, H.; Niiranen, H.; Schols, H.A.; Morra, M.; Stenback, F.; Tuukkanen, J. Pectin-coated titanium implants are well-tolerated in vivo. J. Biomed. Mater. Res. Part A 2010, 93, 1404–1409. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Bunt, C.; Cornish, J.; Quek, S.Y.; Wen, J. Oral Delivery of Bovine Lactoferrin Using Pectin- and Chitosan-Modified Liposomes and Solid Lipid Particles: Improvement of Stability of Lactoferrin. Chem. Biol. Drug Des. 2015, 86, 466–475. [Google Scholar] [CrossRef]
- Yao, X.; Bunt, C.; Cornish, J.; Quek, S.Y.; Wen, J. Preparation, optimization and characterization of bovine lactoferrin-loaded liposomes and solid lipid particles modified by hydrophilic polymers using factorial design. Chem. Biol. Drug Des. 2014, 83, 560–575. [Google Scholar] [CrossRef]
- Li, L.; Liu, Y.; Tan, X.; Teng, F.; Li, Y. Synthesis and characterization of interpenetrating network hydrogels based on sugar beet pectin and heteroprotein complex: Structural characteristics and physicochemical properties. Carbohydr. Polym. 2025, 349, 122959. [Google Scholar] [CrossRef] [PubMed]
- Zafar, N.; Mahmood, A.; Ilyas, S.; Ijaz, H.; Muhammad Sarfraz, R.; Mahdi, W.A.; Salem-Bekhit, M.M.; Ibrahim, M.A.; Benguerba, Y.; Ernst, B. Novel Natrosol/Pectin-co-poly (acrylate) based pH-responsive polymeric carrier system for controlled delivery of Tapentadol Hydrochloride. Saudi Pharm. J. 2023, 31, 101671. [Google Scholar] [CrossRef]
- Work, W.J.; Horie, K.; Hess, M.; Stepto, R.F.T. Definitions of terms related to polymer blends, composites, and multiphase polymeric materials—(IUPAC recommendations 2004). Pure Appl. Chem. 2004, 76, 1985–2007. [Google Scholar] [CrossRef]
- ISO 10993-5:2009; Biological Evaluation of Medical Devices, Part 5: Tests for in vitro Cytotoxicity. International Organization for Standardization: Geneva, Switzerland, 2009.
- Weischenfeldt, J.; Porse, B. Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc. 2008, 3, pdb.prot5080. [Google Scholar] [CrossRef]
- Moya, S.; Azzaroni, O.; Farhan, T.; Osborne, V.L.; Huck, W.T.S. Locking and unlocking of polyelectrolyte brushes: Toward the fabrication of chemically controlled nanoactuators. Angew. Chem.-Int. Ed. 2005, 44, 4578–4581. [Google Scholar] [CrossRef] [PubMed]
- Cona, C.; Bailey, K.; Barker, E. Characterization Methods to Determine Interpenetrating Polymer Network (IPN) in Hydrogels. Polymers 2024, 16, 2050. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Cho, S.K. Microparticle sampling by electrowetting-actuated droplet sweeping. Lab A Chip 2006, 6, 137–144. [Google Scholar] [CrossRef]
- Brown, B.N.; Sicari, B.M.; Badylak, S.F. Rethinking regenerative medicine: A macrophage-centered approach. Front. Immunol. 2014, 5, 510. [Google Scholar] [CrossRef]
- LeBoeuf, R.D.; Raja, R.H.; Fuller, G.M.; Weigel, P.H. Human fibrinogen specifically binds hyaluronic acid. J. Biol. Chem. 1986, 261, 12586–12592. [Google Scholar] [CrossRef]
- Levental, I.; Georges, P.C.; Janmey, P.A. Soft biological materials and their impact on cell function. Soft Matter. 2007, 3, 299–306. [Google Scholar] [CrossRef]
- Swartzlander, M.D.; Barnes, C.A.; Blakney, A.K.; Kaar, J.L.; Kyriakides, T.R.; Bryant, S.J. Linking the foreign body response and protein adsorption to PEG-based hydrogels using proteomics. Biomaterials 2015, 41, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Geelhood, S.J.; Horbett, T.A.; Ward, W.K.; Wood, M.D.; Quinn, M.J. Passivating protein coatings for implantable glucose sensors: Evaluation of protein retention. J. Biomed. Mater. Res. Part B Appl. Biomater. 2007, 81, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.J.; Eaton, J.W.; Ugarova, T.P.; Tang, L. Molecular basis of biomaterial-mediated foreign body reactions. Blood 2001, 98, 1231–1238. [Google Scholar] [CrossRef]
- McNally, A.K.; Jones, J.A.; Macewan, S.R.; Colton, E.; Anderson, J.M. Vitronectin is a critical protein adhesion substrate for IL-4-induced foreign body giant cell formation. J. Biomed. Mater. Res. Part A 2008, 86, 535–543. [Google Scholar] [CrossRef]
- Mariani, E.; Lisignoli, G.; Borzì, R.M.; Pulsatelli, L. Biomaterials: Foreign Bodies or Tuners for the Immune Response? Int. J. Mol. Sci. 2019, 20, 636. [Google Scholar] [CrossRef]
- Vroman, L. The life of an artificial device in contact with blood: Initial events and their effect on its final state. Bull. N. Y. Acad. Med. 1988, 64, 352–357. [Google Scholar] [PubMed]
- Suner, S.S.; Ari, B.; Duygu Sutekin, S.; Sahiner, N. Biocompatible poly(galacturonic acid) micro/nanogels with controllable degradation via tunable chemical crosslinking. Int. J. Biol. Macromol. 2022, 201, 351–363. [Google Scholar] [CrossRef]
- Sheikh, Z.; Brooks, P.; Barzilay, O.; Fine, N.; Glogauer, M. Macrophages, Foreign Body Giant Cells and Their Response to Implantable Biomaterials. Materials 2015, 8, 5269. [Google Scholar] [CrossRef]
- Garcia, A.J. Get a grip: Integrins in cell-biomaterial interactions. Biomaterials 2005, 26, 7525–7529. [Google Scholar] [CrossRef]
- Wilson, C.J.; Clegg, R.E.; Leavesley, D.I.; Pearcy, M.J. Mediation of biomaterial-cell interactions by adsorbed proteins: A review. Tissue Eng. 2005, 11, 1–18. [Google Scholar] [CrossRef]
- Bussy, C.; Verhoef, R.; Haeger, A.; Morra, M.; Duval, J.-L.; Vigneron, P.; Bensoussan, A.; Velzenberger, E.; Cascardo, G.; Cassinelli, C.; et al. Modulating in vitro bone cell and macrophage behavior by immobilized enzymatically tailored pectins. J. Biomed. Mater. Res. Part A 2008, 86A, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Popov, S.V.; Golovchenko, V.V.; Ovodova, R.G.; Smirnov, V.V.; Khramova, D.S.; Popova, G.Y.; Ovodov, Y.S. Characterisation of the oral adjuvant effect of lemnan, a pectic polysaccharide of Lemna minor L. Vaccine 2006, 24, 5413–5419. [Google Scholar] [CrossRef]
- Trout, K.L.; Holian, A. Multinucleated giant cell phenotype in response to stimulation. Immunobiology 2020, 225, 151952. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, T.; Zanker, K.S. Cell fusion in health and disease. Volume II: Cell fusion in disease. Introduction. Adv. Exp. Med. Biol. 2011, 714, 1–3. [Google Scholar] [CrossRef]
- Helming, L.; Gordon, S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009, 19, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Berton, G.; Lowell, C.A. Integrin signalling in neutrophils and macrophages. Cell. Signal. 1999, 11, 621–635. [Google Scholar] [CrossRef]
- McNally, A.K.; Macewan, S.R.; Anderson, J.M. alpha subunit partners to beta1 and beta2 integrins during IL-4-induced foreign body giant cell formation. J. Biomed. Mater. Res. Part A 2007, 82, 568–574. [Google Scholar] [CrossRef]
- Brodbeck, W.G.; Patel, J.; Voskerician, G.; Christenson, E.; Shive, M.S.; Nakayama, Y.; Matsuda, T.; Ziats, N.P.; Anderson, J.M. Biomaterial adherent macrophage apoptosis is increased by hydrophilic and anionic substrates in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 10287–10292. [Google Scholar] [CrossRef]
- MacLauchlan, S.; Skokos, E.A.; Meznarich, N.; Zhu, D.H.; Raoof, S.; Shipley, J.M.; Senior, R.M.; Bornstein, P.; Kyriakides, T.R. Macrophage fusion, giant cell formation, and the foreign body response require matrix metalloproteinase 9. J. Leukoc. Biol. 2009, 85, 617–626. [Google Scholar] [CrossRef]
- Themis, R.; Kyriakides, M.J.F.; Grant, E.; Keeney, A.T.; Cecilia, M.; Giachelli, I.C.-L.; Barrett, J.; Rollins, A.P.B. The CC Chemokine Ligand, CCL2/MCP1, Participates in Macrophage Fusion and Foreign Body Giant Cell Formation. Am. J. Pathol. 2004, 165, 2157–2166. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).