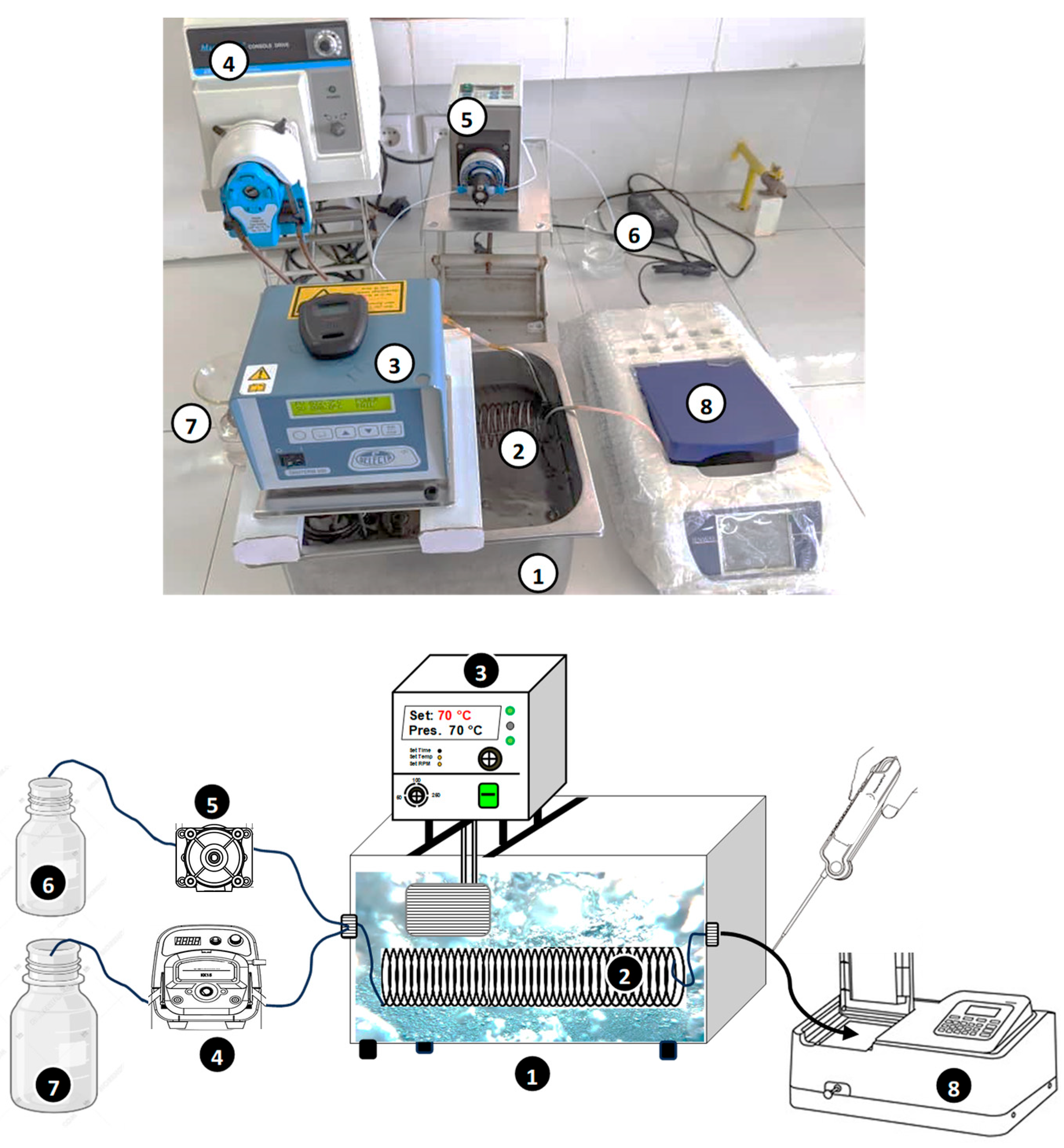
1. Introduction
Water pollution remains a critical environmental concern due to the increasing discharge of hazardous contaminants from industrial activities. Among these pollutants, dye-containing wastewater from the textile, paper, leather, and dyeing industries poses significant ecological and health risks [
1]. Synthetic dyes are widely used due to their stability and resistance to degradation; however, this persistence also makes them difficult to remove through conventional treatment methods. Large volumes of dye wastewater are discharged globally, with estimates exceeding 700,000 tons annually, contributing to severe water pollution. Many dyes and their byproducts exhibit high toxicity, carcinogenicity, and bioaccumulation potential, leading to adverse effects on aquatic ecosystems and human health [
2]. Typically, textile wastewater undergoes a multi-stage treatment process, including coagulation-flocculation, biological treatment, and adsorption onto activated carbon [
3]. While these methods can effectively remove suspended solids and some organic contaminants, they are often inadequate for treating persistent and recalcitrant dyes [
4]. Many synthetic dyes, especially azo, anthraquinone, and sulfur-based dyes, exhibit high chemical stability and resistance to biodegradation, leading to incomplete removal and secondary pollution risks [
5]. Consequently, the development of efficient and sustainable treatment strategies for dye-contaminated wastewater remains a major challenge in environmental engineering.
Advanced oxidation processes (AOPs) have emerged as highly promising solutions for the degradation of refractory and persistent pollutants, particularly synthetic dyes in textile wastewater [
6]. These processes rely on the generation of highly reactive radical species capable of breaking down complex organic molecules into smaller, less harmful compounds, ultimately leading to mineralization [
7]. AOPs are broadly categorized into hydroxyl radical (
•OH)-based and sulfate radical (SO
4•−)-based processes, both of which have demonstrated remarkable efficiency in degrading and mineralizing dyes, even at low concentrations [
8,
9]. Hydroxyl radicals, generated through methods such as Fenton/Fenton-like reactions, UV/H
2O
2, photocatalysis, and ozonation, are known for their high oxidation potential (~2.8 V) and non-selective reactivity with organic contaminants [
10]. On the other hand, sulfate radical-based AOPs [
11], including persulfate (PS) and peroxymonosulfate salts (e.g., K
2S
2O
8 and KHSO
5) activation via heat, transition metals, metal oxides, or radiation, offer a more selective oxidation pathway with a comparable redox potential (~2.5–3.1 V) [
12,
13,
14]. Given their effectiveness, AOPs are often considered as a finishing stage in textile wastewater treatment, particularly for removing residual low-dye contaminants that persist after conventional treatment. For instance, hydroxyl radicals readily degrade azo dyes through cleavage of the N=N bond, while sulfate radicals demonstrate higher efficiency in breaking down anthraquinone dyes due to their electrophilic nature [
15,
16]. The synergy between these radical species enables the comprehensive degradation of complex dye structures, ensuring their complete mineralization into CO
2, H
2O, and inorganic ions [
17].
Among the various strategies for persulfate activation, heat and Fe(II)-catalyzed activation have gained significant attention due to their efficiency in generating reactive sulfate and hydroxyl radicals. In the heat-activated persulfate system, sulfate radicals are primarily generated via the thermal decomposition of persulfate (Equation (1)) [
18]. These radicals undergo rapid hydrolysis, forming hydroxyl radicals (Equations (2) and (3)). Both SO
4•– and
•OH may react with organic pollutants (Equations (4) and (5)) and persulfate itself (Equations (6) and (7)). Radical termination reactions further govern process efficiency, as recombination leads to the formation of peroxymonosulfate (HSO
5−), persulfate, or hydrogen peroxide (Equations (8)–(10)).
Liang and Su [
19] investigated the active radical species generated in thermally activated persulfate (PS) under different pH conditions using a chemical probe method. Their findings indicated that SO
4•− is the dominant oxidant at pH < 7, while both SO
4•− and
•OH coexist at neutral pH. At pH > 9,
•OH becomes the predominant radical species. These results were further supported by an alternative spectroscopic (ESR) spin trapping [
20], which confirmed a similar distribution of radical species across different pH conditions.
Fe(II)-catalyzed persulfate activation follows a more complex pathway but is generally more efficient at lower operating temperatures [
21]. The activation is initiated by Fe(II), which decomposes persulfate into SO
4•− and Fe(III) (Equation (11)), triggering a subsequent reaction series that includes Reactions 2–10. Additionally, Fe(III) can be reduced back to Fe(II) via Reaction 12, though this process is relatively slow. However, Fe(II) also acts as a radical scavenger by reacting with SO
4•− and
•OH (Equations (13) and (14)), thereby reducing radical availability. The solution pH plays a crucial role in the process, with acidic conditions (pH ~3) being the most favorable for optimal reaction performance and preventing iron precipitation.
The contributions of SO
4•– and
•OH in the Fe(II)/KPS system at pH 3 were determined to be 66% and 44%, respectively, using a chemical probing technique with tert-butanol and NaN
3 scavengers on Pararosaniline dye degradation [
21]. This indicates that both radicals coexist and influence the degradation rate at pH 3, in contrast to previous reports on thermally activated persulfate processes, where SO
4•– is typically the dominant oxidant under acidic conditions [
22].
Despite the advanced metrics and high performance of heat- and Fe(II)-catalyzed persulfate activation in pollutant degradation, most studies have been conducted in batch mode. While batch systems provide valuable mechanistic insights, they are not representative of industrial applications, where continuous processes are preferred for scalability and efficiency. Research on continuous-flow systems remains relatively limited but is crucial for translating these activation strategies into practical industrial use. A promising approach for implementing continuous operation is the use of microreactors, which offer enhanced mass and heat transfer, precise control over reaction conditions, and improved energy efficiency [
23]. Microreactors, also known as microfluidic devices, represent advanced systems designed to control chemical reactions and fluid dynamics through the use of microchannels, which typically range in size from micrometers to millimeters (1 µm to 1 mm) [
24]. These devices provide several benefits compared to traditional reactors, including their compact design and large surface-to-volume ratios, which result in shorter diffusion and conduction distances, promoting laminar flow and improved mass transfer efficiency [
23]. Additionally, microreactors can be constructed from transparent materials, allowing for real-time visualization of reaction processes and the integration of external light sources for photocatalytic AOPs [
25]. These features enable efficient radical generation and pollutant degradation in a highly controlled setting, addressing some of the challenges faced by conventional reactor systems. Nevertheless, despite their potential, the use of microreactors in persulfate-based oxidation processes is still in its infancy, with limited research conducted on their applicability for water treatment purposes [
25].
The present study aims to investigate the application of a tubular microreactor (2–6 m in length, 1 mm in diameter) submerged in a temperature-controlled water bath for thermally and Fe(II)-catalyzed persulfate activation, both separately and in combination (hybrid mode), to evaluate their efficiency in degrading Safranin O (SO). SO is a phenazine dye of the quinone-imine class, widely used in textiles and as a photosensitizer in electron- and energy-transfer reactions [
26]. It also serves as a sensitizer in visible light photopolymerization [
27]. Given its known carcinogenic nature, any presence of this dye in wastewater poses significant risks to aquatic life and necessitates effective removal strategies [
27]. The study systematically evaluates process performance under various operating conditions, including potassium persulfate (KPS) and dye flow rates, iron solution concentration (flow rate), and solution pH, across a temperature range of 30–80 °C. Additionally, the effectiveness of the thermal and thermo-catalytic persulfate activation processes was assessed in different water matrices, including seawater, river water, tap water, and mineral water, to determine their practical applicability in real-world scenarios. The benefits and limitations of these processes were highlighted, providing insights into the effective application of microreactors in advanced water treatment systems.
3. Results and Discussion
3.1. Temperature Dynamics and Heat Transfer in a Micro-Tubular Reactor
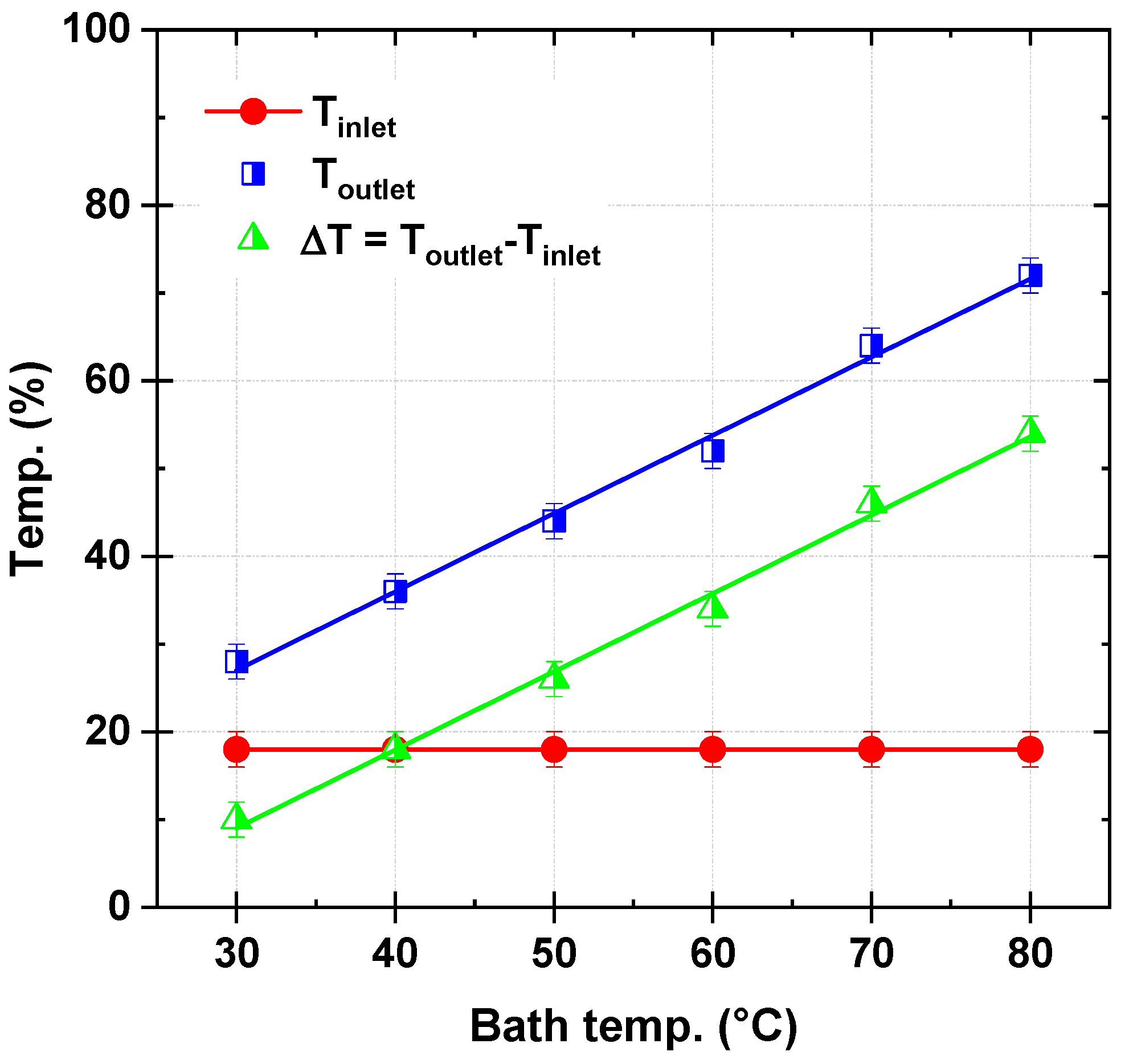
Figure 2 illustrates the recorded reactor outlet temperature (T
outlet) as a function of the bath temperature (T
bath) for a micro-tubular reactor. The reactor inlet temperature (T
inlet) is maintained constant at 18 °C throughout the experiments. The bath temperature is systematically varied from 30 °C to 80 °C, and the corresponding outlet temperatures are measured. The experiments are conducted with a persulfate flow rate of 120 µL/s and a dye solution flow rate of 278 µL/s. The figure also includes the temperature difference (ΔT = T
outlet − T
inlet ) as a function of T
bath, providing a clear depiction of the reactor’s heat transfer performance under steady-state conditions.
The results reveal a strong linear relationship between Toutlet and Tbath, with Toutlet increasing significantly as Tbath rises. At Tbath = 30 °C, Toutlet is recorded as 28 °C, corresponding to a ΔT of 10 °C. In contrast, at Tbath = 80 °C, Toutlet reaches 72 °C, with a ΔT of 54 °C. These results highlight the reactor’s excellent ability to transfer heat efficiently from the external bath to the internal fluid, ensuring a predictable and steady thermal gradient across the reactor length.
The reactor’s exceptional thermal performance, corroborated by a linear correlation observed between Toutlet and Tbath, can be attributed to its unique microscale geometry and the choice of high thermal conductivity material (aluminum). The micro-tubular design, with its 1 mm internal diameter and extended 6 m length, ensures a high surface-area-to-volume ratio. This configuration significantly enhances heat transfer by reducing thermal resistance, facilitating rapid and uniform radial thermal energy distribution along the reactor’s length. For example, the consistent increase in ΔT with Tbath (from 10 °C at Tbath = 30 °C to 54 °C at Tbath = 80 °C) demonstrates the system’s ability to maintain efficient heat exchange even as the operating temperatures increase.
The aluminum material further contributes to the reactor’s heat transfer efficiency due to its high thermal conductivity, which ranges from approximately 237 to 250 W/m K over the tested bath temperature range of 30–80 °C. This property minimizes the temperature drop across the reactor wall, ensuring that the thermal energy from the bath is effectively transferred to the fluid within the reactor. The reactor’s performance is further highlighted by its ability to sustain relatively high flow rates of 120 µL/s for persulfate and 278 µL/s for dye solution without compromising heat transfer efficiency. The data suggest that the reactor design enables near-perfect heat transfer conditions, evidenced by the small temperature gradient (between bath and reactor solution) required to achieve steady-state heat transfer.
The temperature difference (ΔT = T
outlet − T
inlet) as a function of T
bath is another critical indicator of the reactor’s thermal performance. The steady increase in ΔT across the tested temperature range (
Figure 2) suggests minimal thermal resistance and effective convective heat transfer within the reactor. For instance, ΔT values of 10 °C at T
bath = 30 °C, 34 °C at T
bath = 60 °C, and 54 °C at T
bath = 80 °C highlight the proportionality between the bath temperature and the thermal energy transferred to the flowing fluid.
In conclusion, the experimental thermal analysis of
Figure 2 validates the reactor’s high thermal efficiency, emphasizing the importance of its microscale design and material properties. This analysis forms a foundation for further assessments of heat transfer mechanisms in the microreactor, as discussed in the subsequent section of the article.
3.2. Temperature and Persulfate Flow Rate Impact on Dye Conversion
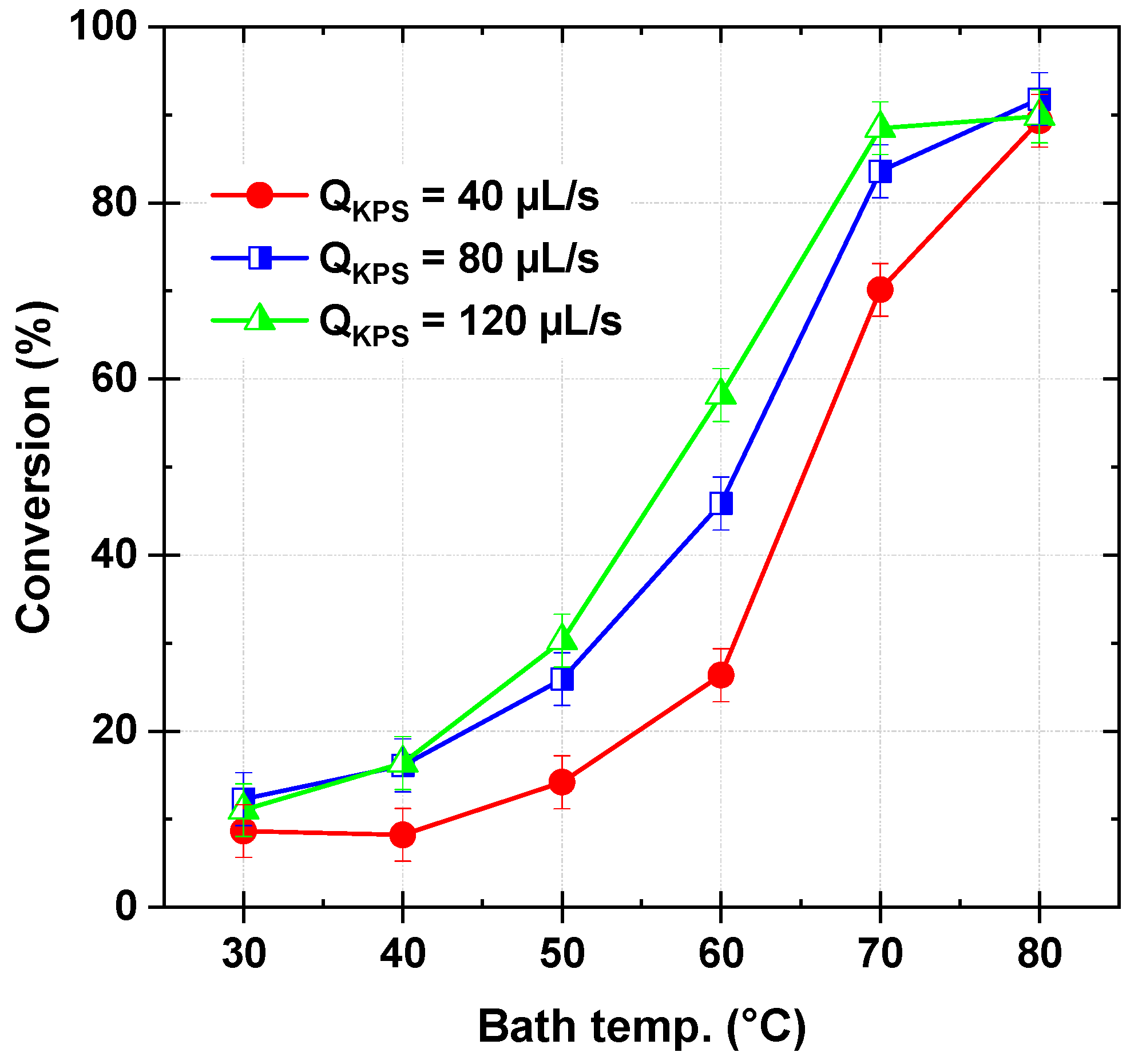
Figure 3 illustrates the dye conversion as a function of bath temperature (30–80 °C) and persulfate flow rate (40–120 µL/s), while maintaining a constant dye flow rate of 278 µL/s and an inlet stream pH of 3. In general, higher persulfate flow rates and temperatures enhance dye conversion, though the effect plateaus beyond certain thresholds. In detail, at the lowest temperature (30 °C) and the lowest persulfate flow rate (40 µL/s), the conversion remains relatively low at 8.65%. A similar trend is observed for higher persulfate flow rates, with conversions of 12.29% at 80 µL/s and 11.03% at 120 µL/s, indicating that, at this temperature (30 °C), increasing the persulfate flow rate does not significantly enhance conversion. This suggests the inability of persulfate to directly oxidize Safranine O, a known persistent and refractory organic pollutant [
26]. As the temperature increases to 40 °C, conversion improves slightly across all flow rates, reaching 8.22%, 16.11%, and 16.37% for 40, 80, and 120 µL/s, respectively. The trend becomes more pronounced at 50 °C, where conversion increases to 14.20%, 25.91%, and 30.27%, demonstrating a greater influence of temperature on dye degradation. At 60 °C, a substantial increase is observed, with conversion reaching 26.37%, 45.86%, and 58.16% for 40, 80, and 120 µL/s, respectively. The highest conversions are recorded at 70 °C, where 70.12%, 83.58%, and 88.47% are achieved. However, at 80 °C, conversion stabilizes or slightly decreases, particularly at the highest flow rate, where it reaches 89.83% at 120 µL/s, a value nearly identical to that at 80 µL/s (91.79%), suggesting a plateau effect.
Since SO
4•– is the primary oxidizing agent under acidic conditions in thermally activated persulfate oxidation processes [
19,
20,
26]. Their generation rate plays a crucial role in determining the dye conversion efficiency. At lower bath temperatures (30–40 °C), persulfate activation (Equation (1)) is limited (k
1 ≤ 9.1 × 10
−4 min
−1 [
28]), resulting in reduced SO
4•− production and a dye conversion below 20%. Under these conditions, the radical’s recombination reaction (SO
4•− + SO
4•− → S
2O
82−, Equation (9)) is negligible due to the low radical concentration, while the scavenging reaction between sulfate radicals and excess persulfate (SO
4•− + S
2O
82− → S
2O
8•− + SO
42−, Equation (6)) remains minimal. As the temperature increases, persulfate activation is enhanced, leading to higher SO
4•− generation and improved dye conversion. At 60 °C, the conversion reaches approximately 60%, highlighting the strong influence of temperature on radical production. Although recombination and scavenging reactions become more significant at elevated temperatures, their impact remains secondary due to the inherently rapid oxidation of dye molecules by sulfate radicals (SO
4•− + S → products, Equation (4)), ensuring efficient degradation.
At 70–80 °C and persulfate flow rates of 40–80 µL/s, high conversion rates are achieved, reaching nearly 89% at 80 µL/s, which originates from effective persulfate activation (k
1 > 10
−3 min
−1 [
28]) under these higher temperature conditions. However, beyond this point, the conversion stabilizes, indicating a plateau effect concerning the persulfate flow rate. This is attributed to increased radical recombination, which competes with the dye oxidation reaction and reduces the availability of SO
4•− for dye degradation. At an excessive persulfate flow rate of 120 µL/s combined with temperatures above 70 °C, conversion efficiency ceases to improve, with the yield at 80 °C remaining identical to that at 70 °C (~89%). This plateau results from two competing inhibitory effects: (i) radical quenching by excess persulfate due to the scavenging reaction (Equation (6)), which depletes SO
4•− before effective dye oxidation occurs, and (ii) enhanced radical self-recombination (Equation (9)), further diminishing the active oxidant concentration. These effects are reflected in temperature measurements, where higher persulfate flow rates lead to increased reaction medium temperatures, accelerating side reactions that ultimately limit dye conversion efficiency.
Therefore, an ideal persulfate flow rate of 120 µL/s at 70 °C is identified, beyond which further increases provide no additional benefit due to radical quenching and recombination. This trend is consistent with experimental findings for Safranine O in batch experiments [
26] and other pollutants under similar conditions, where conversion efficiency peaks at a certain KPS concentration before declining due to excessive radical loss mechanisms.
3.3. Dye Flow Impact Rate on Process Performance
Figure 4 provides three sets of data, each corresponding to different dye flow rates [278 µL/s (3.96 × 10
−3 µmol/s), 565 µL/s (8.05 × 10
−3 µmol/s), and 1100 µL/s (15.68 × 10
−3 µmol/s)], showing the effect of increasing dye flow on conversion efficiency at a fixed oxidant flow rate (120 µL/s). The removal efficiency of SO decreased with increasing dye flow rate, particularly in the activated persulfate zone (T
bath > 50 °C), indicating kinetic and mass transfer limitations. At a lower dye flow rate (Q
dye = 278 µL/s), the conversion efficiency increases significantly from 11.03% at 30 °C to 89.83% at 80 °C. However, as the dye flow rate increases, the removal efficiency declines, especially at higher flow rates. For instance, at Q
dye = 1100 µL/s, the removal efficiency remains below 10% for temperatures up to 60 °C and only reaches 42.20% at 80 °C, significantly lower than the corresponding values for lower Q
dye.
The observed decrease in dye conversion at higher flow rates is primarily attributed to the formation of intermediate degradation products and by-products. These compounds compete with the dye molecules for reactive radicals, particularly SO4•–, leading to a scavenging effect that reduces overall oxidation efficiency. At Qdye = 565 µL/s, for instance, conversion efficiency remains below 15% up to 60 °C and only improves at higher temperatures, indicating a shift in reaction dynamics. Furthermore, kinetic constraints arise as the competition between dye molecules and intermediates alters reaction dynamics, potentially shifting the dominant degradation pathway or requiring higher activation energy for effective removal.
Another crucial factor contributing to this trend is the reduced transit time of dye molecules in the reaction medium. As the dye flow rate increases, the exposure time of each dye molecule to activated persulfate and reactive radicals decreases, limiting the extent of oxidation. This effect is particularly evident at Qdye = 1100 µL/s, where the conversion efficiency remains low across most conditions despite higher oxidant availability. Additionally, mass transfer limitations become more pronounced at higher flow rates, as the reduced contact time between dye molecules and reactive species hinders effective oxidation. In fast-flowing systems, diffusion and mixing effects play a critical role, further influencing degradation efficiency.
Overall, optimizing the balance between dye flow rate, oxidant concentration, and activation conditions is essential to achieving maximum removal efficiency while minimizing radical scavenging effects and incomplete degradation. A careful assessment of operating conditions is necessary to mitigate these limiting factors and enhance process performance.
3.4. Solutions pH Impact on Process Performance
The effect of solution pH on dye conversion was investigated within the pH range of 3–12 and a bath temperature range of 30–80 °C for each pH value. The pH values correspond to those of the inlet streams (dye and persulfate solutions).
Figure 5 presents the dye conversion data as a function of pH and temperature. The results indicate an unappreciable difference in dye conversion across the pH range of 3–9. However, a moderate improvement in conversion is observed under strongly basic conditions (pH 12). This was later attributed to the combined effects of thermal activation and alkaline activation of persulfate. Under basic conditions, persulfate activation becomes more effective, particularly at lower temperatures, leading to enhanced generation of reactive radicals, primarily hydroxyl radicals, which contribute to improved dye degradation. The same outcomes have been retrieved by Ding et al. [
29] for the removal of acyclovir using thermally activated persulfate. The base-catalyzed hydrolysis of persulfate follows two key reactions, resulting in the formation of sulfate radicals (SO
4•−) and superoxide radical anions (O
2•–), the conjugate base of HO
2• [
30]:
According to Equation (3), the generated sulfate radicals are converted into hydroxyl radicals (the major oxidant in the basic medium). Thereafter, these non-selective radicals (i.e.,
•OH) attack the deprotonated molecules of Safranin O (pK
a = 5.8). The observed improvement in dye degradation at pH 12, compared to that at lower pH values (where SO is protonated in an acidic solution), is attributed to its high reactivity with hydroxyl radicals compared to sulfate radicals (
Table 1). According to
Table 1, the reactivity of SO (second-order rate constant) with
•OH is 260-fold greater than with SO
4•–. It is worth mentioning that at lower solution pH, the positively charged SO is attracted to the negatively charged SO
4•–, which may enhance dye degradation under these conditions. However, the effect of this attraction is negligible compared to the higher reactivity of SO with hydroxyl radicals.
The comparable degradation rates observed between pH 3 and 9 can be explained by the progressive conversion of SO
4•– into
•OH as the solution transitions from acidic to basic conditions. According to Liang and Su’s [
19] chemical probing study and ESR spin trapping results [
20]. In heat-activated persulfate systems, SO
4•– is the dominant oxidant under acidic conditions (pH < 7, SO (pK
a = 5.8) is protonated), while at neutral pH, both SO
4•– and
•OH coexist. Under more alkaline conditions (pH > 9, SO is deprotonated),
•OH becomes the predominant radical species. While
•OH has a higher oxidation potential (2.8 V) than SO
4•– (2.5–2.6 V), its recombination rate is significantly higher (
•OH +
•OH → H
2O
2, Equation (10)), with a second-order rate constant of 5.5 × 10
9 M
−1s
−1. In contrast, SO
4•– exhibits slower recombination kinetics (Equation (9),
k = 4 × 10
8 M
−1s
−1), leading to a longer lifetime in solution. This trade-off between oxidation potential and radical self-scavenging leads to a relatively stable dye conversion across the pH range of 3–9 while maintaining the overall increase in dye conversion with activation temperature. All these observations are consistent with those reported in batch-mode systems by Merouani et al. [
26] for safranine O degradation in a heated KPS process.
3.5. Reactor Length Impact on Process Performance
Figure 6 depicts the dye conversion as a function of bath temperature (30–80 °C) and reactor length (L = 2, 4, and 6 m), while maintaining a constant dye and KPS flow rate of 278 µL/s and 120 µL/s, respectively (pH 3). The impact of reactor length on dye conversion follows a clear trend, with longer reactors generally providing higher conversion. At 30 °C, increasing the length from 2 m to 4 m improves conversion from 6.76% to 11.40%, but further extending it to 6 m does not significantly enhance performance (11.03%). A similar pattern is observed at 40 °C, where the 6 m reactor shows the highest conversion (16.38%), but the 4 m reactor performs slightly worse than the 2 m one (8.76% vs. 9.54%). At 50 °C, the conversion increases progressively with length, reaching 30.27% for the longest reactor. These results suggest that at lower temperatures, a longer reactor can compensate for slower reaction kinetics by increasing residence time.
At 60 °C, the positive effect of reactor length persists, with conversions of 41.45%, 54.08%, and 58.17% for 2 m, 4 m, and 6 m, respectively. However, as the temperature increases beyond 70 °C, the difference between reactor lengths diminishes. At 70 °C, all three reactors exhibit high conversion (82.87% for 2 m, 88.54% for 4 m, and 88.47% for 6 m), and at 80 °C, the values converge further (88.72%, 92.32%, and 89.83%). This suggests that above a certain threshold, the process reaches near-complete conversion regardless of reactor length. Similarly, Gupta and Samanta [
31] reported a maximum degradation efficiency of 99% for CIP (ciprofloxacin) at a solution temperature of 70 °C. Moreover, Shen et al. [
32] demonstrated efficient degradation of difloxacin under conditions of 2 mM PDS and 60 °C, achieving a degradation rate of approximately 80–100% within a concentration range of 2–20 mg/L.
Overall, the results indicate that reactor length plays a significant role in dye conversion at bath temperatures up to 70 °C, as it increases residence time and enhances reaction efficiency. However, beyond 70 °C, the effect becomes negligible, likely due to the reaction reaching complete conversion to the effective heat exchange at 80 °C. These findings highlight the importance of optimizing reactor dimensions based on operational conditions to maximize process efficiency.
3.6. Dual-Activation Strategy: Thermal and Fe2⁺-Catalyzed Persulfate
As discussed in the introduction, both heat and dissolved Fe2⁺ cations effectively activate KPS, leading to the formation of SO4•– (Equations (1) and (11)), which subsequently undergoes rapid hydrolysis to produce •OH radicals (Equation (2)). The Fe2⁺ consumed during the reaction can be regenerated through the reduction of Fe3⁺ by KPS (Equation (12)), although this process occurs at a relatively slow rate (k12 < 0.7 M−1s−1). Additionally, Fe2⁺ may compete for reactions with the generated radicals (Equations (13) and (14)), acting as a radical scavenger. These competing reactions become more significant at higher Fe2⁺ concentrations. Operating the Fe2⁺/KPS system at elevated temperatures combines thermal and iron-catalyzed activation, referred to as a thermocatalytic activation process. While higher radical production can be achieved, the effectiveness of these radicals in pollutant degradation depends on several factors, including temperature, KPS concentration, iron concentration, and pollutant load. Excessive radical production may favor radical-radical recombination over radical-organic oxidation, reducing the overall efficiency of the process. Therefore, assessing both the benefits and limitations of the thermo-catalytic process is crucial to optimizing its application. However, this hybrid system must operate within an acidic pH range, similar to the Fe2⁺/KPS process, to prevent iron precipitation. In this study, the solution pH was maintained at 3, ensuring consistency with the Fe2⁺/KPS system.
In the following sections, Fe2⁺ ions were added to the dye solution at concentrations of 0.01, 0.05, 0.1, and 0.2 mM, with a pH adjusted to 3 for all conditions. The pH of the KPS solution (second stream) was also pre-adjusted to 3 to ensure the stability of ferrous ions in the solution mixed streams at the reactor inlet. The impact of varying Fe2⁺ concentrations was examined over a range of bath temperatures (30–80 °C) to gain a broader understanding of the process and identify potential areas for improvement and optimization. Additionally, the system was first tested using deionized water as the matrix, followed by experiments with different water matrices under the same range of iron concentrations and bath temperatures. This approach allows us to identify the most influential factors, clearly highlighting the benefits and limitations of the hybrid thermo-catalytic system in the microreactor setup.
3.6.1. Performance of the Thermo-Catalytic Process in Deionized Water
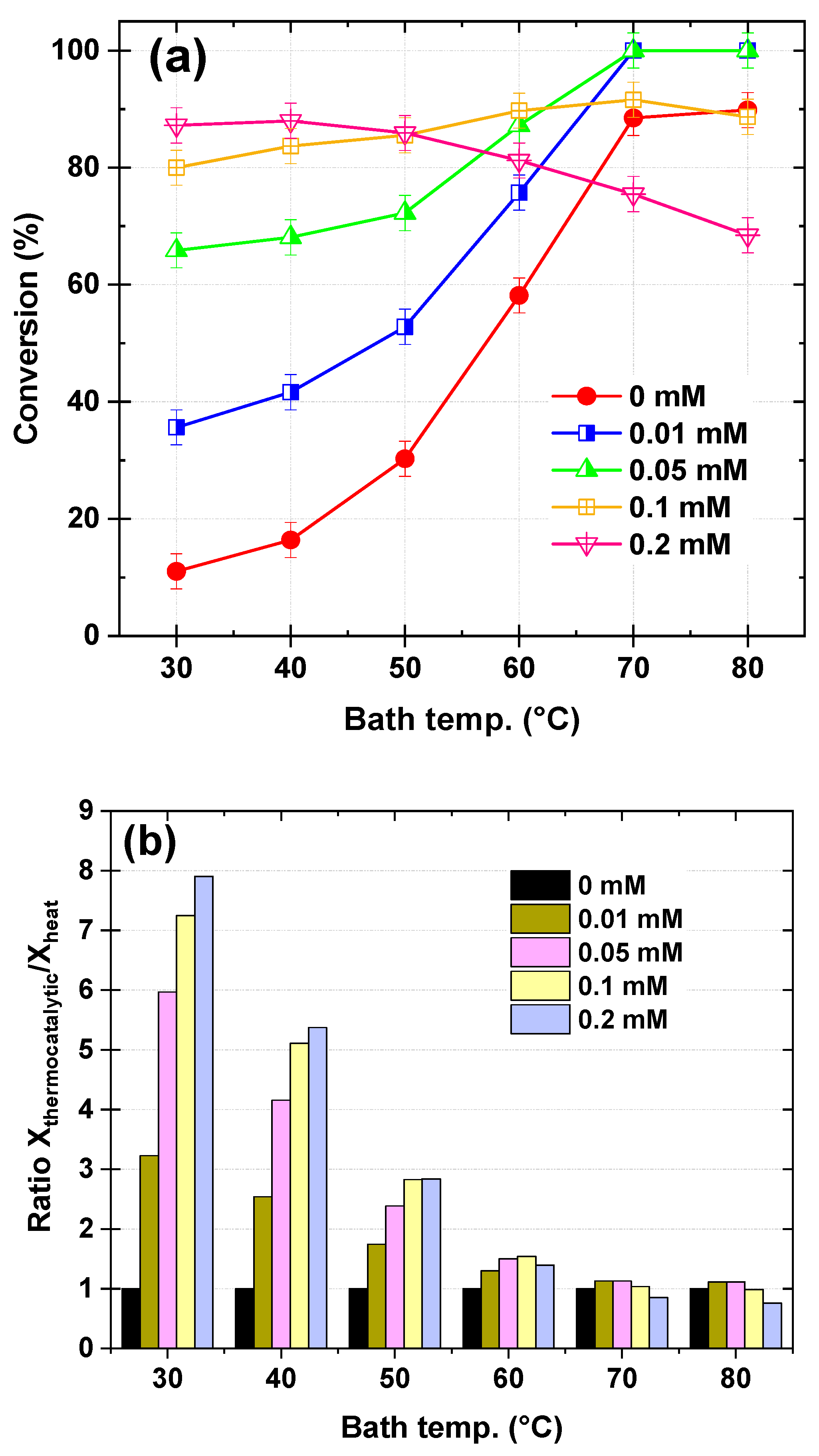
Figure 7a presents the effect of Fe
2⁺ concentration in the dye solution on the conversion of Safranin O at different bath temperatures, with a fixed KPS flow rate of 120 µL/s and a dye solution flow rate of 278 µL/s. The Fe
2⁺ concentration was varied from 0 to 0.2 mM, corresponding to an inlet Fe
2⁺ molar flow rate of 0, 2.78 × 10
−3, 13.9 × 10
−3, 27.8 × 10
−3, and 55.6 × 10
−3 µmol/s, respectively. At low temperatures (30–40 °C), Safranin O conversion increased significantly with Fe
2⁺ concentration, rising from 11.03% (0 mM Fe
2⁺) to 87.22% (0.2 mM Fe
2⁺) at 30 °C and from 16.38% (0 mM Fe
2⁺) to 88.00% (0.2 mM Fe
2⁺) at 40 °C. In the temperature range 40–50 °C, the conversion continued to improve, reaching 30.27% (0 mM Fe
2⁺) to 85.91% (0.2 mM Fe
2⁺) at 50 °C. At high temperatures (60–80 °C), the conversion peaked at 70 °C for lower Fe
2⁺ concentrations (100% for 0.01 mM and 0.05 mM Fe
2⁺), while higher Fe
2⁺ concentrations (0.1 mM and 0.2 mM) showed a decline in conversion at temperatures above 60 °C, dropping to 68.44% for 0.2 mM Fe
2⁺ at 80 °C.
Figure 7b provides additional insight by dividing the conversion efficiency of the thermo-catalytic process (Fe
2⁺/Heat/KPS) by that of the thermal activation of KPS (Heat/KPS) alone, expressed as a ratio. This ratio highlights the beneficial effect of Fe
2⁺ in enhancing the degradation of Safranin O. At low temperatures (30–40 °C), the presence of Fe
2⁺ significantly boosts the conversion ratio, with values increasing from 3.23 (0.01 mM Fe
2⁺) to 7.90 (0.2 mM Fe
2⁺) at 30 °C and from 2.54 (0.01 mM Fe
2⁺) to 5.37 (0.2 mM Fe
2⁺) at 40 °C. In the temperature range 50–60 °C, the conversion ratio remains elevated but begins to decline, with values ranging from 1.74 (0.01 mM Fe
2⁺) to 2.84 (0.2 mM Fe
2⁺) at 50 °C and from 1.30 (0.01 mM Fe
2⁺) to 1.54 (0.1 mM Fe
2⁺) at 60 °C. At high temperatures (70–80 °C), the ratio decreases further, particularly for higher Fe
2⁺ concentrations (0.1 mM and 0.2 mM), where the ratio drops to 0.76 (0.2 mM Fe
2⁺) at 80 °C. In this case, the combination of heat and catalytic persulfate activation leads to an antagonistic effect under high-temperature and high Fe
2⁺ dosage conditions, where excessive radical generation promotes parasitic reactions, reducing overall efficiency.
The results of
Figure 7 demonstrate the dependence of the Fe
2⁺ impact on the operating bath temperature, highlighting the interplay between Fe
2⁺-catalytic activation and thermal activation of KPS toward SO removal. At low temperatures (30–40 °C), the activation of KPS is primarily driven by Fe
2⁺ catalysis, as described by the reaction of Equation (11). Thermal activation of KPS is negligible in this range, making Fe
2⁺ the dominant contributor to sulfate radical (SO
4•–) generation. Increasing the Fe
2⁺ concentration significantly enhances Safranin O degradation, with the conversion ratio reaching up to 7.90 at 30 °C for 0.2 mM Fe
2⁺. This is attributed to the direct proportionality between Fe
2⁺ concentration and SO
4•– generation, which drives the degradation process. Importantly, no plateau or optimum Fe
2⁺ concentration is observed in this range, unlike in batch systems. This can be explained by the relatively low impact of competitive reactions, such as Fe(II) + SO
4•– → SO
42− + Fe(III) (Equation (13)), which consume SO
4•– radicals. In this temperature range, the degradation reaction dominates, leading to a continuous increase in conversion with Fe
2⁺ concentration.
In the bath temperature range of 50–60 °C, thermal activation of KPS becomes more significant, contributing to SO4•– generation alongside Fe2⁺ catalysis. The beneficial effect of increasing Fe2⁺ concentration persists but is limited to concentrations up to 0.1 mM, beyond which a plateau in SO conversion is observed. This plateau is attributed to the increased significance of competitive radical consumption reactions: Fe(II) + SO4•– → SO42− + Fe(III) (Equation (13)) and the self-recombination of SO4•– radicals: SO4•– + SO4•– → S2O82− (Equation (9)). These reactions become more pronounced at higher Fe2⁺ concentrations, leading to a balance between radical generation and consumption. As a result, the conversion efficiency plateaus above 0.1 mM Fe2⁺, indicating an optimal Fe2⁺ dosage for this temperature range.
At higher temperatures (above 60 °C), thermal activation of KPS becomes highly efficient, generating a large number of SO4•– radicals. When combined with Fe2⁺ catalysis, the excess radical concentration accelerates parasitic reactions, i.e., Equations (9) and (13). This leads to a shift in the optimal Fe2⁺ dosage to 0.05 mM, beyond which further increases in Fe2⁺ concentration (e.g., 0.2 mM) become detrimental to the degradation efficiency. The conversion ratio decreases significantly at higher Fe2⁺ concentrations, dropping to 0.76 at 80 °C for 0.2 mM Fe2⁺. This highlights the need to balance thermal and catalytic activation processes, as excessive radical generation at high temperatures exacerbates parasitic reactions, reducing the overall efficiency.
3.6.2. Thermal and Thermo-Catalytic Process Performance in Real/Complex Water Matrices
Figure 8a,b illustrates the performance of the heat-activated persulfate and thermo-catalytic systems for Safranin O conversion across different water matrices (tap water, mineral water, seawater, secondary treated effluent from a wastewater treatment plant (SEWWTP), and river water) at bath temperatures ranging from 30 to 80 °C. Deionized water was used as a baseline for comparison. Over the entire temperature range, no significant difference in performance was observed between deionized water, tap water, and mineral water for both systems. This is attributed to the relatively low mineral content in these matrices, as certain anions (e.g., chloride, bicarbonate, nitrate, nitrite) are known to scavenge reactive radicals and oxidants (SO
4•– and KPS in this case in this system).
Table 3 summarizes some of these scavenging reactions. The impact of these reactions depends on the concentration of anions, which is lower in tap and mineral water, as detailed in
Table 2, which provides the main characteristics of each water matrix.
Notably, all matrices, without exception, demonstrated higher treatment performance, similar to deionized water, at temperatures of 70 and 80 °C for both the heat-activated persulfate and thermo-catalytic systems. However, the impact of matrices such as river water, SEWWTP, and seawater was particularly pronounced in the lower temperature range (30–60 °C). For the heat-activated persulfate system, the reduced performance in river water and SEWWTP at lower temperatures (30–40 °C, non-activating temperatures) was primarily attributed to the consumption of persulfate molecules by organic constituents in these matrices, particularly fulvic and humic acids, which are typical natural organic matter (NOM, k
•OH/NOM = 2.23 × 10
8 M
−1s
−1, k
SO4•−/NOM = 6.0 × 10
6 M
−1s
−1 [
33,
34]) present at concentrations of several ppm. Similarly, the high chloride content in seawater, which also consumes persulfate (Equations (18) and (19) in
Table 3), exerted a comparable impact on NOM in river water and SEWWTP. At 50–60 °C, where persulfate is activated into SO
4•– as the primary oxidant, both NOM and chloride effectively compete for SO
4•–, leading to reduced dye conversion. This observation aligns with previous studies by Merouani et al. [
26], Gu et al. [
35], and Chen et al. [
36], who reported retardation in the heat-activated KPS oxidation of Safranin O, diclofenac, and 1,1,1-trichloroethane in the presence of humic acids in batch mode operation. Merouani et al. [
26] also noted a decline in the performance of heat-activated persulfate at 50 °C in the presence of excessive chloride, attributed to the scavenging of sulfate radicals by chloride, as described by Reactions 18–30 in
Table 3. The secondary chlorine radicals (Cl
•, Cl
2•–…) generated from these reactions are less reactive than SO
4•–, thereby resulting in retardation of the process.
Table 3.
Reactions between mineral anions and free radicals (
•OH and SO
4•−) generated in the Fe(II)/KPS system [
37,
38,
39]. (Indices:
d: direct and
r: reverse).
Table 3.
Reactions between mineral anions and free radicals (
•OH and SO
4•−) generated in the Fe(II)/KPS system [
37,
38,
39]. (Indices:
d: direct and
r: reverse).
| Chloride Reactions |
|---|
| Cl− + SO4•− ⇌ SO42− + Cl• | kd = 4.7 × 108 M−1s−1, kr = 2.5 × 108 M−1s−1 | (18) |
| Cl− + •OH ⇌ HClO•− | k d = 4.7 × 108 M−1s−1, kr = 2.5 × 108 M−1s−1 | (19) |
| Cl• + Cl− ⇌ Cl2•− | k d = 8.5 × 109 M−1s−1, kr = 6 × 104 M−1s−1 | (20) |
| Cl• + H2O → HClO•− + H+ | k = 2.5 × 105 s−1 | (21) |
| Cl• + OH−→ HClO•− | k = 1.8 × 1010 s−1 | (22) |
| Cl2•− + H2O → Cl− + HClO•− + H+ | k = 1 × 105 s−1 | (23) |
| HClO•− ⇌ •OH + Cl− | k d = 6.1 × 109 M−1s−1, kr = 4.3 × 109 M−1s−1 | (24) |
| HClO•− + H+ → Cl• + H2O•− | k = 2.1 × 1010 M−1s−1 | (25) |
| HClO•− + Cl− → Cl2•− + OH− | k = 1 × 105 M−1s−1 | (26) |
| Cl2•− + Cl2•− → Cl2 + 2Cl− | k = 6.3 × 108 M−1s−1 | (27) |
| Cl2•− + •OH → HClO + Cl− | k = 1 × 109 M−1s−1 | (28) |
| Cl2•− + Cl• → Cl2 + Cl− | k = 2.1 × 109 M−1s−1 | (29) |
| Cl•+ Cl•→ Cl2 | k = 8.8 × 107 M−1s−1 | (30) |
| Nitrite reactions |
| NO3− + SO4•− → NO3• + SO42− | k = 2.1 × 109 M−1s−1 | (31) |
| NO3− + •OH →N3• + OH− | | (32) |
| Nitrite reactions |
| NO2− + SO4•− → SO42− + NO2• | k S1 = 8.8 × 108 M−1s−1 | (33) |
| NO2− + •OH → OH− + NO2• | k S1 S1 = 1 × 1010 M−1s−1 | (34) |
| Bromide reactions |
| SO4•− + Br− → Br• + SO42− | k = 3.5 × 109 M−1s−1 | (35) |
| •OH + Br− → BrOH•− | k = 1.1 × 1010 M−1s−1 | (36) |
| BrOH•− + H+ → Br• + H2O | k = 4.4 × 1010 M−1s−1 | (37) |
| Br• + Br− ⇌ Br2•− | kd = 1.2×1010 M−1s−1, kr = 1.9 × 104 M−1s−1 | (38) |
| BrOH•− + Br− → Br2•− + OH− | k =1.9 × 108 M−1s−1 | (39) |
| Br2•− + •OH → HOBr + Br− | k = 1 × 109 M−1s−1 | (40) |
| Br2•− + Br2•− → Br2 + 2Br− | k = 1.9 × 109 M−1s−1 | (41) |
| Br2•− + Br• → Br2 + Br− | k = 2 × 109 M−1s−1 | (42) |
| BrO• + BrO• + H2O → BrO2− + OBr− + 2H+ | k = 5 × 109 M−1s−1 | (43) |
| Other Br−/Cl−-induced reactions in seawater |
| Cl• + Br− ⇌ ClBr•− | kd = 1.2 × 1010 M−1s−1, kr = 2 × 103 s−1 | (44) |
| Cl2•− + Br− ⇌ ClBr•− + Cl− | kd =1.2 × 1010 M−1s−1, kr = 1.9 × 103 s−1 | (45) |
| ClOH•− + Br− ⇌ ClBr•− + OH− | kd =4.0 × 109 M−1s−1, kr = 1.1 × 102 s−1 | (46) |
| Br2•− + Cl2•− → Br2 + 2Cl− | k = 4 × 109 M−1s−1 | (47) |
| BrCl•− + •OH → BrCl + OH− | k = 1 × 109 M−1s−1 | (48) |
| BrCl•− + Cl2•− → BrCl + 2Cl− | k = 2 × 109 M−1s−1 | (49) |
| BrCl•− + Br2•− → Br2 + Cl− +Br− | k = 4 × 109 M−1s−1 | (50) |
| BrCl•− + BrCl•− → BrCl + Br− + Cl− | k = 1 × 109 M−1s−1 | (51) |
Similar reaction mechanisms explain the reduced performance of seawater in the thermo-catalytic activation process (30–60 °C), where both sulfate and
•OH radicals contribute to the dye degradation. However, the reductive impact of matrices (seawater, river water, and SEWWTP) on dye conversion in the thermo-catalytic process (heat/Fe
2⁺/KPS) is more pronounced than in the heat-activated KPS system over the 30–60 °C range. In fact, according to Meoruani et al.’s recent investigation [
21]. The Fe(II)/KPS system (at pH 3) is very sensitive to the matrix component. NOM, natural mineral water, seawater, river water, and secondary effluent of wastewater treatment plant (SEWWTP) appreciably reduced the degradation of Pararosaniline dye [
21]. The author attributed the reductive effect to the quenching of oxidative species (radicals and KPS) by the matrix components (mineral anions and NOM), with some reactions reported in
Table 3. Herein, we should add that the potential consumption of ferrous ions (Fe
2⁺) by NOM and chloride may also reduce the available Fe
2⁺ for KPS activation and subsequent SO
4•– generation. Finally, the negligible influence of matrix type on dye conversion at 70 and 80 °C is attributed to the overwhelming quantity of radicals generated under these high-temperature activation conditions. Thus, even in the presence of NOM (river water and SEWWTP) or excess chloride (seawater), the radical concentration is sufficient to react efficiently with both SO and other constituents in the solution without significantly affecting the oxidation of the target organic pollutant (SO).
Figure 8c provides a comparative analysis (ratio) of the thermo-catalytic process (Fe
2⁺/Heat/KPS) relative to the heat-activated KPS system across different water matrices. At lower temperatures (30–40 °C), the ratio of conversion (thermo-catalytic to heat-activated) is significantly higher, particularly in seawater, SEWWTP, and river water, with values reaching up to 13.52 for SEWWTP at 30 °C. This highlights the enhanced efficiency of the thermo-catalytic process at low temperatures, where Fe
2⁺ activation plays a dominant role in generating sulfate radicals. However, the ratio decreases as the temperature increases, approaching unity at 70–80 °C for all matrices. This indicates that the contribution of Fe
2⁺ catalysis diminishes at higher temperatures, where thermal activation becomes the primary driver of radical generation. The higher ratios observed in seawater, SEWWTP, and river water at low temperatures (30–60 °C) can be attributed to the ability of Fe
2⁺ to partially mitigate the inhibitory effects of chloride and natural organic matter (NOM) on persulfate activation. However, as the temperature increases, the overwhelming generation of radicals in the heat-activated system reduces the relative advantage of the thermo-catalytic process. Notably, the ratio remains close to or slightly above 1 at high temperatures (70–80 °C), suggesting that the thermo-catalytic process neither significantly enhances nor hinders performance under these conditions. This aligns with the earlier observation that the radical concentration at high temperatures is sufficient to overcome the scavenging effects of chloride and NOM, regardless of the presence of Fe
2⁺. Overall,
Figure 8 underscores the importance of Fe
2⁺ catalysis in improving degradation efficiency at lower temperatures, particularly in complex water matrices.
3.7. Mineralization Assessment
To follow the mineralization of the dye along with its removal, TOC analysis was conducted at the outlet stream of the reactor for two bath temperatures (50 °C and 70 °C) in the absence and presence of Fe
2⁺ (0.05 mM). The KPS and dye inlet flow rates were maintained at 120 µL/s and 278 µL/s, respectively. The results, presented in
Table 4 in terms of removal percentages, show that while higher dye conversion was achieved, the TOC removal was significantly lower, indicating that mineralization kinetics occur at a slower rate compared to dye degradation. This trend is logical, as mineralization involves the conversion of dye degradation byproducts into mineral species (e.g., CO
2, H
2O), which is a more complex and slower process. Compared to the literature works shown in
Table 5, our obtained results are in good concordance in terms of pollutant removal as well as the extent of mineralization. Considering the difference in the reaction mode (continuous for our study and batch for literature), the degradation kinetics in the present study (in terms of SO removal and mineralization) are relatively faster compared to those reported in
Table 5 (literature). These findings clearly demonstrate the efficacy of our system for the thermal activation of persulfate, even the very short residence time in our microtubular reactor.
At 50° C, in the absence of Fe2⁺, the TOC removal was 15 ± 2%, while the dye removal reached 30.27 ± 2%. When Fe2⁺ was added, the TOC removal increased to 37.5 ± 2%, and the dye removal significantly improved to 72 ± 2%. This demonstrates that Fe2⁺ enhances both dye degradation and mineralization, likely due to its role in activating persulfate and generating more reactive radicals.
At 70 °C, the TOC removal in the absence of Fe2⁺ was 42 ± 2%, with dye removal reaching 88.47 ± 2%. With the addition of Fe2⁺, the TOC removal increased to 52.5 ± 2%, and the dye removal reached 100%. These results further confirm that higher temperatures and the presence of Fe2⁺ significantly improve both dye degradation and mineralization. However, the TOC removal remains lower than the dye removal, highlighting the slower kinetics of mineralization compared to dye degradation. This trend is expected, as mineralization involves not only breaking down the dye structure but also converting its intermediate by-products into final mineral species such as CO2 and H2O. The observed gap between dye disappearance and TOC removal underscores the stepwise nature of organic compound oxidation, where complete mineralization requires prolonged reaction times (longer reactor length or using lower flow rates) or additional oxidative conditions. These findings emphasize the need for extended residence times or supplementary treatment steps when targeting both pollutant degradation and total organic removal in practical applications.
Overall, the results underscore the importance of the thermocatalytic KPS activation process (temperature and Fe2⁺) in enhancing both dye removal and mineralization, while also emphasizing the inherent complexity and slower rate of mineralization compared to initial dye degradation.
3.8. Conclusions
This study highlights the effectiveness of a tubular microreactor system for thermally and Fe2⁺-catalyzed persulfate activation in the degradation of the persistent dye Safranin O. The research demonstrates the potential of this approach as an AOP for wastewater treatment, emphasizing the interplay between thermal activation, catalytic enhancement, and reactor design in optimizing pollutant removal efficiency.
Key findings reveal that dye conversion improves significantly with increasing bath temperature, reaching a maximum at 70 °C, beyond which radical recombination limits further improvement. Reactor length plays a crucial role at lower temperatures but has a diminished effect at higher temperatures due to efficient heat activation. Additionally, increasing persulfate flow rates enhances dye degradation, though a plateau effect occurs at 80 °C. The introduction of Fe2⁺ substantially improves conversion at moderate temperatures (40–50 °C), with operable performance observed at 0.01–0.05 mM Fe2⁺. However, excessive Fe2⁺ concentrations (>0.1 mM) lead to radical scavenging effects at higher temperatures, reducing efficiency.
Water matrix composition affects SO degradation, particularly at lower temperatures (30–60 °C), where secondary treated effluent (SEWWTP), river water, and seawater exhibit greater inhibitory effects compared to deionized, tap, and mineral water. Nonetheless, at higher temperatures (70–80 °C), all matrices exhibit comparable removal efficiencies (higher), indicating that thermal activation can mitigate matrix effects. Moreover, the thermo-catalytic system demonstrates superior mineralization performance, significantly enhancing overall treatment efficiency.
All the above findings establish the thermo-catalytic persulfate activation process as a viable and scalable strategy for water treatment applications, particularly in flow-based microreactor systems. Future research should focus on optimizing reactor design, exploring alternative catalysts, and evaluating long-term operational stability for practical implementation in industrial and environmental remediation settings. Additionally, the observed mass transport limitations in the present work should be thoroughly addressed in future studies to improve the efficacy of the tubular system for efficient activation of persulfate and other oxidants in the presence of various catalysts. Additionally, while the present study provides experimental data demonstrating the performance of the micro plug-flow reactor for degrading Safranin O, future work will focus on modeling the system using material and thermal balances to determine concentration and temperature profiles along the reactor length, thereby enabling optimization of reactor dimensions for fixed inlet flow conditions.




 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}