
Probing Heterolytic H2 Dissociation on Heterogeneous Catalysts: A Brief Review of Experimental Strategies


Abstract
1. Introduction
2. Fundamental Mechanisms of Hydrogen Dissociation on Heterogeneous Catalysts
2.1. Hydrogen Adsorption on Catalyst Surfaces
2.2. Homolytic Dissociation vs. Heterolytic Dissociation of H2
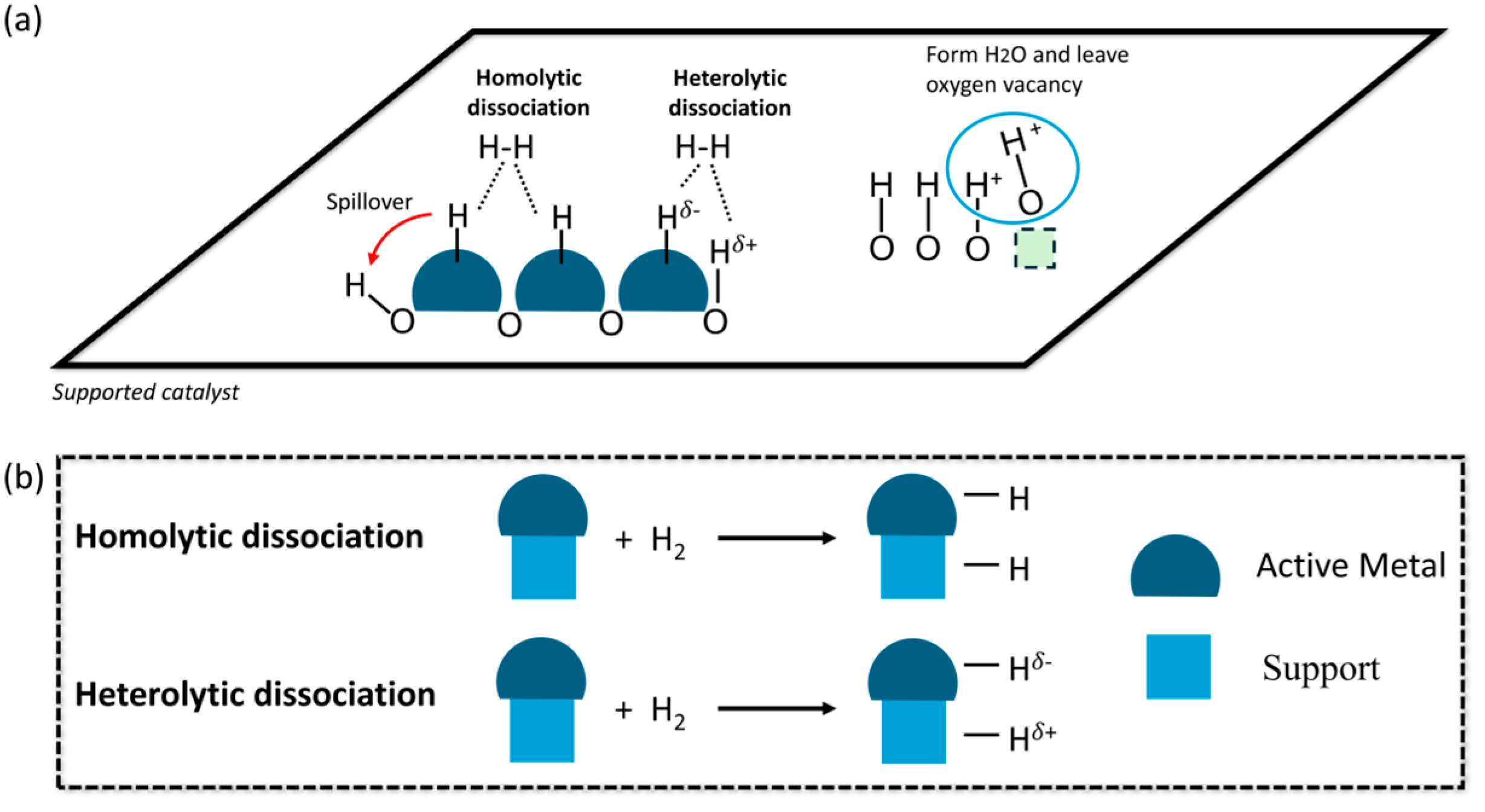
2.3. Classification of Catalyst for H2 Heterolytic Dissociation
3. Experimental Methods for Studying H2 Adsorption on Metal Oxides
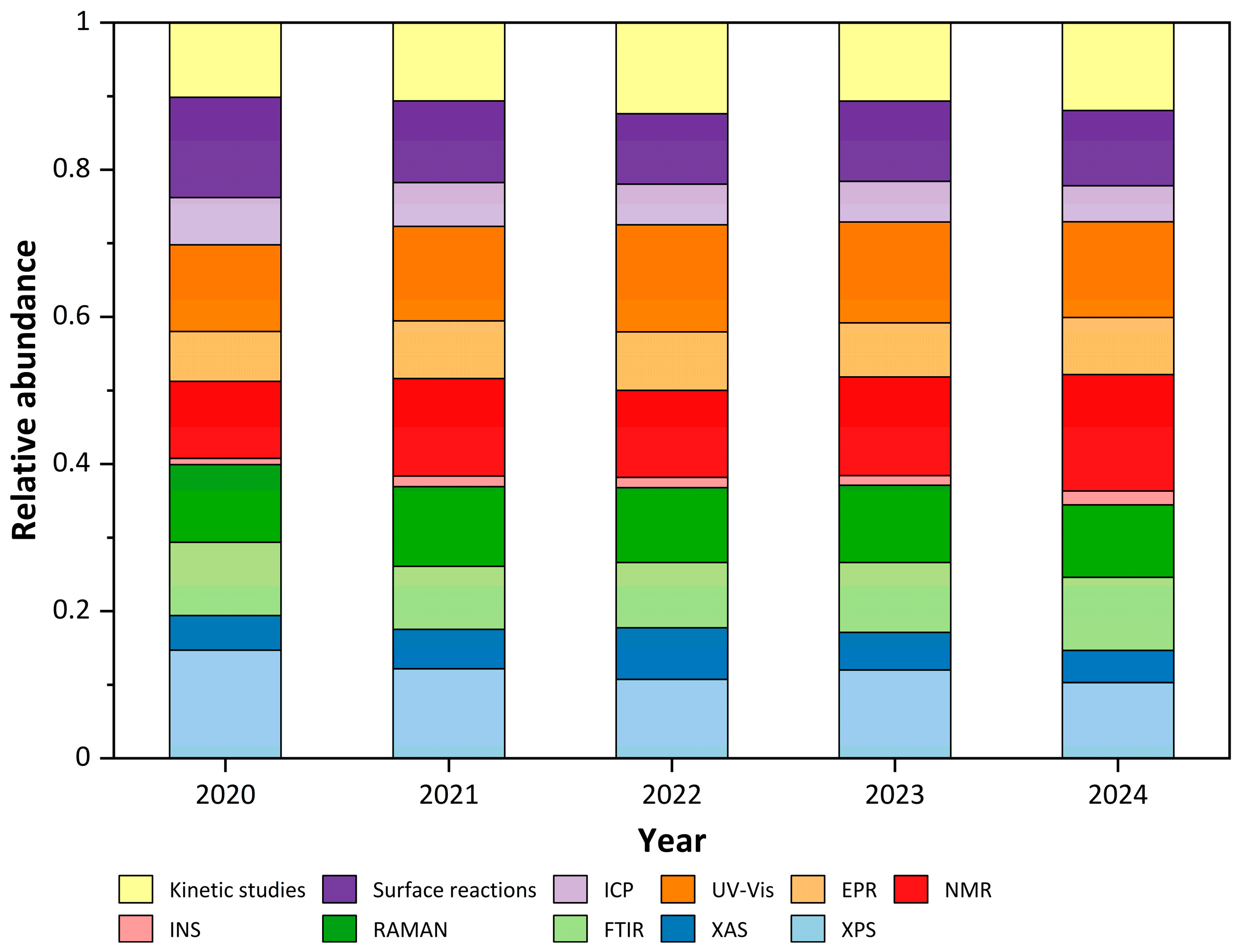
3.1. Spectroscopic Techniques for Catalyst Characterization
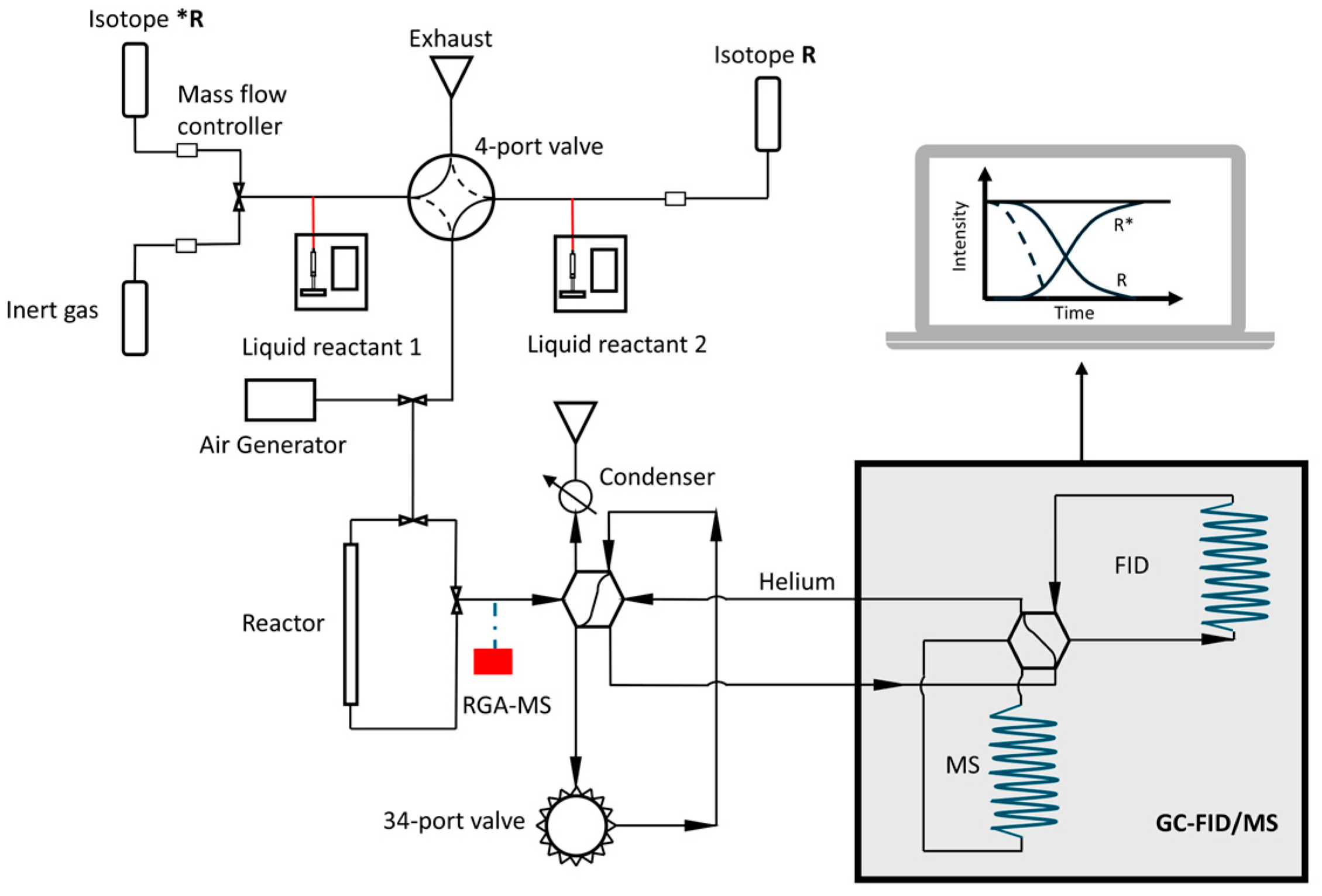
3.2. Surface Reaction Probing: Temperature-Programmed Techniques

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Common Applications | Coupled Method | Example Studies |
|---|---|---|---|
| Temperature-Programmed Desorption (TPD) | Titrate active sites with corresponding titrants; qualify the adsorption strength | ICP-OES + CO2-TPD | [48] |
| XPS + H2-TPD | [48] | ||
| NH3-FTIR + NH3-TPD | [83] | ||
| Temperature-Programmed Reduction (TPR) | Quantify the reducible elements or sites in a material; qualify the interaction strength | in situ IR + in situ electron energy loss spectroscopy (EELS) + H2-TPR | [54] |
| [H2S + H2]-TPD + H2-TPR | [84] | ||
| H2-TPR | [85] | ||
| in situ electron paramagnetic resonance (EPR) + H2-TPR | [86] | ||
| Temperature-Programmed Oxidation (TPO) | Investigate the oxidation state changes | TPR-TPO cycles | [87] |
| Temperature-Programmed Surface Reaction (TPSR) | Monitor and quantify the consumption of reactants and the generation of products | mass spectroscopy (MS) + TPSR | [88] |
| [O2 + CO + H2]-TPSR | [89] |
3.3. Kinetic Studies for Mechanistic Insights
4. Challenges and Future Perspectives
- The instrument must withstand elevated temperature, pressures, and a hydrogen-rich environment.
- The instrument or the group of instruments can identify the active intermediates and substrates.
- The characterization tools have enough time resolution to capture the formation of transient intermediates or surface changes.
- The combination of these real-time or operando techniques and transient analysis is crucial to building meaningful correlations between surface chemistry and catalytic kinetics. Thus, it can draw a complete mechanism for the reaction.
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, S.; Salim, O.; Piri, M. The Effects of Pore Shape and Geometry on the Storage of CO2 in Mesoporous Media. Mater. Today Sustain. 2025, 29, 101076. [Google Scholar] [CrossRef]
- Lou, X.; Chakraborty, N.; Karpyn, Z. Experimental Investigation of Shale Rock Properties Altering In-Situ Gas Density and Storage. Front. Earth Sci. 2022, 10, 877551. [Google Scholar] [CrossRef]
- Zheng, L.; Wei, P.; Zhang, Z.; Nie, S.; Lou, X.; Cui, K.; Fu, Y. Joint exploration and development: A self-salvation road to sustainable development of unconventional oil and gas resources. Nat. Gas Ind. B 2017, 4, 477–490. [Google Scholar] [CrossRef]
- Nazir, H.; Louis, C.; Jose, S.; Prakash, J.; Muthuswamy, N.; Buan, M.E.; Flox, C.; Chavan, S.; Shi, X.; Kauranen, P.; et al. Is the H2 economy realizable in the foreseeable future? Part I: H2 production methods. Int. J. Hydrogen Energy 2020, 45, 13777–13788. [Google Scholar] [CrossRef]
- Aireddy, D.R.; Ding, K. Heterolytic dissociation of H2 in heterogeneous catalysis. ACS Catal. 2022, 12, 4707–4723. [Google Scholar] [CrossRef]
- Jing, Y.; Wang, Y. Heterolytic dissociation of H2 and bond activation: Spotting new opportunities from a unified view. Chem Catal. 2023, 3, 100515. [Google Scholar] [CrossRef]
- Wan Kim, T.; Kim, D.; Hyun Kim, S.; Suh, Y.W. Heterolytic H2 Activation in Heterogeneous Hydrogenation/Hydroprocessing Catalysis. ChemCatChem 2024, 16, e202301581. [Google Scholar] [CrossRef]
- Tang, C.; Tang, S.; Sha, F.; Han, Z.; Feng, Z.; Wang, J.; Li, C. Insights into the selectivity determinant and rate-determining step of CO2 hydrogenation to methanol. J. Phys. Chem. C 2022, 126, 10399–10407. [Google Scholar] [CrossRef]
- Saeys, M.; Reyniers, M.F.; Thybaut, J.W.; Neurock, M.; Marin, G.B. First-principles based kinetic model for the hydrogenation of toluene. J. Catal. 2005, 236, 129–138. [Google Scholar] [CrossRef]
- Jeong, H.; Kim, T.W.; Kim, M.; Han, G.B.; Jeong, B.; Suh, Y.W. Mesoporous acidic SiO2–Al2O3 support boosts nickel hydrogenation catalysis for H2 storage in aromatic LOHC compounds. ACS Sustain. Chem. Eng. 2022, 10, 15550–15563. [Google Scholar] [CrossRef]
- Kim, T.W.; Jeong, H.; Kim, D.; Jo, Y.; Jung, H.J.; Park, J.H.; Suh, Y.W. Feasible coupling of CH4/H2 mixtures to H2 storage in liquid organic hydrogen carrier systems. J. Power Sources 2022, 541, 231721. [Google Scholar] [CrossRef]
- Nie, L.; Resasco, D.E. Kinetics and mechanism of m-cresol hydrodeoxygenation on a Pt/SiO2 catalyst. J. Catal. 2014, 317, 22–29. [Google Scholar] [CrossRef]
- Tadepalli, S.; Halder, R.; Lawal, A. Catalytic hydrogenation of o-nitroanisole in a microreactor: Reactor performance and kinetic studies. Chem. Eng. Sci. 2007, 62, 2663–2678. [Google Scholar] [CrossRef]
- Jin, P.; Luo, N.; Wang, F. Analogy in the Mechanism of Heterolytic H2 Dissociation. ACS Catal. 2024, 14, 18639–18650. [Google Scholar] [CrossRef]
- Broom, D.P.; Webb, C.J.; Hurst, K.E.; Parilla, P.A.; Gennett, T.; Brown, C.M.; Zacharia, R.; Tylianakis, E.; Klontzas, E.; Froudakis, G.E.; et al. Outlook and challenges for hydrogen storage in nanoporous materials. Appl. Phys. A 2016, 122, 151. [Google Scholar] [CrossRef]
- Nobuhara, K.; Kasai, H.; Diño, W.A.; Nakanishi, H. H2 dissociative adsorption on Mg, Ti, Ni, Pd and La surfaces. Surf. Sci. 2004, 566, 703–707. [Google Scholar] [CrossRef]
- Shi, H.; Yuan, H.; Li, Z.; Wang, W.; Li, Z.; Shao, X. Low-temperature heterolytic adsorption of H2 on ZnO (1010) surface. J. Phys. Chem. C 2019, 123, 13283–13287. [Google Scholar] [CrossRef]
- Nour Ghassemi, E. Hydrogen dissociation on metal surfaces: A semi-empirical approach. Catalysis 2004, 20, 679–694. [Google Scholar]
- Christmann, K. Interaction of hydrogen with solid surfaces. Surf. Sci. Rep. 1988, 9, 1–163. [Google Scholar] [CrossRef]
- Yang, C.; Ma, S.; Liu, Y.; Wang, L.; Yuan, D.; Shao, W.P.; Zhang, L.; Yang, F.; Lin, T.; Ding, H.; et al. Homolytic H2 dissociation for enhanced hydrogenation catalysis on oxides. Nat. Commun. 2024, 15, 540. [Google Scholar] [CrossRef]
- Boronat, M.; Illas, F.; Corma, A. Active sites for H2 adsorption and activation in Au/TiO2 and the role of the support. J. Phys. Chem. A 2009, 113, 3750–3757. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.N.; Li, J.; Kitano, M.; Hosono, H. Unique nanocages of 12CaO· 7Al2O3 boost heterolytic hydrogen activation and selective hydrogenation of heteroarenes over ruthenium catalyst. Green Chem. 2017, 19, 749–756. [Google Scholar] [CrossRef]
- Fang, M.; Machalaba, N.; Sánchez-Delgado, R.A. Hydrogenation of arenes and N-heteroaromatic compounds over ruthenium nanoparticles on poly(4-vinylpyridine): A versatile catalyst operating by a substrate-dependent dual site mechanism. Dalton Trans. 2011, 40, 10621–10632. [Google Scholar] [CrossRef]
- ZZhang, Z.; Wang, Z.Q.; Li, Z.; Zheng, W.B.; Fan, L.; Zhang, J.; Hu, Y.M.; Luo, M.F.; Wu, X.P.; Gong, X.P.; et al. Metal-free ceria catalysis for selective hydrogenation of crotonaldehyde. ACS Catal. 2020, 10, 14560–14566. [Google Scholar] [CrossRef]
- Tamura, M.; Tokonami, K.; Nakagawa, Y.; Tomishige, K. Rapid synthesis of unsaturated alcohols under mild conditions by highly selective hydrogenation. Chem. Commun. 2013, 49, 7034–7036. [Google Scholar] [CrossRef] [PubMed]
- Mitsudome, T.; Matoba, M.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Core–shell AgNP@ CeO2 nanocomposite catalyst for highly chemoselective reductions of unsaturated aldehydes. Chemistry 2013, 19, 5255–5258. [Google Scholar] [CrossRef]
- Ye, R.P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q.; et al. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 2019, 10, 5698. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Ross, J.R. Heterogeneous catalysts for hydrogenation of CO2 and bicarbonates to formic acid and formates. Catal. Rev. 2018, 60, 566–593. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, P.; Jiang, L.; Zhang, Z.; Deng, K.; Zhang, Y.; Zhao, Y.; Li, J.; Bottle, S.; Zhu, H. Selective deoxygenation of carbonyl groups at room temperature and atmospheric hydrogen pressure over nitrogen-doped carbon supported Pd catalyst. J. Catal. 2018, 368, 207–216. [Google Scholar] [CrossRef]
- Lee, J.; Christopher, P. Does H2 Temperature-Programmed Reduction Always Probe Solid-State Redox Chemistry? The Case of Pt/CeO2. Angew. Chem. Int. Ed. 2025, 64, e202414388. [Google Scholar] [CrossRef]
- Bai, S.-T.; De Smet, G.; Liao, Y.; Sun, R.; Zhou, C.; Beller, M.; Maes, B.U.W.; Sels, B.F. Homogeneous and heterogeneous catalysts for hydrogenation of CO2 to methanol under mild conditions. Chem. Soc. Rev. 2021, 50, 4259–4298. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, O.; Crabtree, R.H. Outer sphere hydrogenation catalysis. New J. Chem. 2013, 37, 21–27. [Google Scholar] [CrossRef]
- Bullock, R.M.; Rappoli, B.J. Ionic hydrogenations using transition metal hydrides. Rapid hydrogenation of hindered alkenes at low temperature. J. Chem. Soc. Chem. Commun. 1989, 1447–1448. [Google Scholar] [CrossRef]
- Bullock, R.M. Catalytic ionic hydrogenations. Chemistry 2004, 10, 2366–2374. [Google Scholar] [CrossRef]
- Moore, E.J.; Sullivan, J.M.; Norton, J.R. Kinetic and thermodynamic acidity of hydrido transition-metal complexes. 3. Thermodynamic acidity of common mononuclear carbonyl hydrides. J. Am. Chem. Soc. 1986, 108, 2257–2263. [Google Scholar] [CrossRef] [PubMed]
- Luan, L.; Song, J.S.; Bullock, R.M. Ionic Hydrogenation of Alkynes by HOTf and Cp (CO) 3WH. J. Org. Chem. 1995, 60, 7170–7176. [Google Scholar] [CrossRef]
- Zhang, F.H.; Liu, C.; Li, W.; Tian, G.L.; Xie, J.H.; Zhou, Q.L. An efficient ruthenium catalyst bearing tetradentate ligand for hydrogenations of carbon dioxide. Chin. J. Chem. 2018, 36, 1000–1002. [Google Scholar] [CrossRef]
- Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G.A.; Prakash, G.S. Conversion of CO2 from air into methanol using a polyamine and a homogeneous ruthenium catalyst. J. Am. Chem. Soc. 2016, 138, 778–781. [Google Scholar] [CrossRef]
- Everett, M.; Wass, D.F. Highly productive CO2 hydrogenation to methanol–a tandem catalytic approach via amide intermediates. Chem. Commun. 2017, 53, 9502–9504. [Google Scholar] [CrossRef]
- Kumar, A.; Janes, T.; Espinosa-Jalapa, N.A.; Milstein, D. Manganese catalyzed hydrogenation of organic carbonates to methanol and alcohols. Angew. Chem. Int. Ed. 2018, 57, 12076–12080. [Google Scholar] [CrossRef]
- Kar, S.; Goeppert, A.; Kothandaraman, J.; Prakash, G.S. Manganese-catalyzed sequential hydrogenation of CO2 to methanol via formamide. Acs Catal. 2017, 7, 6347–6351. [Google Scholar] [CrossRef]
- Lane, E.M.; Zhang, Y.; Hazari, N.; Bernskoetter, W.H. Sequential hydrogenation of CO2 to methanol using a pincer iron catalyst. Organometallics 2019, 38, 3084–3091. [Google Scholar] [CrossRef]
- Khan, M.N.; van Ingen, Y.; Boruah, T.; McLauchlan, A.; Wirth, T.; Melen, R.L. Advances in CO2 activation by frustrated Lewis pairs: From stoichiometric to catalytic reactions. Chem. Sci. 2023, 14, 13661–13695. [Google Scholar] [CrossRef]
- Zhao, X.; Stephan, D.W. Bis-boranes in the frustrated Lewis pair activation of carbon dioxide. Chem. Commun. 2011, 47, 1833–1835. [Google Scholar] [CrossRef] [PubMed]
- Luiz Fiorio, J.; Guerra, R.R.G.; Martín-Matute, B.; Rossi, L.M. Gold Catalysts for Selective Hydrogenations: The Role of Heterolytic H2 Dissociation. ChemCatChem 2024, 16, e202400207. [Google Scholar] [CrossRef]
- Wang, Y.; Arandiyan, H.; Scott, J.; Bagheri, A.; Dai, H.; Amal, R. Recent advances in ordered meso/macroporous metal oxides for heterogeneous catalysis: A review. J. Mater. Chem. A 2017, 5, 8825–8846. [Google Scholar] [CrossRef]
- Lu, J.; Aydin, C.; Browning, N.D.; Gates, B.C. Hydrogen activation and metal hydride formation trigger cluster formation from supported iridium complexes. J. Am. Chem. Soc. 2012, 134, 5022–5025. [Google Scholar] [CrossRef]
- Kim, T.W.; Kim, M.; Kim, S.K.; Choi, Y.N.; Jung, M.; Oh, H.; Suh, Y.W. Remarkably fast low-temperature hydrogen storage into aromatic benzyltoluenes over MgO-supported Ru nanoparticles with homolytic and heterolytic H2 adsorption. Appl. Catal. B Environ. 2021, 286, 119889. [Google Scholar] [CrossRef]
- Kim, T.W.; Jeong, H.; Jo, Y.; Kim, D.; Park, J.H.; Kim, S.K.; Suh, Y.W. Advanced heterolytic H2 adsorption of K-added Ru/MgO catalysts for accelerating hydrogen storage into aromatic benzyltoluenes. J. Energy Chem. 2022, 71, 333–343. [Google Scholar] [CrossRef]
- Qin, R.; Zhou, L.; Liu, P.; Gong, Y.; Liu, K.; Xu, C.; Zhao, Y.; Gu, L.; Fu, G.; Zheng, N. Alkali ions secure hydrides for catalytic hydrogenation. Nat. Catal. 2020, 3, 703–709. [Google Scholar] [CrossRef]
- Wilson, N.M.; Flaherty, D.W. Mechanism for the direct synthesis of H2O2 on Pd clusters: Heterolytic reaction pathways at the liquid–solid interface. J. Am. Chem. Soc. 2016, 138, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Doudin, N.; Yuk, S.F.; Marcinkowski, M.D.; Nguyen, M.T.; Liu, J.C.; Wang, Y.; Novotny, Z.; Kay, B.D.; Li, J.; Glezakou, V.-A.; et al. Understanding heterolytic H2 cleavage and water-assisted hydrogen spillover on Fe3O4 (001)-supported single palladium atoms. ACS Catal. 2019, 9, 7876–7887. [Google Scholar] [CrossRef]
- Nelson, N.C.; Szanyi, J. Heterolytic hydrogen activation: Understanding support effects in water–gas shift, hydrodeoxygenation, and CO oxidation catalysis. ACS Catal. 2020, 10, 5663–5671. [Google Scholar] [CrossRef]
- Lee, J.; Tieu, P.; Finzel, J.; Zang, W.; Yan, X.; Graham, G.; Pan, X.; Christopher, P. How Pt influences H2 reactions on high surface-area Pt/CeO2 powder catalyst surfaces. Jacs Au 2023, 3, 2299–2313. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Wu, G.; Liu, X.; Ren, Y.; Dai, W.; Wang, C.; Xie, Z.; Guan, N.; Li, L. Acetylene-selective hydrogenation catalyzed by cationic nickel confined in zeolite. J. Am. Chem. Soc. 2019, 141, 9920–9927. [Google Scholar] [CrossRef]
- Song, B.; Xie, L.H. H2 Activation Mechanisms on ZnO-Based Catalysts. J. Phys. Chem. C 2025, 129, 4825–4840. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, T.; Yang, S.; Sun, K.; Yan, H.; Feng, X.; Yang, C.; Yan, N. Intensifying hydrogen heterocracking via regulating the ZnO overlayer for enhanced fatty acid ester hydrogenation. ACS Catal. 2023, 13, 16126–16135. [Google Scholar] [CrossRef]
- Tang, S.; Feng, Z.; Han, Z.; Sha, F.; Tang, C.; Zhang, Y.; Wang, J.; Li, C. Mononuclear Re sites on In2O3 catalyst for highly efficient CO2 hydrogenation to methanol. J. Catal. 2023, 417, 462–472. [Google Scholar] [CrossRef]
- Zhang, M.; Dou, M.; Yu, Y. Theoretical study of the promotional effect of ZrO2 on In2O3 catalyzed methanol synthesis from CO2 hydrogenation. Appl. Surf. Sci. 2018, 433, 780–789. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, G.; Zhu, J.; Zhang, X.; Ding, F.; Zhang, A.; Guo, X.; Song, C. CO2 hydrogenation to methanol over In2O3-based catalysts: From mechanism to catalyst development. Acs Catal. 2021, 11, 1406–1423. [Google Scholar] [CrossRef]
- Ruiz Puigdollers, A.; Illas, F.; Pacchioni, G. ZrO2 Nanoparticles: A density functional theory study of structure, properties and reactivity. Rend. Lincei 2017, 28, 19–27. [Google Scholar] [CrossRef]
- Liu, Z.P.; Wang, C.M.; Fan, K.N. Single Gold Atoms in Heterogeneous Catalysis: Selective 1, 3-Butadiene Hydrogenation over Au/ZrO2. Angew. Chem. Int. Ed. 2006, 45, 6865–6868. [Google Scholar] [CrossRef]
- Abdel-Mageed, A.M.; Büsselmann, M.; Wiese, K.; Fauth, C.; Behm, R.J. Influence of water vapor on the performance of Au/ZnO catalysts in methanol synthesis from CO2 and H2: A high-pressure kinetic and TAP reactor study. Appl. Catal. B Environ. 2021, 297, 120416. [Google Scholar] [CrossRef]
- Whittaker, T.; Kumar, K.S.; Peterson, C.; Pollock, M.N.; Grabow, L.C.; Chandler, B.D. H2 oxidation over supported Au nanoparticle catalysts: Evidence for heterolytic H2 activation at the metal–support interface. J. Am. Chem. Soc. 2018, 140, 16469–16487. [Google Scholar] [CrossRef] [PubMed]
- Hirunsit, P.; Luadthong, C.; Faungnawakij, K. Effect of alumina hydroxylation on glycerol hydrogenolysis to 1, 2-propanediol over Cu/Al2O3: Combined experiment and DFT investigation. RSC Adv. 2015, 5, 11188–11197. [Google Scholar] [CrossRef]
- Nishimura, S. Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis; Wiley: New York, NY, USA, 2001; pp. 213–215. [Google Scholar]
- Bond, J.Q.; Stangland, E.E.; Cybulskis, V.J. Best practices in the characterization of bulk catalyst properties. J. Catal. 2024, 433, 115487. [Google Scholar] [CrossRef]
- Christensen, C.H.; Nørskov, J.K. A molecular view of heterogeneous catalysis. J. Chem. Phys. 2008, 128, 182503. [Google Scholar] [CrossRef] [PubMed]
- Koichumanova, K. In Situ Infrared Spectroscopy Under Hydrothermal Conditions: Application for Aqueous Phase Reforming. Ph.D. Thesis, University of Twente, Enschede, The Netherlands, 2015. [Google Scholar]
- Wu, Z.; Cheng, Y.; Tao, F.; Daemen, L.; Foo, G.S.; Nguyen, L.; Zhang, X.; Beste, A.; Ramirez-Cuesta, A.J. Direct neutron spectroscopy observation of cerium hydride species on a cerium oxide catalyst. J. Am. Chem. Soc. 2017, 139, 9721–9727. [Google Scholar] [CrossRef]
- Wang, L.; Yan, T.; Song, R.; Sun, W.; Dong, Y.; Guo, J.; Zhang, Z.; Wang, X.; Ozin, G.A. Room-temperature activation of H2 by a surface frustrated Lewis pair. Angew. Chem. Int. Ed. 2019, 58, 9501–9505. [Google Scholar] [CrossRef]
- Wei, X.; Jiao, Y.; Zou, X.; Guo, Y.; Li, W.; Ai, T. P vacancy-induced electron redistribution and phase reconstruction of CoFeP for overall water splitting at industrial-level current density. Inorg. Chem. Front. 2025, 12, 2678–2690. [Google Scholar] [CrossRef]
- Mun, B.S.; Liu, Z.; Motin, M.A.; Roy, P.C.; Kim, C.M. In situ observation of H2 dissociation on the ZnO (0001) surface under high pressure of hydrogen using ambient-pressure XPS. Int. J. Hydrogen Energy 2018, 43, 8655–8661. [Google Scholar] [CrossRef]
- Ledbetter, K.; Larsen, C.B.; Lim, H.; Zoric, M.R.; Koroidov, S.; Das Pemmaraju, C.; Gaffney, K.J.; Cordones, A.A. Dissociation of Pyridinethiolate Ligands during Hydrogen Evolution Reactions of Ni-Based Catalysts: Evidence from X-Ray Absorption Spectroscopy. Inorg. Chem. 2022, 61, 9868–9876. [Google Scholar] [CrossRef]
- Scarano, D.; Bertarione, S.; Spoto, G.; Zecchina, A.; Areán, C.O. FTIR spectroscopy of hydrogen, carbon monoxide, and methane adsorbed and co-adsorbed on zinc oxide. Thin Solid Films 2001, 400, 50–55. [Google Scholar] [CrossRef]
- Lindgren, J.; Olbert-Majkut, A.; Pettersson, M.; Kiljunen, T. Raman spectroscopy and crystal-field split rotational states of photoproducts CO and H2 after dissociation of formaldehyde in solid argon. J. Chem. Phys. 2012, 137, 164310. [Google Scholar] [CrossRef] [PubMed]
- Polo-Garzon, F.; Luo, S.; Cheng, Y.; Page, K.L.; Ramirez-Cuesta, A.J.; Britt, P.F.; Wu, Z. Neutron scattering investigations of hydride species in heterogeneous catalysis. ChemSusChem 2019, 12, 93–103. [Google Scholar] [CrossRef]
- Coperet, C.; Estes, D.P.; Larmier, K.; Searles, K. Isolated surface hydrides: Formation, structure, and reactivity. Chem. Rev. 2016, 116, 8463–8505. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Tohjo, Y.; Takahashi, T.; Sawada, H.; Ono, Y. Properties of chemisorbed hydrogen species on Ag-A zeolite partially reduced with hydrogen as studied by 1H MAS NMR. Catal. Today 2001, 66, 81–89. [Google Scholar] [CrossRef]
- Bu, Y.; Er, S.; Niemantsverdriet, J.H.; Fredriksson, H.O. Preferential oxidation of CO in H2 on Cu and Cu/CeOx catalysts studied by in situ UV–Vis and mass spectrometry and DFT. J. Catal. 2018, 357, 176–187. [Google Scholar] [CrossRef]
- Kim, T.W.; Kim, D.; Jo, Y.; Jung, H.J.; Park, J.H.; Suh, Y.W. Potassium as the best alkali metal promoter in boosting the hydrogenation activity of Ru/MgO for aromatic LOHC molecules by facilitated heterolytic H2 adsorption. J. Catal. 2023, 419, 112–124. [Google Scholar] [CrossRef]
- Zhu, R.; Liu, B.; Wang, S.; Huang, X.; Schuarca, R.L.; He, W.; Cybulskis, V.J.; Bond, J.Q. Understanding the mechanism (s) of ketone oxidation on VOx/γ-Al2O3. J. Catal. 2021, 404, 109–127. [Google Scholar] [CrossRef]
- Wu, Q.; Qin, R.; Zhu, M.; Shen, H.; Yu, S.; Zhong, Y.; Fu, G.; Yi, X.; Zheng, N. Frustrated Lewis pairs on pentacoordinated Al 3+-enriched Al2O3 promote heterolytic hydrogen activation and hydrogenation. Chem. Sci. 2024, 15, 3140–3147. [Google Scholar] [CrossRef] [PubMed]
- Li, X.S.; Xin, Q.; Guo, X.X.; Grange, P.; Delmon, B. Reversible hydrogen adsorption on MoS2 studied by temperature-programmed desorption and temperature-programmed reduction. J. Catal. 1992, 137, 385–393. [Google Scholar] [CrossRef]
- Wang, X.; Xiao, T.; Liu, Y.; Zhang, C.; Zhao, F. Heterolytic hydrogenation and H–migration-assisted hydrodeoxygenation reaction under mild conditions over Pt/TiO2-D. ACS Catal. 2024, 14, 13800–13813. [Google Scholar] [CrossRef]
- Wang, W.; Huo, K.; Wang, Y.; Xie, J.; Sun, X.; He, Y.; Li, M.; Liang, J.; Gao, X.; Yang, G.; et al. Rational control of oxygen vacancy density in In2O3 to boost methanol synthesis from CO2 hydrogenation. ACS Catal. 2024, 14, 9887–9900. [Google Scholar] [CrossRef]
- Rynkowski, J.; Rajski, D.; Szyszka, I.; Grzechowiak, J.R. Effect of platinum on the hydrogenation activity of nickel catalysts. Catal. Today 2004, 90, 159–166. [Google Scholar] [CrossRef]
- Yang, F.; Zhou, W.; Yang, C.; Zhang, T.; Huang, Y. Enhanced methanol selectivity in CO2 hydrogenation by decoration of K on MoS2 catalyst. Acta Phys.-Chim. Sin. 2024, 40, 2308017. [Google Scholar] [CrossRef]
- Liu, J.; Hensley, A.J.; Giannakakis, G.; Therrien, A.J.; Sukkar, A.; Schilling, A.C.; Groden, K.; Ulumuddin, N.; Hannagan, R.T.; Ouyang, M.; et al. Developing single-site Pt catalysts for the preferential oxidation of CO: A surface science and first principles-guided approach. Appl. Catal. B Environ. 2021, 284, 119716. [Google Scholar] [CrossRef]
- Burch, R.; Shestov, A.A.; Sullivan, J.A. A transient kinetic study of the mechanism of the NO+ H2 reaction over Pt/SiO2 catalysts: 1. isotopic transient kinetics and temperature programmed analysis. J. Catal. 1999, 186, 353–361. [Google Scholar] [CrossRef]
- Agnelli, M.; Swaan, H.M.; Marquez-Alvarez, C.; Martin, G.A.; Mirodatos, C. CO hydrogenation on a nickel catalyst: II. A mechanistic study by transient kinetics and infrared spectroscopy. J. Catal. 1998, 175, 117–128. [Google Scholar] [CrossRef]
- Bundhoo, A.; Schweicher, J.; Frennet, A.; Kruse, N. Chemical transient kinetics applied to CO hydrogenation over a pure nickel catalyst. J. Phys. Chem. C 2009, 113, 10731–10739. [Google Scholar] [CrossRef]
- Ali, S.H.; Goodwin, J.G., Jr. Isotopie Transient Kinetic Analysis of the Induction Phenomenon for Methanol Synthesis on Pd/SiO2. J. Catal. 1997, 170, 265–274. [Google Scholar] [CrossRef]
- Hu, Y.H.; Ruckenstein, E. Multiple transient response methods to identify mechanisms of heterogeneous catalytic reactions. Acc. Chem. Res. 2003, 36, 791–797. [Google Scholar] [CrossRef]
- Ledesma, C.; Yang, J.; Chen, D.; Holmen, A. Recent approaches in mechanistic and kinetic studies of catalytic reactions using SSITKA technique. Acs Catal. 2014, 4, 4527–4547. [Google Scholar] [CrossRef]
- Ali, S.H.; Goodwin, J.G., Jr. SSITKA investigation of palladium precursor and support effects on CO hydrogenation over supported Pd catalysts. J. Catal. 1998, 176, 3–13. [Google Scholar] [CrossRef]
- Schweicher, J.; Bundhoo, A.; Frennet, A.; Kruse, N.; Daly, H.; Meunier, F.C. DRIFTS/MS studies during chemical transients and SSITKA of the CO/H2 reaction over Co-MgO catalysts. J. Phys. Chem. C 2010, 114, 2248–2255. [Google Scholar] [CrossRef]
- Frøseth, V.; Storsæter, S.; Borg, Ø.; Blekkan, E.A.; Rønning, M.; Holmen, A. Steady state isotopic transient kinetic analysis (SSITKA) of CO hydrogenation on different Co catalysts. Appl. Catal. A Gen. 2005, 289, 10–15. [Google Scholar] [CrossRef]
- Otroshchenko, T.; Kondratenko, V.A.; Zanina, A.; Zhang, Q.; Kondratenko, E.V. Progress through Temporal Analysis of Products and Steady-state Isotopic Transient Kinetic Analysis to Elucidate Oxidation, CO2 Hydrogenation and Lower Olefins Production Reactions. ChemCatChem 2024, 16, e202400081. [Google Scholar] [CrossRef]
- Hevia, M.A.; Bridier, B.; Pérez-Ramírez, J. Mechanistic study of the palladium-catalyzed ethyne hydrogenation by the Temporal Analysis of Products technique. Appl. Catal. A Gen. 2012, 439, 163–170. [Google Scholar] [CrossRef]
- Hohmeyer, J.; Kondratenko, E.V.; Bron, M.; Kröhnert, J.; Jentoft, F.C.; Schlögl, R.; Claus, P. Activation of dihydrogen on supported and unsupported silver catalysts. J. Catal. 2010, 269, 5–14. [Google Scholar] [CrossRef]
- Vilé, G.; Albani, D.; Nachtegaal, M.; Chen, Z.; Dontsova, D.; Antonietti, M.; López, N.; Pérez-Ramírez, J. Ein stabiler “Single-site”-Palladiumkatalysator für Hydrierungen. Angew. Chem. 2015, 127, 11417–11422. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, Y.; Qin, R.; Mo, S.; Chen, G.; Gu, L.; Chevrier, D.M.; Zhang, P.; Guo, Q.; Zang, D.; et al. Photochemical route for synthesizing atomically dispersed palladium catalysts. Science 2016, 352, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Liu, X.; Wang, A.; Zhang, L.; Qiao, B.; Yang, X.; Huang, Y.; Miao, S.; Liu, J.; Zhang, T. FeOx-supported platinum single-atom and pseudo-single-atom catalysts for chemoselective hydrogenation of functionalized nitroarenes. Nat. Commun. 2014, 5, 5634. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, S.; Tourvieille, J.N.; Sommier, A.; Batsale, J.C.; Beccard, B.; Pradère, C. Thermal camera-based fourier transform infrared thermospectroscopic imager. Appl. Spectrosc. 2021, 75, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Knöpke, L.R.; Nemati, N.; Köckritz, A.; Brückner, A.; Bentrup, U. Reaction monitoring of heterogeneously catalyzed hydrogenation of imines by coupled ATR-FTIR, UV/Vis, and Raman spectroscopy. ChemCatChem 2010, 2, 273–280. [Google Scholar] [CrossRef]
- Yang, Y.; Disselkamp, R.S.; Szanyi, J.; Peden, C.H.; Campbell, C.T.; Goodwin, J.G. Design and operating characteristics of a transient kinetic analysis catalysis reactor system employing in situ transmission Fourier transform infrared. Rev. Sci. Instrum. 2006, 77, 094104. [Google Scholar] [CrossRef]
- Kong, X.; Chen, Y.; Bao, X.; Zhu, Y. Integrating operando spectroscopies and transient analysis for dynamic catalytic insights. Sci. China Chem. 2025, 1–16. [Google Scholar] [CrossRef]


| Catalyst | Features | Application | Examples |
|---|---|---|---|
| Ionic hydrogenation catalyst | Metal serves as a hydride acceptor with a base as a proton acceptor | Hydrogenation of chemicals containing polar bonds, such as C=O, C=N, etc. | HMo-(CO)3(C2H5) [33] |
| [Mo(Cp)(CO3)]H] [34,35] | |||
| [W(Cp)(CO)3H)] [34,36] | |||
| [W(CP)(CO)2(PR3)(O=CEt2)]+ [35] | |||
| Bifunctional catalyst | Contains a proton acceptor (N or O) and a hydride acceptor (Ru, Ir, Fe) | Hydrogenation of CO2 to form methanol under mild conditions | Ru-PtBuNPyNPyNEt [31,37] |
| Ru-MACHO-BH [31,38] | |||
| Ru-bisPN [31,39] | |||
| Mn-PtBuNPy’NPy/Mn-PtBuNPyNtBu [31,40] | |||
| Mn-PiPrNPiPr [31,41] | |||
| Fe-PiPrNPiPr [31,42] | |||
| Frustrated Lewis pairs catalyst | Contains bulky Lewis acids and bases, which hinder full interaction with each other, enabling the activity and cooperative H2 activation | Metal-free hydrogenation reactions; CO2 activation; reduction in ketones | tBu3P/B(C6F5)3 FLP [43] |
| (Me3Si)3P-CO2-B(p-C6F4H)3 [43] | |||
| 1,1-bis-(C6F5)2BOB(C6F-)2 [43,44] |
| Catalyst System | Additives/Substrates | Target Reactions | Ref. |
|---|---|---|---|
| Ru/MgO | Aromatics | Hydrogenation | [48,49] |
| Ru (III)/γ-Al2O3 | Unsaturated hydrocarbons | Selective hydrogenation | [50] |
| Pd/SiO2 | H2 + O2 (with solvents) | Direct H2O synthesis | [51] |
| Fe3O4 (001)-supported single Pd atoms | H2 + H2O | H2 dissociation | [52] |
| Pd-/nitrogen-doped carbon | Aromatic carbonyl compounds | Selective deoxygenation of carbonyl groups | [29] |
| Pd/Al2O3; Pd/CeO2; Pd/CeO2-Al2O3 | CO2 + H2 | Reverse water–gas shift (rWGS) | [53] |
| Pt/CeO2 | H2 | H2 activation | [54] |
| Ni (II)@Chabazite | Acetylene + H2 | Selective hydrogenation | [55] |
| Ni/ZnO (>500 °C) | Fatty acid ester + H2 | Hydrogenation to fatty alcohols | [56,57] |
| Re/In2O3 | CO2 + H2 | Methanol synthesis | [58] |
| In2O3/ZrO2 | CO2 + H2 | Methanol synthesis | [59,60] |
| Au/ZrO2 | Butadiene | Selective hydrogenation | [61,62] |
| Au/ZnO | CO2 + H2 | Methanol synthesis | [63] |
| Au/TiO2; Au/Al2O3 | H2 + O2, H2O | H2 oxidation | [64] |
| Cu/Al2O3 | Glycerol + H2 | Glycerol hydrogenolysis to 1,2-propanediol | [65] |
| Technique | Type of Probe | Detected Information | Role in H2 Dissociation Studies | Ref. |
|---|---|---|---|---|
| X-ray Photoelectron Spectroscopy (XPS) | X-ray photons (photons) | Surface elemental composition and oxidation state | Investigate the interaction between H2 and solid surface; monitor changes in oxidation state | [73] |
| X-ray Absorption Spectroscopy (XAS/XANES/EXAFS) | X-ray photons (photons) | Bulk oxidation states of metal or metal ions; coordination environments | Tracks metal–support interactions and redox/structure changes during H2 activation | [74] |
| Infrared Spectroscopy (IR) | Infrared light (photons) | Vibrational modes of surface-chemisorbed species | Differentiates between homolytic/heterolytic pathways based on intermediates according to surface chemistry | [54,64,75] |
| Raman Spectroscopy | Monochromatic laser (photons) | Vibration of metal oxides; organic residuals | A great complement to IR and EPR, which can provide insights into formed complexes | [76] |
| Inelastic Neutron Scattering (INS) | Beams of neutrons (neutrons) | Surface M–H species or H-H species | Identify chemical nature of hydrogen content and quantify surface hydrogen species and bulk hydrides | [77] |
| 1H Nuclear Magnetic Resonance Spectroscopy (1H NMR) | Strong magnetic field (field) | Hydrogen bonding, diffusion, surface hydrides | Investigate formed chemisorbed hydrogen species during reduction | [78,79] |
| Electron Paramagnetic Resonance (EPR) | Magnetic field (field) | Radicals, transition metal states, defects | Determine whether heterolytically dissociated H+/H− are exhibited | [71] |
| UV-Vis Diffuse Reflectance | Ultraviolet or visible light (photons) | d-d transitions of metal ions | Monitor changes in metal centers during reduction | [74,80] |
| Inductively Coupled Plasma Optical Emission Spectroscopy/Mass Spectrometry (ICP-OES/MS) | Ions or charged particles (ions) | Analyze composition of catalyst | Quantify dispersed metal component in catalysts | [81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Lou, X.; Liu, B. Probing Heterolytic H2 Dissociation on Heterogeneous Catalysts: A Brief Review of Experimental Strategies. Processes 2025, 13, 1465. https://doi.org/10.3390/pr13051465
Wang S, Lou X, Liu B. Probing Heterolytic H2 Dissociation on Heterogeneous Catalysts: A Brief Review of Experimental Strategies. Processes. 2025; 13(5):1465. https://doi.org/10.3390/pr13051465
Chicago/Turabian StyleWang, Siwen, Xuanqing Lou, and Bowei Liu. 2025. "Probing Heterolytic H2 Dissociation on Heterogeneous Catalysts: A Brief Review of Experimental Strategies" Processes 13, no. 5: 1465. https://doi.org/10.3390/pr13051465
APA StyleWang, S., Lou, X., & Liu, B. (2025). Probing Heterolytic H2 Dissociation on Heterogeneous Catalysts: A Brief Review of Experimental Strategies. Processes, 13(5), 1465. https://doi.org/10.3390/pr13051465