1. Introduction
circRNAs are a class of non-coding RNAs characterized by covalently closed-loop structures, which are integrated into the linear mRNAs of eukaryotic cells [
1,
2,
3,
4]. They are not methylated, they are not 5′ capped, and they do not contain the 3′ poly(A) tail [
4,
5,
6]. The most important feature of circRNAs is that they are mostly formed in the nucleus through a mechanism known as back splicing [
7]. circRNAs were initially described more than three decades ago and were considered as non-coding RNAs with no specific function, or even as products of random transcription or splicing mistakes [
8]. High-throughput sequencing and bioinformatics studies have shown that these circRNAs have important functions in gene regulation processes, including transcription regulation and even protein synthesis in some instances [
4,
9,
10,
11]. This has changed the view of circRNAs as just transcriptional noise, but rather they are involved in multiple biological processes, such as cancer and other diseases [
12,
13]. microRNAs (miRNAs), specifically mature microRNAs, are naturally occurring small molecules of non-coding RNA with 17-25-nucleotide sizes [
8,
14,
15]. They are significant post-transcriptional modulators of gene expression that affect target sites in messenger RNAs’ untranslated regions via direct base pairing [
16]. Knowledge of circRNA–miRNA interactions (CMIs) can reveal new approaches for diagnosing and treating complex human diseases [
17]. In CMI analysis, circRNA interacts with miRNA at a particular site called the miRNA response element (MRE) [
9]. These MREs include sequences that can form a circRNA–miRNA complex. This interaction has been shown to be crucial in the process of gene regulation and is well illustrated in various biological phenomena, such as cancers [
9]. This form of binding confers the ability of circRNAs to influence one or more miRNAs in an extensive array of downstream pathways. For instance, ciRS-7 is reported to interact with miR-7 and suppress the function of the latter, hence regulating the genes controlled by the latter [
12,
13,
18].
Because of the scarcity of resources and materials, as well as manpower, it is practically impossible to carry out large-scale experiments to validate all the conceivable CMIs. Consequently, the use of computational methods to filter out the likely-to-interact CMIs in advance has become a standard strategy. These methods rely on an algorithm and model to predict the interactions based on prior knowledge, and it helps to down-sample the experiment space by offering a smaller list of potential candidates for validation in a wet laboratory. This synergism of computation and experiment conductively augments the discovery and identification of CMIs, enabling more purposeful and cost-effective study [
9].
In the biomedical field, computational models for predicting associations between various entities, such as genes, proteins, microRNAs, and diseases, are becoming increasingly common [
19,
20,
21,
22]. These models leverage large datasets, including genomic, proteomic, and transcriptomic data, to infer potential interactions or associations. They rely on machine learning algorithms, statistical methods, and network-based approaches to predict these relationships, which can then be experimentally validated in the lab. While prediction models for CMIs are gaining attention, this field is still relatively underexplored compared to other areas of biomedical research. Most existing models for predicting CMIs rely on the nearest-neighbor relationship in biological networks [
9,
12]. These approaches assume that circRNAs and miRNAs that are close to each other in a biological network, such as a protein–protein interaction network or a regulatory network, are more likely to interact.
By using various computing methods, including advanced artificial intelligence and machine learning, as well as deep learning approaches, there are several approaches that have been proposed for the prediction of CMIs [
23]. The deep learning-based methods include KGDCMI introduced by Wang X-F et al. [
17] and WSCD by Guo L-X et al. [
24]. KGDCMI employs a deep neural network technique to predict CMIs by fusing multi-source information through a deep neural network. On the other hand, WSCD employs Word2vec, structural deep network embedding (SDNE), convolutional neural networks (CNNs), and deep neural networks (DNNs) to extract and fuse both attribute and behavioral features for CMI predictions. Also, there are several graph-based deep learning methods that have been proposed for CMI prediction. These methods include SGCNCMI by Yu C-Q et al. [
25] and GCNCMI by He J et al. [
26], which are based on graph convolutional networks (GCNs). SGCNCMI utilizes GCNs to identify CMIs with the help of node similarity, while GCNCMI utilizes node information by employing graph convolutional layers and integrating embedded representations for more accurate predictions. When it comes to methods that use boosting techniques, the authors Qian Y et al. [
27] introduced CMASG and Wang X-F et al. [
28] also proposed BioDGW-CMI. CMASG uses GNNs to learn node and feature representations and singular value decomposition (SVD) for feature extraction while a light gradient boosting machine (LightGBM) is used for the final prediction. In the same manner, BioDGW-CMI combines bio-text mining using BioBERT with a heat wavelet diffusion pattern analysis for node embedding, with LightGBM for realistic predictions. These models are based on learning latent biology and incorporation of thin and complex network representations. The most recent development, namely, BEROLECMI [
9], introduces a new technique whereby CMI is predicted by presenting the role attributes of a molecule. This model uses bidirectional encoder representations from DNABERT [
29], a transformer-based model for DNA sequences, to extract molecular attribute features. It employs Gaussian and sigmoid kernel functions to construct self-similarity networks and utilizes a sparse autoencoder (SAE) to learn high-level representations. It additionally extracts role similarity from the CMI network and combines all these features for prediction purposes. In the case of CMI pairs, BEROLECMI used LightGBM for final prediction and was able to predict 14 out of 15 CMI pairs and outperformed other models on common datasets. Further, KS-CMI introduced by Wang X-F et al. [
30] and JSNDCMI by Wang X-F et al. [
31] combine boosting approach with feature extraction techniques. KS-CMI realizes feature representation improvement in circRNA–miRNA–cancer networks by employing a CatBoost classifier and a denoising autoencoder (DAE). However, JSNDCMI combines the functional and topological structure similarities with a gradient boosting decision tree (GBDT) classifier to support the identification of CMIs in sparse networks. The existing studies on CMI prediction like BEROLECMI have contributed significantly to the development of CMI prediction using molecular characteristics and biological networks. However, most of these models are mainly based on conventional machine learning approaches or boosting schemes which may not effectively characterize the relationships and dependencies of the nodes within the biological networks. Furthermore, feature fusion methods are not designed to make full use of graph-based structural information.
In this paper, we used molecular attributes, self-similarity networks, and role-based network embeddings to determine the interaction between circRNA and miRNA. We constructed a graph attention network (GAT), which models the intricate relationships between circRNA and miRNA nodes within a biological interaction network. This is inspired by the fact that GAT can capture the intricate dependencies between nodes in a graph using attention mechanisms that help it filter out the most salient interactions in the context of the circRNA–miRNA biological graph. In contrast to the traditional classifiers, the GAT model can efficiently distinguish between the structural and functional properties of nodes in the network by imposing various degrees of attention weights on the neighboring nodes. This allows the model to not only include local topology information but also distinguish between the types of molecular interactions, thus making the predictions more accurate. The results obtained show that our GAT model significantly outperforms the existing CMI prediction methods, providing an effective approach to address the complexities inherent in biological networks.
The key contributions of our study are as follows:
We proposed a novel approach based on GAT which can utilize the attention mechanism to model the complex dependencies in the biological network for CMI predictions.
The role-based graph embeddings generated using Role2Vec are used as graph structures for the GAT model.
Our proposed GAT method obtained state-of-the-art performance, surpassing the existing models like BEROLECMI and KS-CMI based on the performance metrics accuracy, precision, recall, F1-score, AUROC, and AUPR.
Our GAT approach is different from the existing methods in that it utilizes the known interaction data to perform two tasks: first, it generates the RNA feature based on role definition through Role2Vec, and then it guides the graph attention network (GAT) through its graph structure input. The GAT model processes biologically meaningful graph data consisting of circRNA and miRNA nodes. This enables our model to capitalize on the interaction network to produce features and construct the structural framework which enhances biological interpretation during predictions.
2. Materials and Methods
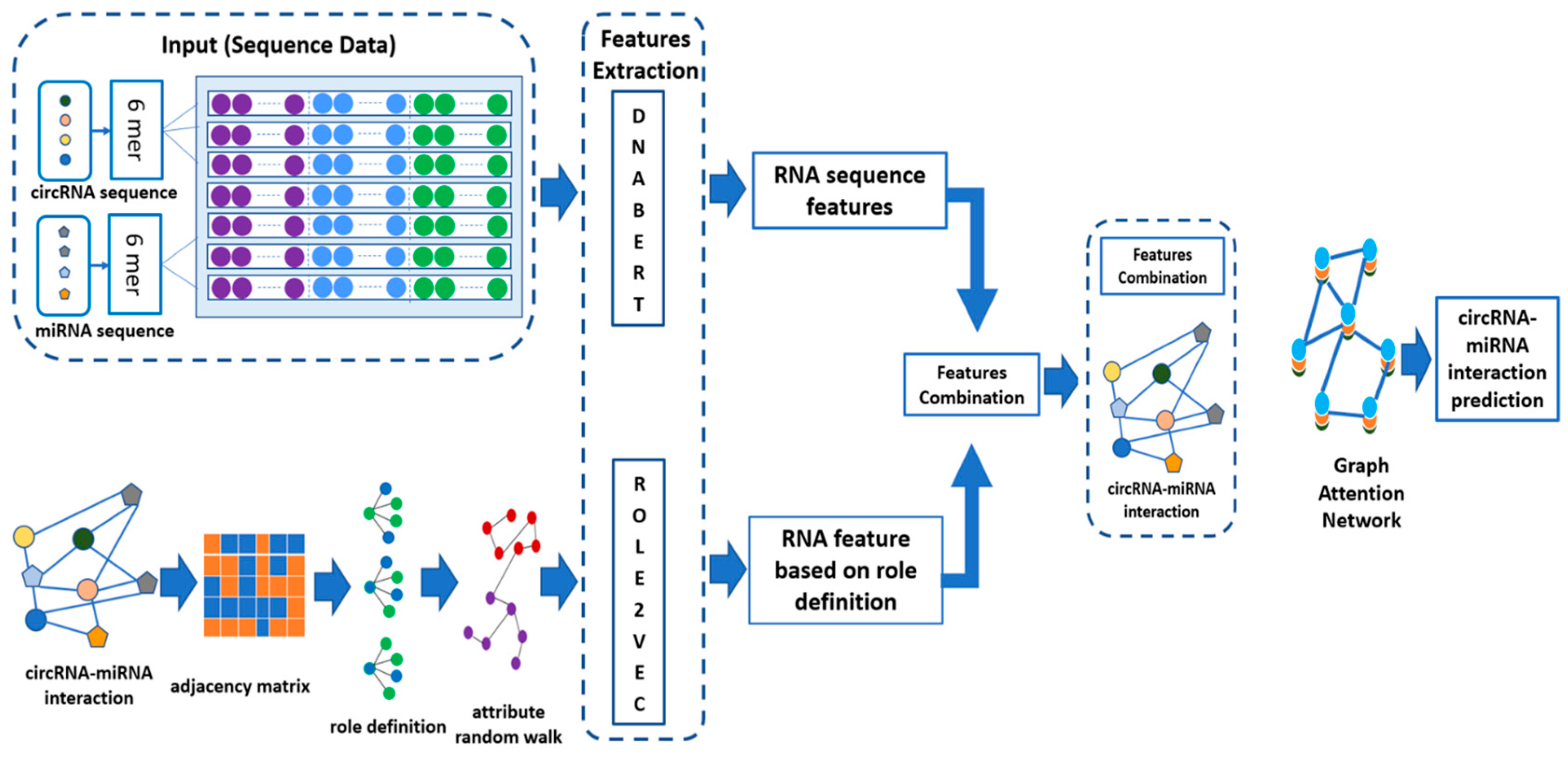
The approach described in this study combines the sequence of circRNAs and miRNAs and their molecular structures to predict circRNA–miRNA interaction using the GAT model. As illustrated in
Figure 1 below, the circRNA and miRNA sequences are first passed through DNABERT [
29], which is used to extract RNA sequence features utilizing 6-mer encoding. In 6-mer encoding, each RNA sequence is fragmented into smaller units, which are subsequences of 6 nucleotides. A DNABERT tokenizer is then employed to convert these 6-mer fragments into high-dimensional feature representations by passing them through a pre-trained DNABERT model. These features are then integrated with the structural role-based features obtained using the method Role2Vec, which is a graph embedding approach that captures the topological data extracted from the interaction network based on an adjacency matrix. This adjacency matrix reveals the known CMIs. To construct the negative samples, we used random sampling in which we generated all the possible combinations of circRNA and miRNA pairs via Cartesian products. We removed the interacting pairs that are found in the CMI dataset, and the remaining pairs were considered as negative samples. The feature vectors concatenate each individual RNA sequence and the corresponding role-based feature. These feature vectors are fed into the GAT model that takes graph structure into account and learns complicated relationships between circRNAs and miRNAs. By examining the features of the CMI lists, the constructed GAT model can focus on the relevant features and optimize the attention mechanism for the interaction patterns of new potential CMIs. This approach, assessed by five-fold cross-validation, shows that enriching interaction features with graph structure information improves the prediction.
2.1. Datasets
In this study, we utilized two key datasets: CMI-9905 and CMI-9589 [
17,
31]. CMI-9905 contains 9905 circRNA–miRNA interactions including 2346 circRNAs and 962 miRNAs, and it is based on high-confidence interaction data. CMI-9905 is one of the largest and most used benchmarks for CMI prediction because of its broad coverage and experimental validation. CMI-9589 has 9589 interacting pairs of 2115 circRNAs and 821 miRNAs downloaded from the circBank database [
32], which is another benchmark for assessing the generalizability of models in different biological settings. These datasets can be used to assess the performance of circRNA–miRNA prediction models that are well established with strict benchmarks and proven to be effective in various situations.
2.2. Features Extraction Using DNABERT for circRNA and miRNA Sequences
In this paper, we employed DNABERT, a BERT model for DNA sequences pre-trained on GENCODE, to extract features from both circRNA and miRNA sequences [
33]. The circRNA and miRNA sequence files were downloaded from
https://github.com/1axin (accessed on 18 August 2024) [
9]. The first step in the methodology involves breaking down each e circRNA and miRNA sequence into 6-base pairs known as 6-mers with a sliding window of length 6. This results in each sequence becoming part of a set of overlapping 6-mer subsequences, wherein the subsequences are tokenized and passed to the DNABERT model. The 6-mer subsequences were then passed through the DNABERT tokenizer which converts them to inputs that the model can process. These tokens are then input into the DNABERT model, which outputs embedding vectors that represent each 6-mer fragment based on the number of hidden units in the model’s architecture. The means of these embeddings are then calculated to generate a feature vector for each sequence. After the feature vectors of the sequences have been obtained, they are made into vectors of equal sizes and then stacked into a single array for input. These are then stored in an h5 file for future use in downstream analysis. This process takes advantage of DNABERT to capture the inherent relations within its circRNA/miRNA sequences which helps in improving the reliability of any predictions/analysis made on extracted features.
2.3. Role-Based Graph Embeddings (Role2Vec)
Role2Vec is a graph embedding approach for learning the structural roles of nodes in a network by contrast with node centrality [
34,
35,
36]. With an aim of capturing proximal relationships that are more focused, Role2Vec learns the embeddings of nodes that contribute to and exercise the functional roles within a graph such as hubs, bridges, or more commonly, articulation points, which are paramount in cases where node roles are more important than their connectivity. The method makes use of a random walk strategy, which received much attention in learning network embeddings [
37,
38]. Instead of relying on the nodes in the immediate neighborhood to convey information about the node, the random walk strategy gathers information about the nodes that play similar structural roles in different parts of the graph. This is based on the Markov process where the probability of moving from one node to another depends on the node at current and on the chosen walk. The equation to derive this probability is as follows [
34]:
where
is the neighbor of node
, and
is the weight of the edge that links node
and
.
These walks are then employed to further train a skip-gram model with the aim of acquiring the node representation. The objective function of the skip-gram model is to maximize the log-probability of the context nodes
, ….,
and it can be given by the following equation [
30].
Here,
represents the window size, defining the size of the context around the target node. By integrating the random walk with the skip-gram model, the goal of Role2Vec is to bring similar nodes together in the embedding space. This can be expressed as follows:
represents the probability of node appearing in the context of node and is the set of node pairs generated by the random walks.
This approach helps in generalizing the node roles present across various networks and improves Role2Vec when used in applications such as link prediction roles, identification of roles, and combined transfer learning in various networks. Role2Vec is, thus, useful in tasks where it is important to identify node functions or jobs instead of just connections.
2.4. Graph Attention Network
GATs are an extension of graph neural networks (GNNs) where nodes and their generated features incorporate attention coefficients that decide the importance of each neighboring node to modify the model’s flexibility for capturing the structure of graph-structured data [
39,
40,
41,
42]. This attention mechanism may filter out the neighboring nodes that are most important to GATs during the feature aggregation stage, which is important to obtain significant node representations. For each node
, the feature vector
(with
features) is first linearly transformed by a shared weight matrix
, resulting in a new representation
. After that, the attention mechanism calculates the weight (attention score) assigned to each of the neighboring node
by applying a shared attention function to the transformed features of each pair of nodes (u and v) in the neighborhood. Using a self-attention mechanism, the model computes unnormalized attention weights for each edge, reflecting the relevance of the connection between nodes. The equation that computes the attention score is as follows [
43]:
These raw scores are then normalized, in proportion to all neighbors of the target node, using a softmax function to produce the attention coefficients. The softmax function across all the neighbors of node
is as follows [
41]:
The last node representation is then obtained by summing the neighbors’ features, but the weights are given by the attention coefficients. This architecture makes GATs more capable of adapting to the local structure of the graph as compared to the traditional GNNs.
The GAT demonstrates superior benefits over GCNs when it comes to modeling circRNA–miRNA interactions. All the traditional GCNs conduct neighborhood information aggregation through uniform weight systems without considering different network interaction strengths. The attention mechanism within GAT enables the model to determine customizable weights for each neighbor, thus permitting it to concentrate on crucial nodes. The capability serves essential purposes when studying biological graphs that contain functional interactions of differing significance.
2.5. Performance Measures
We used the evaluation metrics accuracy, precision, recall, and F1-score to evaluate the performance of our GAT model based on five-fold cross-validation [
44]. These performance metrics are calculated using the following values: true positive (TP), which refers to the positive instances that are correctly classified as positive; true negative (TN), which refers to the negative instances that are correctly classified as negative; false positive (FP), which refers to the negative instances that are incorrectly classified as positive; and false negative (FN), which refers to the positive instances that are incorrectly classified as negative.
Accuracy is one of the most widely used metrics that is defined as the percentage of correctly classified instances [
45,
46,
47] and can be calculated as follows:
Precision measures the proportion of correctly predicted positive instances to the total number of instances predicted as positive and thus, how often the proposed model is right with its positive predictions, and it can be calculated as follows:
Recall is the ratio of correctly predicted positive instances to the total number of actual positive instances emphasizing the model’s ability to identify all the relevant positive cases. It can be calculated as follows:
Since precision and recall as the two measures are not solely sufficient for providing results, the F1-score is, therefore, calculated as the harmonic mean of those two in order to obtain a single value that enhances the two. F1-score can be calculated as follows:
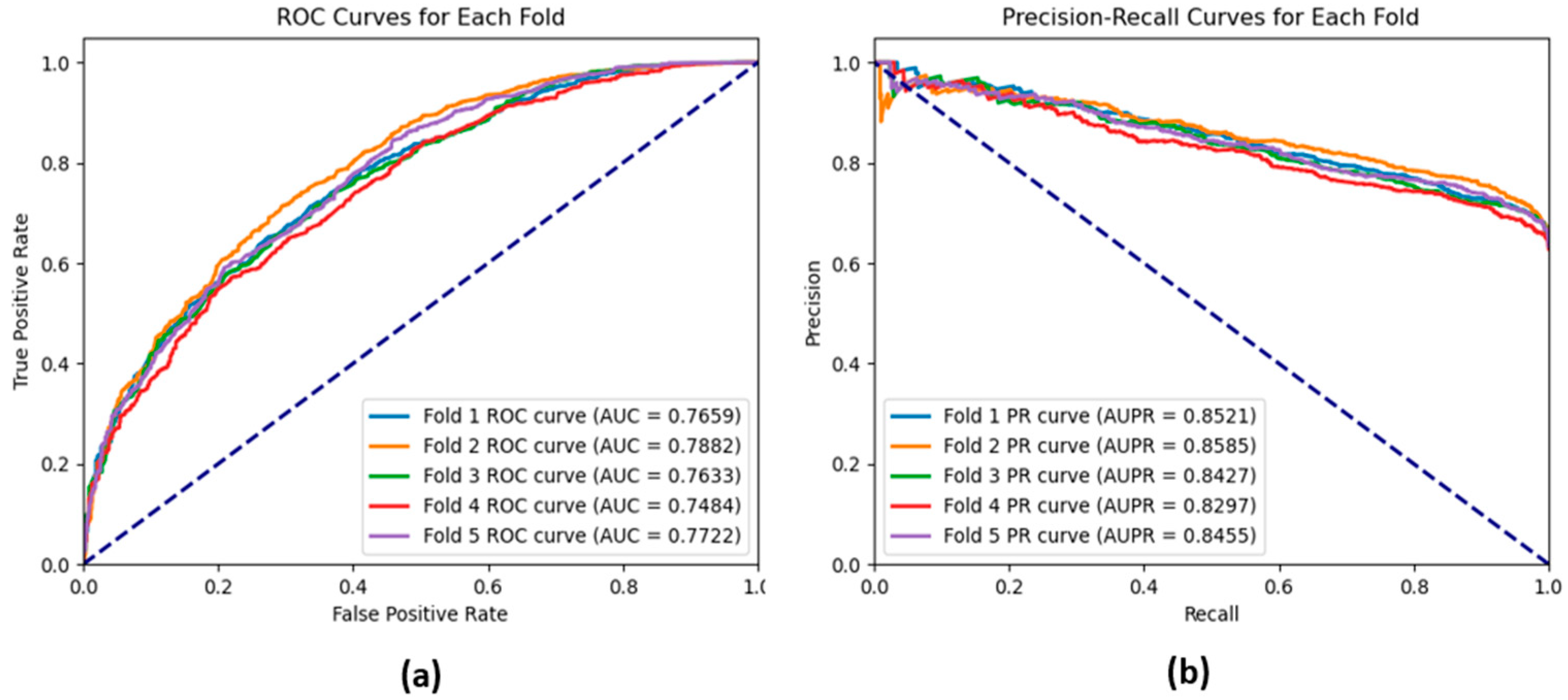
In addition to the performance measures, we use the area under the receiver operating characteristics (AUROC), which is considered a threshold-independent measure. The ROC curve compares the true positive against the false positive. It has a value between 0.5, which reflects the random guess classification, to 1, which means high classification quality. This metric is especially relevant for the binary classification problem when the classes are almost equiproportional. Also, we used the area under the precision–recall curve (AUPR) which focuses on the trade-off between precision and recall. AUPR is especially important when the classes are not equiproportional; therefore, it reflects the model in predicting the minority class without being influenced by the majority class.
4. Conclusions
circRNAs and miRNAs play critical roles in gene regulation, and they have an association with various diseases, including cancer. circRNA–miRNA interaction (CMI) prediction can provide researchers with insights into disease mechanisms and uncover potential therapeutic targets. In this paper, we propose a novel approach for predicting CMI using a graph attention network (GAT). The utilized features are extracted from circRNA and miRNA sequences using DNABERT. Also, we extracted CMI features using Role2Vec. The GAT model leverages attention mechanisms and CMI graph structure to learn complex node dependencies in CMI networks. Our approach outperformed both the traditional deep neural networks and existing CMI prediction methods. We evaluated our model using the commonly used datasets CMI-9905 and CMI-9589. It achieved accuracies of 0.8762 and 0.8837 on the CMI-9905 and CMI-9589 datasets, respectively. The precision, recall, F1-score, AUROC, and AUPR achieved by our model on the benchmark dataset (CMI-9905) are 0.9007, 0.9056, 0.9031, 0.9500, and 0.9729, respectively. These values are the highest compared to the existing CMI prediction methods.
Nevertheless, our approach has limitations and directions for future development. The high-quality datasets, including CMI-9905 and CMI-9589, limit their applicability to diverse biological contexts. In future work, building an efficiently extensible theoretical model and an easily accessible interface can enhance usability and enable scientists to predict and validate CMIs from various sets of data and in various diseases.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}