
A Review of Machine Learning in Organic Solar Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
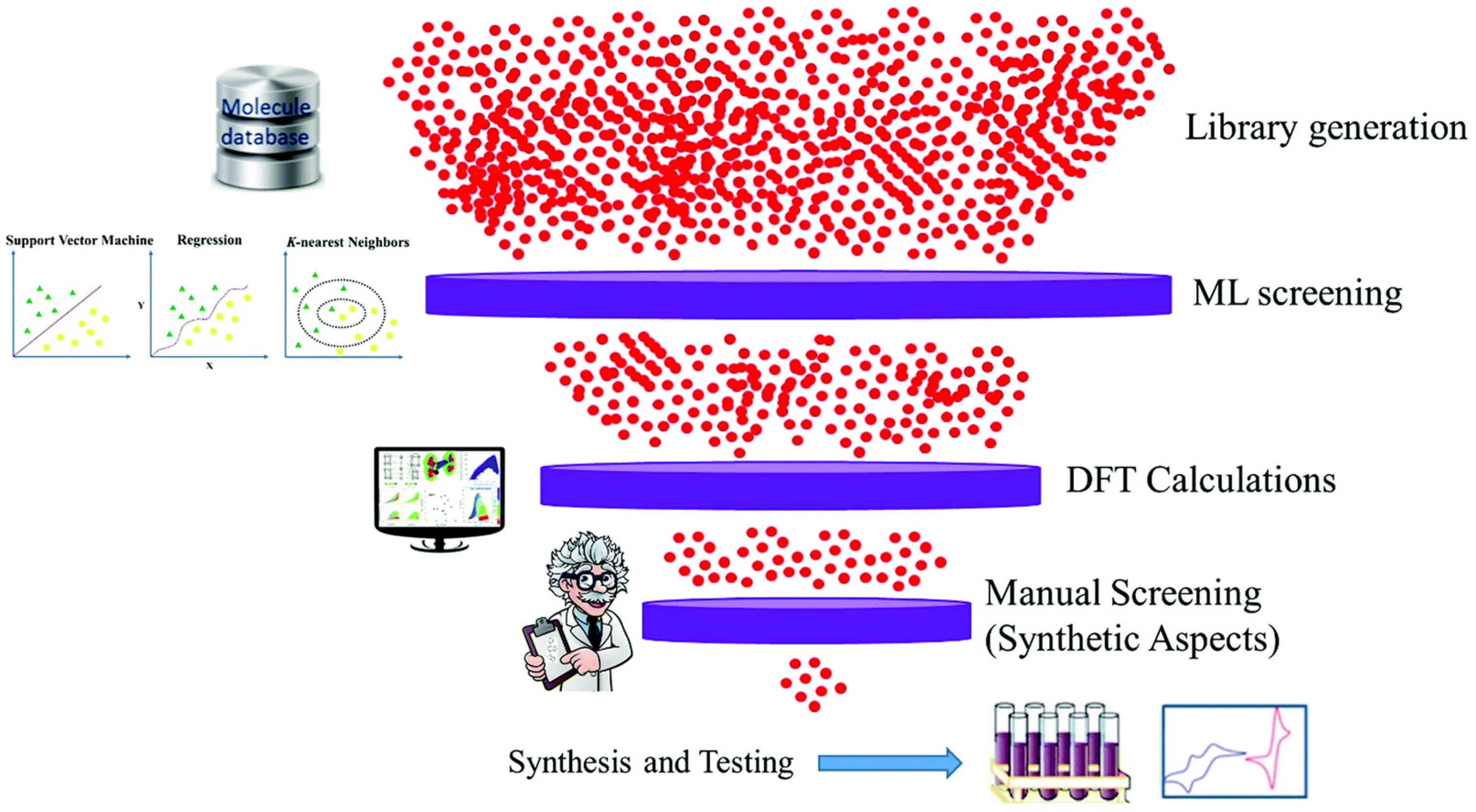
2. The Use of ML in OSCs
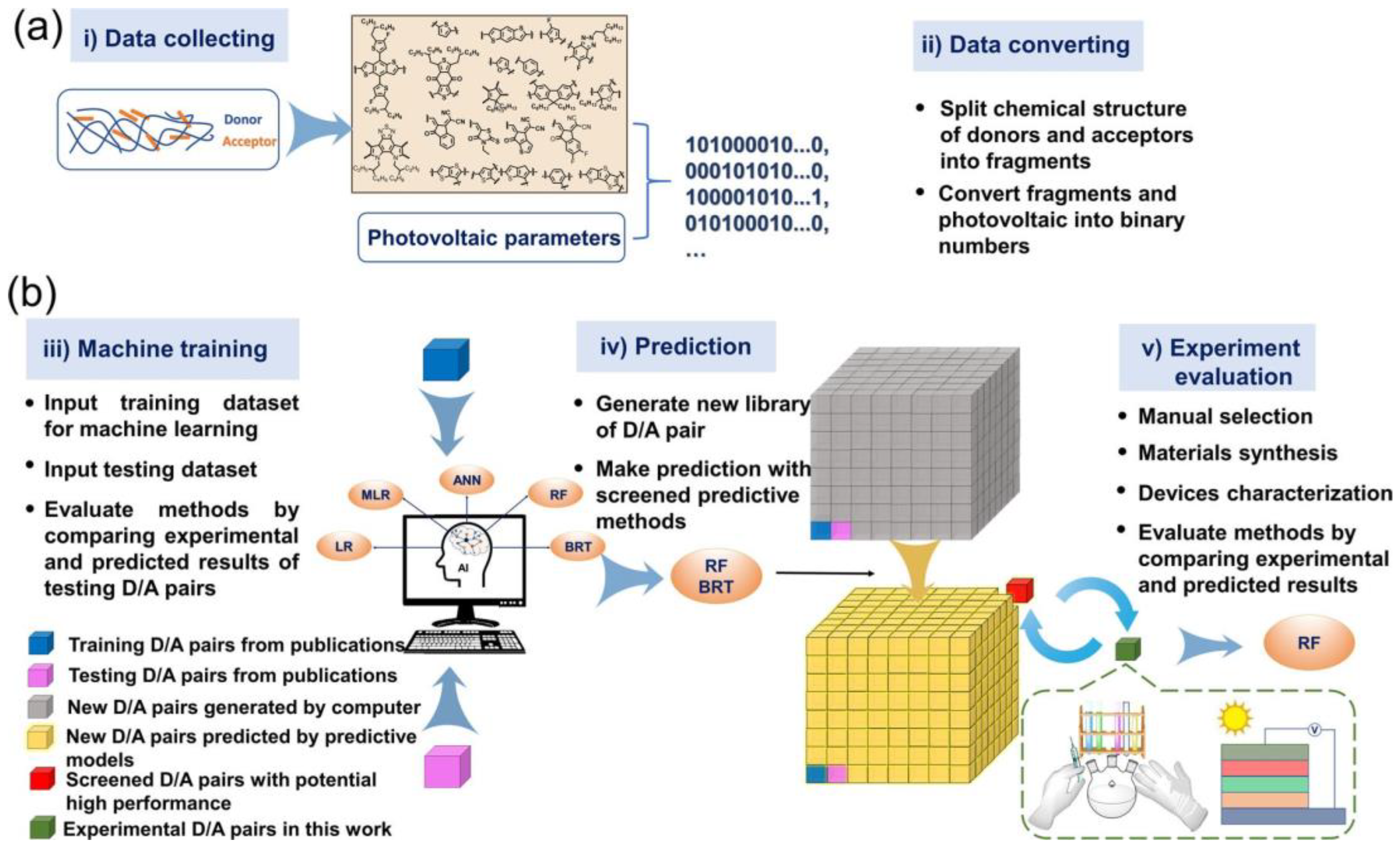
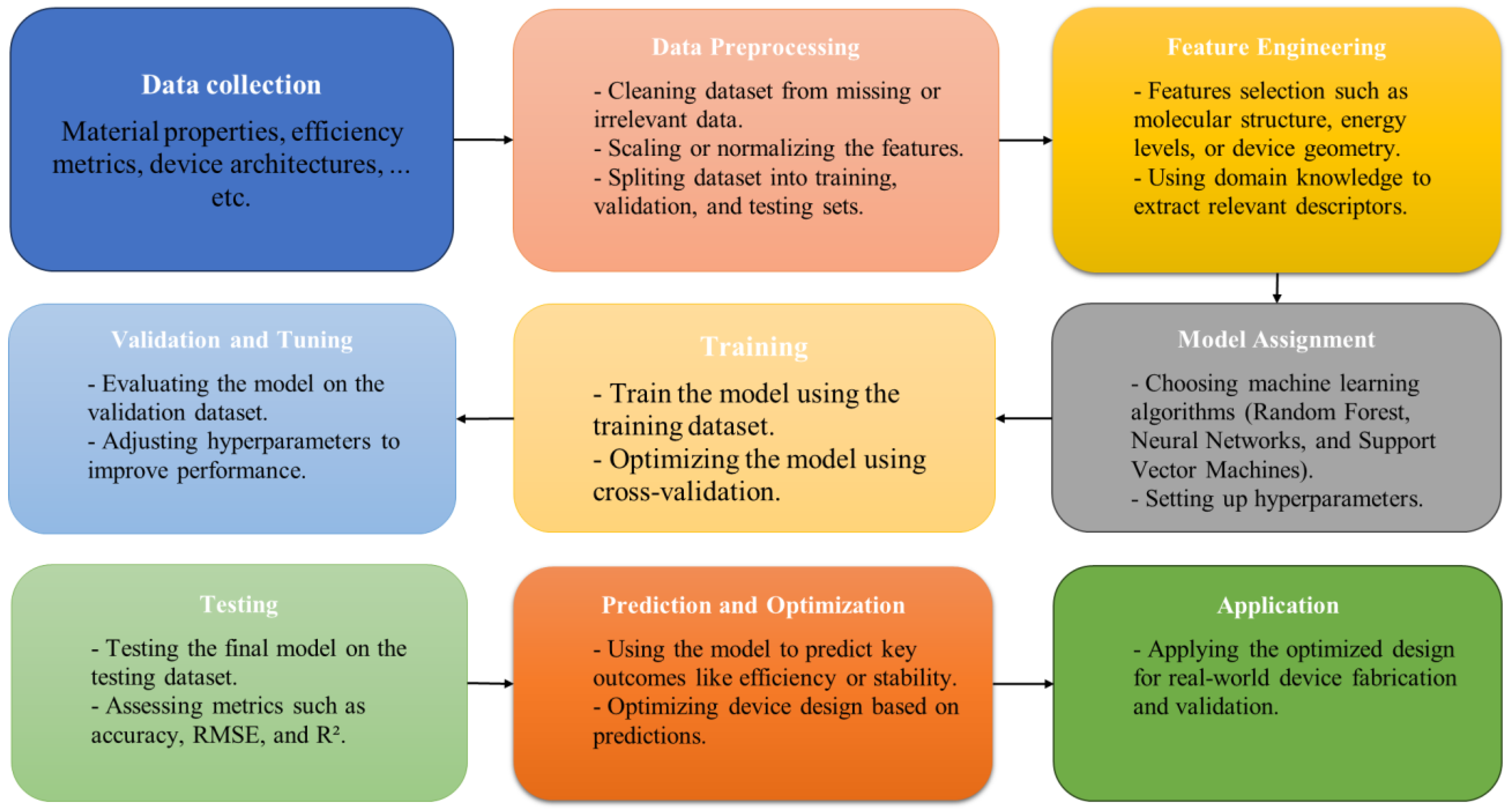
3. Steps of ML Applications
3.1. Sample Collection
3.2. Data Processing
3.3. Model Training
- Support Vector Machines (SVMs): SVMs have been used for classifying OSC materials into high- and low-performing categories. For example, they can predict whether a new donor/acceptor pair will result in a device with high PCE by analyzing molecular descriptors.
- Random Forests (RFs): RF models are commonly applied to regression tasks, such as predicting the PCE of OSCs based on input features like bandgap, chemical composition, and solvent properties. Their ability to handle high-dimensional data and provide feature importance rankings makes them valuable for identifying key parameters.
- Neural Networks (NNs): Deep learning approaches, including feedforward neural networks, have been applied to predict OSC performance metrics. These models can capture non-linear relationships in large datasets but require careful tuning to avoid overfitting.
- Gaussian Process Regression (GPR): GPR models are useful for predicting OSC properties when data are scarce. They provide uncertainty estimates, making them ideal for guiding experimental design and reducing the number of necessary experiments.
- k-Means Clustering: This unsupervised learning technique groups materials with similar characteristics, which can aid in identifying novel donor/acceptor combinations or processing conditions.
3.4. Model Testing
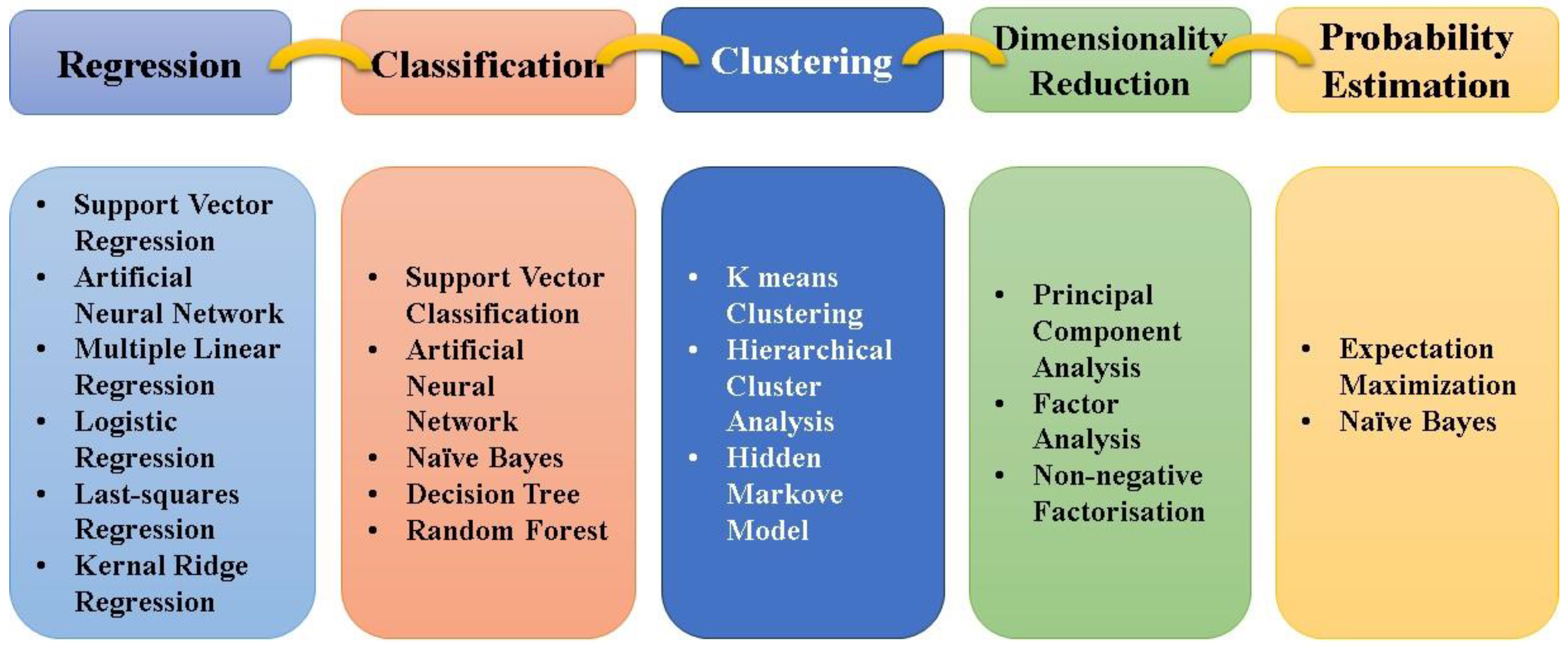
4. Types of ML Algorithm
- Supervised Learning: Supervised learning algorithms are trained on labeled data, meaning each training example is paired with an output label. These algorithms learn to map inputs to outputs, which is critical for predicting the properties of new materials [30,31]. They are mostly used to categorize data into predefined classes. For example, in OSCs, classification algorithms can predict whether a new material will act as a donor or acceptor based on its molecular structure [32]. In supervised learning, accuracy is a metric that measures how well a model correctly predicts the target variable, calculated as the ratio of correct predictions to the total number of predictions. It is widely used in classification tasks to evaluate performance but can be misleading for imbalanced datasets, where one class dominates; in such cases, metrics like precision (the proportion of correct positive predictions), recall (the ability to identify all actual positives), F1-score (the harmonic mean of precision and recall), or ROC-AUC (the area under the curve representing true positive versus false positive rates) are more informative. Below are the key algorithms of supervised learning.
- Support Vector Machines (SVM): SVMs are effective in classifying materials based on their electronic properties. For instance, they can help determine which molecular structures are likely to result in high-efficiency donor or acceptor materials for OSCs [4].
- Decision Trees and Random Forests: These algorithms identify critical structural features that determine material performance. They can be used to analyze various molecular descriptors and pinpoint which attributes are most influential in achieving high PCE [33].
- Linear Regression: Linear regression is often used to model the relationship between molecular descriptors and PCE. For example, linear regression can help establish how changes in molecular structure affect the efficiency of OSCs [34].
- Neural Networks: Neural networks can capture more complex, non-linear relationships between structure and efficiency. They are particularly useful in modeling the intricate dependencies between various molecular features and the overall performance of OSCs [35].
- Unsupervised Learning: Unsupervised learning algorithms deal with data without labeled responses. They are useful for discovering hidden patterns or intrinsic structures in the data [36]. Below are the key algorithms of unsupervised learning.
- Clustering Algorithms: Clustering algorithms, such as k-Means, can group materials with similar properties, aiding in the identification of promising material families. For instance, clustering can reveal which sets of molecular structures consistently yield high-efficiency OSCs [37].
- Dimensionality Reduction Techniques: Techniques like PCA reduce the complexity of data while retaining essential patterns, which is crucial when dealing with high-dimensional datasets in material sciences. PCA can help identify the most influential factors in determining OSC performance, streamlining the design process [11].
- Semi-supervised Learning: Semi-supervised learning strikes a balance between supervised and unsupervised methods, making it especially useful when labeled data are hard to come by but unlabeled data are abundant. Imagine having a small set of data points with labels and a much larger set without them. Semi-supervised learning uses the labeled data to guide the learning process and make sense of the unlabeled data. Techniques like self-training allow a model to start learning with labeled data, then predict labels for the unlabeled data and improve itself iteratively. Graph-based approaches also come into play, where relationships between data points are mapped to spread labels from known points to unknown ones [38].
- Reinforcement Learning: Reinforcement learning involves training models through trial and error, using feedback from their actions. This approach can optimize material synthesis processes or experimental procedures to maximize efficiency or yield. For instance, Q-Learning and Deep Q-Networks (DQN) can optimize the sequence of synthesis steps to produce materials with desired properties efficiently, thereby refining the fabrication process of OSCs to enhance their stability and efficiency [11].
5. ML Analysis of OSCs
- Hybrid and Multiscale Modeling: These approaches integrate different modeling techniques to provide a comprehensive understanding of material behavior across various scales [11].
- Atomistic or Molecular-Level Models: These models focus on the interactions at the molecular level, which are crucial for understanding the fundamental properties of materials. For instance, molecular dynamics simulations can reveal how molecular vibrations and rotations affect the electronic properties of OSCs [41].
- Continuum or Device-Level Models: These models help in understanding how molecular-level properties translate to macroscopic device performance. For example, continuum models can simulate the charge transport properties in OSCs, providing insights into how molecular arrangements affect overall efficiency.
- 2.
- Performance Prediction and Optimization: Performance prediction and optimization involve using computational models, statistical methods, or ML techniques to forecast and improve the performance of a system, device, or process.
- Performance Prediction: In the context of OSCs, performance prediction involves using models or algorithms to estimate and forecast the characteristics and efficiency of the solar cell based on various factors. This prediction may encompass the expected PCE, short-circuit current density (Jsc), open-circuit voltage (Voc), fill factor (FF), or other key metrics that quantify the effectiveness of the solar cell in converting sunlight into electricity. For example, ML models can predict how different material compositions and device architectures will perform under specific operating conditions [42].
- Optimization Strategies: Optimization involves adjusting parameters such as material composition, device architecture, layer thicknesses, interfaces, or manufacturing processes to maximize efficiency, increase stability, or enhance other desirable characteristics. ML algorithms can be used to identify the optimal combinations of these parameters, significantly reducing the need for extensive trial-and-error experimentation. For instance, genetic algorithms can be employed to explore a vast parameter space and find the best configuration for high-efficiency OSCs [11].
- 3.
- Materials Discovery and Design: Materials discovery and design involve the systematic search, identification, and development of new materials or the optimization of existing materials with desired properties for specific applications.
- Property Prediction and Screening: ML models can predict the properties of potential materials, allowing researchers to screen large databases and identify promising candidates quickly. For example, predictive models can estimate the electronic properties of new organic molecules, aiding in the discovery of high-performance materials for OSCs [37,43].
- Database Mining and High-Throughput Screening: ML algorithms can mine existing databases of materials to identify patterns and correlations that may not be apparent through traditional analysis. High-throughput screening techniques can rapidly evaluate a vast number of materials, accelerating the discovery process [44].
- Structure–Property Relationships: Understanding the relationships between molecular structure and material properties is crucial for designing new materials. ML can help elucidate these relationships, guiding the rational design of materials with desired characteristics.
- Design and Synthesis: Once promising materials are identified, ML can aid in optimizing the synthesis processes to ensure reproducibility and scalability. For example, ML models can suggest optimal reaction conditions to synthesize high-purity materials efficiently.
- 4.
- Process and Manufacturing Optimization: Process and manufacturing optimization in the context of OSCs involves improving and refining the procedures, techniques, and production methods used in fabricating these photovoltaic devices [31].
- Process Control and Standardization: ML can be used to develop standardized protocols that ensure consistent quality and performance of OSCs. For example, ML algorithms can monitor production processes in real time, adjusting parameters to maintain optimal conditions.
- Yield Improvement: By analyzing production data, ML can identify factors that influence yield and suggest modifications to improve it. This can lead to higher efficiency and lower costs in OSC manufacturing.
- Scaling Production and Cost Reduction: ML techniques can optimize manufacturing processes to make them more scalable and cost-effective. For instance, predictive models can help in planning resource allocation and minimizing waste.
- Robustness and Reliability: ML can enhance the robustness and reliability of OSCs by identifying and mitigating factors that lead to device degradation. This can result in longer-lasting and more stable solar cells.
- 5.
- Pattern Recognition and Data Analysis: Pattern recognition and data analysis involve the systematic process of identifying meaningful patterns, structures, or relationships within datasets, enabling the extraction of valuable insights or information [37].
- Data Collection and Preprocessing: Efficient data collection and preprocessing are crucial for ensuring high-quality inputs for ML models. This includes cleaning data, handling missing values, and normalizing data to make it suitable for analysis.
- Exploratory Data Analysis (EDA): EDA techniques help in understanding the underlying patterns and distributions in the data. Visualization tools can provide insights into how different variables interact and influence OSC performance.
- Feature Extraction and Selection: Identifying the most relevant features or descriptors is essential for building accurate ML models. Techniques like PCA can reduce the dimensionality of the data, focusing on the most significant variables.
- Clustering and Classification: Clustering algorithms can group similar data points, helping to identify patterns in material properties. Classification algorithms can categorize materials based on their predicted performance.
- Regression and Prediction: Regression techniques can model the relationships between variables, providing predictions for new data points. These predictions can guide the development of new materials and the optimization of OSCs.
- Anomaly Detection and Outlier Analysis: Identifying anomalies and outliers in the data can reveal potential issues or novel phenomena that warrant further investigation. This can lead to new discoveries and improvements in OSC technology.
- Correlation and Relationship Analysis: Understanding the correlations and relationships between different variables helps in identifying key factors that influence OSC performance. This knowledge can inform the design and optimization of new materials [44].
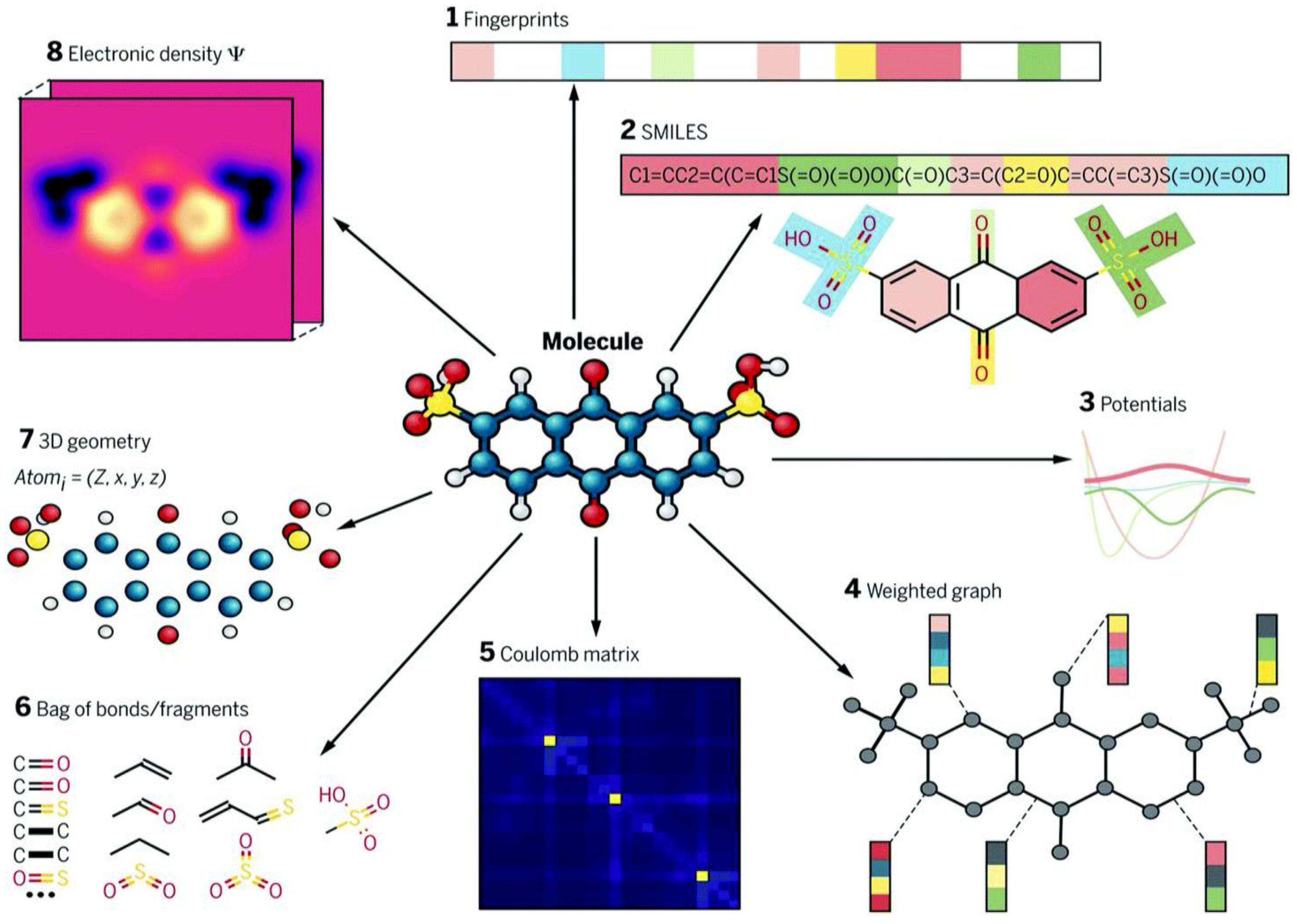
5.1. Molecular Descriptors
- Zero-dimensional descriptors provide basic molecular information, such as atomic number, molecular weight, and atom types, without including topological or connectivity information.
- One-dimensional descriptors capture counts and types of chemical fragments, representing molecular composition.
- Two-dimensional descriptors include topological and topo-chemical properties, reflecting atom connectivity and chemical bonding patterns.
- Three-dimensional descriptors capture geometric and conformational information, such as molecular volume, surface area, and partial charges.
5.2. Comparison of Prediction Accuracies
- Descriptors for Energy Level Predictions: When predicting LUMO and HOMO, studies have demonstrated that 3D descriptors generally outperform 2D descriptors due to their inclusion of geometric information. The RF model achieved an MAE of 0.16 eV using combined 2D and 3D descriptors, whereas models relying solely on 2D descriptors showed an MAE of approximately 0.24 eV [47].
- Descriptors for PCE Predictions: Sui et al. developed a cascaded support vector machine (CasSVM) model using a combination of 0D, 1D, and 2D descriptors to predict key device parameters such as JSC, VOC, and FF, which were then correlated to PCE [48,49]. Their model achieved an MAE of 0.35% for PCE predictions, corresponding to about 10% of the average PCE value (3.89%). In contrast, earlier models that excluded 2D descriptors showed higher MAE values, often exceeding 0.50%. The R2 value of 0.96 for the CasSVM model highlights its superior predictive capability when utilizing a diverse set of molecular descriptors.
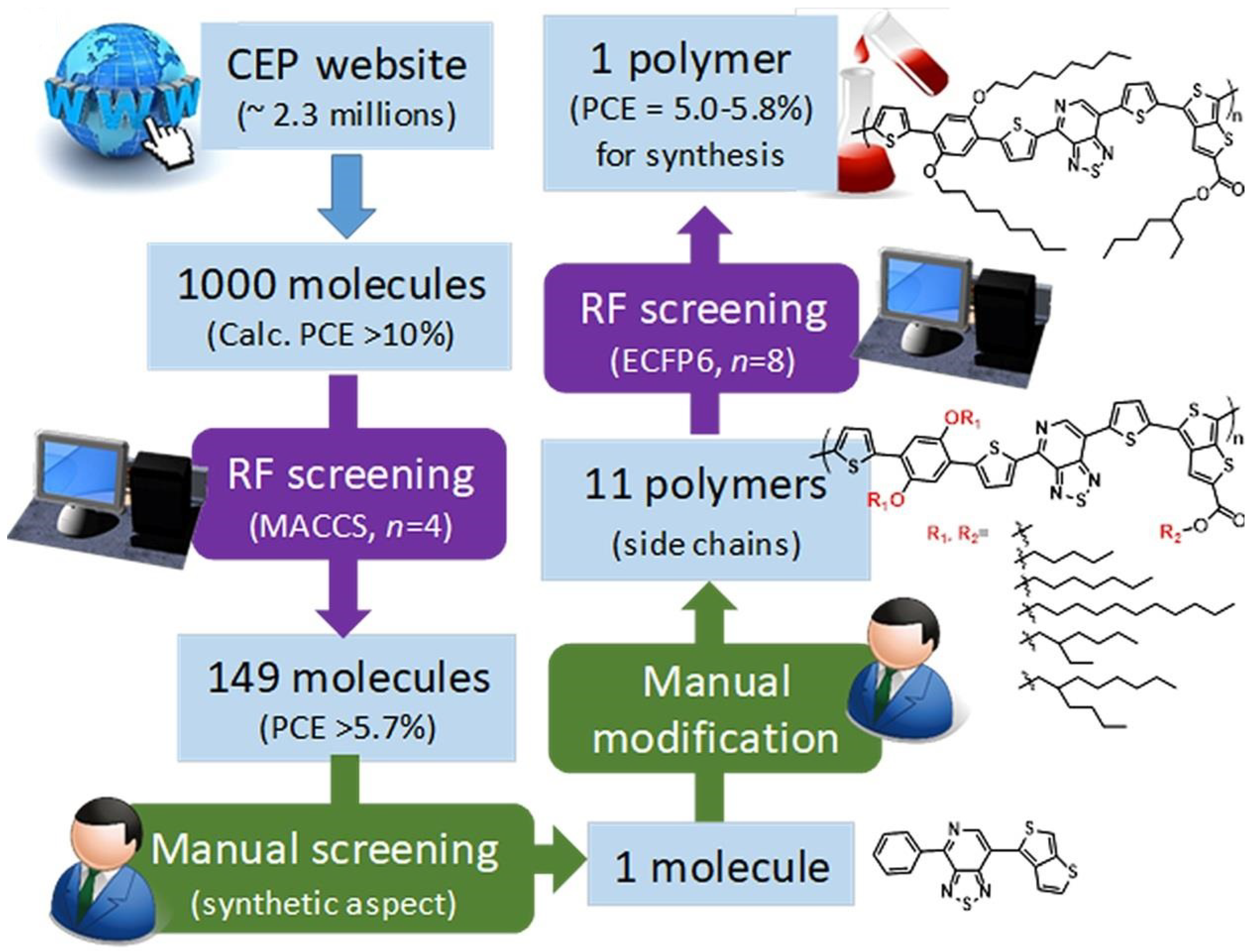
- High-Throughput Screening: Omar et al. compared 0D and 3D descriptors for high-throughput screening of organic semiconductors [50]. They found that 3D descriptors, incorporating molecular volume and partial charges, improved the classification accuracy of high- versus low-performing materials by 15% compared to 0D descriptors alone.
5.3. Efficiency Versus Complexity
5.4. Molecular Fingerprints
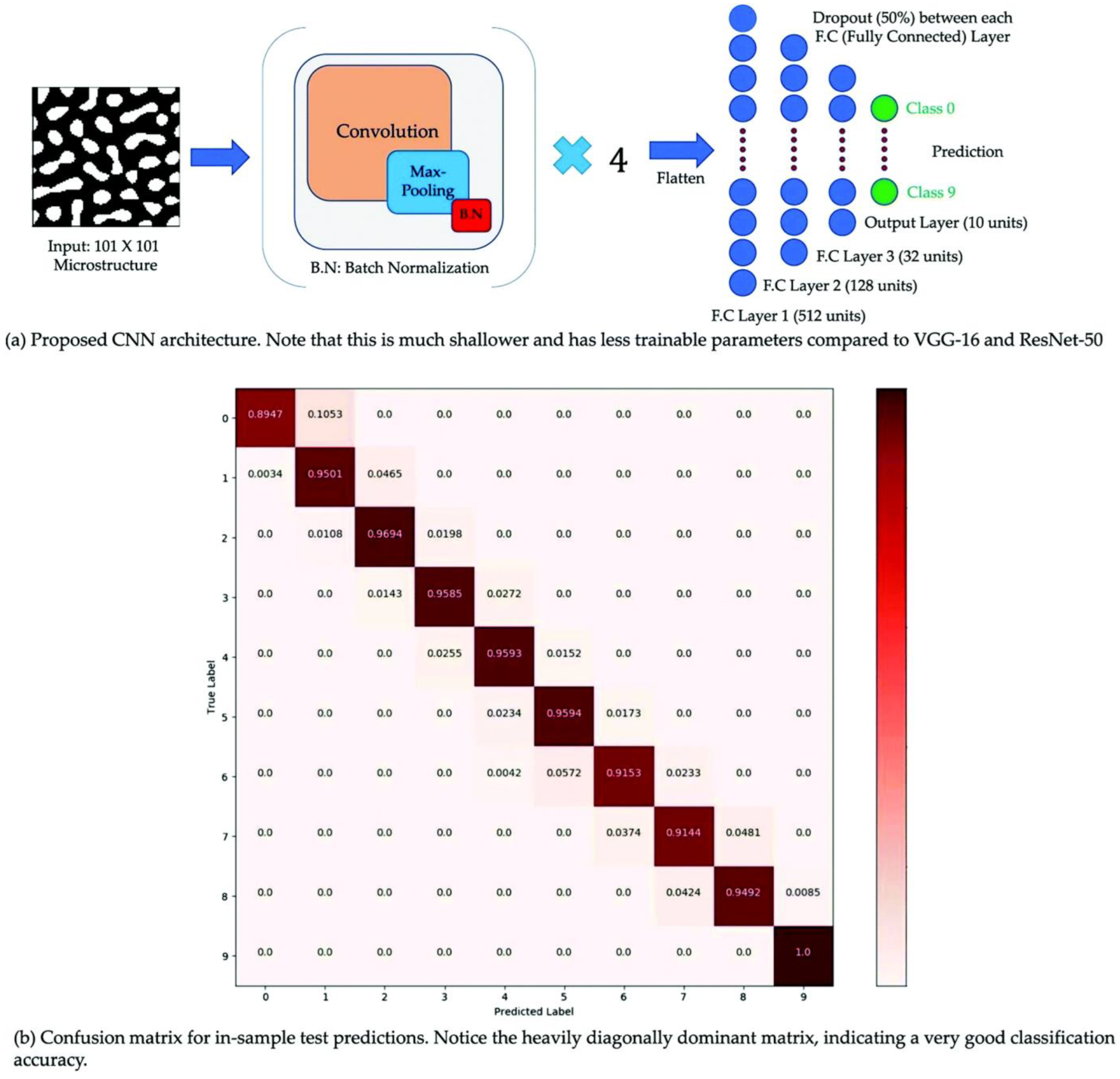
5.5. Images
5.6. Microscopic Properties
5.7. Energy Levels
5.8. Simulated Properties
6. Challenges and Future Prospects
6.1. Data Infrastructure
6.2. Descriptor Selection
6.3. Multidimensional Design
- GISAXS: Useful for probing nanoscale morphology with a resolution typically around 1–100 nm. Data processing often involves advanced fitting procedures to extract domain spacing and orientation information.
- AFM: Provides surface morphology details at a resolution of ~1 nm but requires noise reduction techniques to mitigate surface irregularities.
- TEM: Offers atomic to nanoscale resolution (~0.1 nm) but demands complex sample preparation and interpretation.
- GIWAXS: Captures crystallographic information with sub-nanometer resolution, requiring extensive data modeling to distinguish between amorphous and crystalline phases.
6.4. Experimental Validation
6.5. Development of Better Software
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ML | Machine Learning |
| OSCs | Organic Solar Cells |
| PCE | Power Conversion Efficiency |
| HOMO | Highest Occupied Molecular Orbital |
| LUMO | Lowest Unoccupied Molecular Orbital |
| BHJ | Bulk Heterojunction |
| HCEP | Harvard Clean Energy Project |
| DoF | Degree of Freedom |
| D/A | Donor/Acceptor |
| DFT | Density Functional Theory |
| NFAs | Non-Fullerene Acceptors |
| HPC | High-Performance Computing |
| PCA | Principal Component Analysis |
| DA | Discriminant Analysis |
| ICA | Independent Component Analysis |
| Voc | Open-circuit Voltage |
| Jsc | Short-circuit Current Density |
| FF | Fill Factor |
| RF | Random Forest |
| LR | Linear Regression |
| BRT | Boosted Regression Trees |
| GBRT | Gradient Boosting Regression Tree |
| ANN | Artificial Neural Networks |
| k-NN | k-Nearest Neighbors |
| KRR | Kernel Ridge Regression |
| SVR | Support Vector Regression |
| DNN | Deep Neural Network |
| CNNs | Convolutional Neural Networks |
| BAE | Bistricyclic Aromatic Compounds |
| CasSVM | Cascaded Support Vector Machine |
| MAE | Mean Absolute Error |
| Si | Silicon |
| GaAs | Gallium Arsenide |
| GVA | Grammar Variational Autoencoder |
| MD | Molecular Dynamics |
| AFM | Atomic Force Microscopy |
| TEM | Transmission Electron Microscopy |
| GIWAXS | Grazing Incidence Wide-Angle X-ray Scattering |
References
- Du, X.; Heumueller, T.; Gruber, W.; Classen, A.; Unruh, T.; Li, N.; Brabec, C.J. Efficient polymer solar cells based on non-fullerene acceptors with potential device lifetime approaching 10 years. Joule 2019, 3, 215–226. [Google Scholar] [CrossRef]
- Wan, X.; Li, C.; Zhang, M.; Chen, Y. Acceptor–donor–acceptor type molecules for high performance organic photovoltaics–chemistry and mechanism. Chem. Soc. Rev. 2020, 49, 2828–2842. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Tang, W.; Bi, P.; Liu, Z.; Lu, W.; Wang, X.; Hao, X.; Wong, W.-K.; Zhu, X. Enhanced light-harvesting of benzodithiophene conjugated porphyrin electron donors in organic solar cells. J. Mater. Chem. C 2019, 7, 380–386. [Google Scholar] [CrossRef]
- Wang, H.; Feng, J.; Dong, Z.; Jin, L.; Li, M.; Yuan, J.; Li, Y. Efficient screening framework for organic solar cells with deep learning and ensemble learning. npj Comput. Mater. 2023, 9, 200. [Google Scholar] [CrossRef]
- Danladi, E.; Dogo, D.S.; Michael, S.U.; Uloko, F.O.; Salawu, A.A.O. Recent advances in modeling of perovskite solar cells using scaps-1d: Effect of absorber and etm thickness. East Eur. J. Phys. 2021, 4, 5–17. [Google Scholar] [CrossRef]
- Hussain, S.S.; Riaz, S.; Nowsherwan, G.A.; Jahangir, K.; Raza, A.; Iqbal, M.J.; Sadiq, I.; Hussain, S.M.; Naseem, S. Numerical Modeling and Optimization of Lead-Free Hybrid Double Perovskite Solar Cell by Using SCAPS-1D. J. Renew. Energy 2021, 2021, 6668687. [Google Scholar] [CrossRef]
- Saha, N.; Brunetti, G.; Armenise, M.N.; Carlo, A.D.; Ciminelli, C. Modeling Highly Efficient Homojunction Perovskite Solar Cells With Graphene-TiO2 Nanocomposite as the Electron Transport Layer. IEEE J. Photovolt. 2023, 13, 705–710. [Google Scholar] [CrossRef]
- Mahmood, A.; Wang, J.-L. Machine learning for high performance organic solar cells: Current scenario and future prospects. Energy Environ. Sci. 2021, 14, 90–105. [Google Scholar] [CrossRef]
- Wadsworth, A.; Moser, M.; Marks, A.; Little, M.S.; Gasparini, N.; Brabec, C.J.; Baran, D.; McCulloch, I. Critical review of the molecular design progress in non-fullerene electron acceptors towards commercially viable organic solar cells. Chem. Soc. Rev. 2019, 48, 1596–1625. [Google Scholar] [CrossRef] [PubMed]
- Scharber, M.C.; Mühlbacher, D.; Koppe, M.; Denk, P.; Waldauf, C.; Heeger, A.J.; Brabec, C.J. Design rules for donors in bulk-heterojunction solar cells—Towards 10% energy-conversion efficiency. Adv. Mater. 2006, 18, 789–794. [Google Scholar] [CrossRef]
- Padula, D.; Simpson, J.D.; Troisi, A. Combining electronic and structural features in machine learning models to predict organic solar cells properties. Mater. Horiz. 2019, 6, 343–349. [Google Scholar] [CrossRef]
- Gu, G.H.; Noh, J.; Kim, I.; Jung, Y. Machine learning for renewable energy materials. J. Mater. Chem. A 2019, 7, 17096–17117. [Google Scholar] [CrossRef]
- Rodríguez-Martínez, X.; Pascual-San-José, E.; Campoy-Quiles, M. Accelerating organic solar cell material’s discovery: High-throughput screening and big data. Energy Environ. Sci. 2021, 14, 3301–3322. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.; Yang, D.; Yang, S.; Ge, Z. Stability of organic solar cells: Toward commercial applications. Chem. Soc. Rev. 2024, 53, 2350–2387. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luke, J.; Privitera, A.; Rolland, N.; Labanti, C.; Londi, G.; Lemaur, V.; Toolan, D.T.; Sneyd, A.J.; Jeong, S. The critical role of the donor polymer in the stability of high-performance non-fullerene acceptor organic solar cells. Joule 2023, 7, 810–829. [Google Scholar] [CrossRef]
- França, R.P.; Monteiro, A.C.B.; Arthur, R.; Iano, Y. An overview of deep learning in big data, image, and signal processing in the modern digital age. In Trends in Deep Learning Methodologies; Academic Press: Cambridge, MA, USA, 2021; pp. 63–87. [Google Scholar]
- Dong, S.; Wang, P.; Abbas, K. A survey on deep learning and its applications. Comput. Sci. Rev. 2021, 40, 100379. [Google Scholar] [CrossRef]
- Cova, T.; Pais, A. Deep learning for deep chemistry: Optimizing the prediction of chemical patterns. Front. Chem. 2019, 7, 809. [Google Scholar] [CrossRef] [PubMed]
- Schleder, G.R.; Padilha, A.C.; Acosta, C.M.; Costa, M.; Fazzio, A. From DFT to machine learning: Recent approaches to materials science—A review. J. Phys. Mater. 2019, 2, 032001. [Google Scholar] [CrossRef]
- Zhou, T.; Song, Z.; Sundmacher, K. Big data creates new opportunities for materials research: A review on methods and applications of machine learning for materials design. Engineering 2019, 5, 1017–1026. [Google Scholar] [CrossRef]
- Géron, A. Hands-On Machine Learning with Scikit-Learn, Keras, and TensorFlow; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2022. [Google Scholar]
- Kim, K.G. Book review: Deep learning. Healthc. Inform. Res. 2016, 22, 351–354. [Google Scholar] [CrossRef]
- Pyzer-Knapp, E.O.; Li, K.; Aspuru-Guzik, A. Learning from the harvard clean energy project: The use of neural networks to accelerate materials discovery. Adv. Funct. Mater. 2015, 25, 6495–6502. [Google Scholar] [CrossRef]
- Pyzer-Knapp, E.O.; Pitera, J.W.; Staar, P.W.; Takeda, S.; Laino, T.; Sanders, D.P.; Sexton, J.; Smith, J.R.; Curioni, A. Accelerating materials discovery using artificial intelligence, high performance computing and robotics. npj Comput. Mater. 2022, 8, 84. [Google Scholar] [CrossRef]
- Taffese, W.Z.; Espinosa-Leal, L. Unveiling non-steady chloride migration insights through explainable machine learning. J. Build. Eng. 2024, 82, 108370. [Google Scholar] [CrossRef]
- Guyon, I.; Elisseeff, A. An introduction to variable and feature selection. J. Mach. Learn. Res. 2003, 3, 1157–1182. [Google Scholar]
- Alwadai, N.; Khan, S.U.-D.; Elqahtani, Z.M.; Ud-Din Khan, S. Machine learning assisted prediction of power conversion efficiency of all-small molecule organic solar cells: A data visualization and statistical analysis. Molecules 2022, 27, 5905. [Google Scholar] [CrossRef] [PubMed]
- Hußner, I.; Lazarides, R.; Symes, W.; Richter, E.; Westphal, A. Reflect on your teaching experience: Systematic reflection of teaching behaviour and changes in student teachers’ self-efficacy for reflection. Z. Für Erzieh. 2023, 26, 1301–1320. [Google Scholar] [CrossRef]
- Lombardo, D.; Calandra, P.; Pasqua, L.; Magazù, S. Self-assembly of organic nanomaterials and biomaterials: The bottom-up approach for functional nanostructures formation and advanced applications. Materials 2020, 13, 1048. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Alvarez-Gonzaga, O.A.; Rodriguez, J.I. Machine learning models with different cheminformatics data sets to forecast the power conversion efficiency of organic solar cells. arXiv 2024, arXiv:2410.23444. [Google Scholar]
- Chen, G.; Tang, D.-M. Machine Learning as a “Catalyst” for Advancements in Carbon Nanotube Research. Nanomaterials 2024, 14, 1688. [Google Scholar] [CrossRef]
- Zhang, C.-R.; Li, M.; Zhao, M.; Gong, J.-J.; Liu, X.-M.; Chen, Y.-H.; Liu, Z.-J.; Wu, Y.-Z.; Chen, H.-S. Machine learning study on organic solar cells and virtual screening of designed non-fullerene acceptors. J. Appl. Phys. 2023, 134, 153104. [Google Scholar] [CrossRef]
- Rosenblatt, F. The perceptron: A probabilistic model for information storage and organization in the brain. Psychol. Rev. 1958, 65, 386. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, I. Deep Learning; MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Ain, A. Data clustering: 50 years beyond K-means. Pattern Recognit. Lett. 2010, 31, 651–666. [Google Scholar]
- Sahu, H.; Yang, F.; Ye, X.; Ma, J.; Fang, W.; Ma, H. Designing promising molecules for organic solar cells via machine learning assisted virtual screening. J. Mater. Chem. A 2019, 7, 17480–17488. [Google Scholar] [CrossRef]
- Greenstein, B.L.; Hutchison, G.R. Screening efficient tandem organic solar cells with machine learning and genetic algorithms. J. Phys. Chem. C 2023, 127, 6179–6191. [Google Scholar] [CrossRef]
- Sun, W.; Zheng, Y.; Yang, K.; Zhang, Q.; Shah, A.A.; Wu, Z.; Sun, Y.; Feng, L.; Chen, D.; Xiao, Z. Machine learning–assisted molecular design and efficiency prediction for high-performance organic photovoltaic materials. Sci. Adv. 2019, 5, eaay4275. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, S.; Al-Naamani, E.; Saeki, A. Computer-aided screening of conjugated polymers for organic solar cell: Classification by random forest. J. Phys. Chem. Lett. 2018, 9, 2639–2646. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications; Elsevier: Amsterdam, The Netherlands, 2023. [Google Scholar]
- Afzal, M.A.F.; Hachmann, J. High-throughput computational studies in catalysis and materials research, and their impact on rational design. In Handbook on Big Data and Machine Learning in the Physical Sciences: Volume 1. Big Data Methods in Experimental Materials Discovery; World Scientific: Singapore, 2020; pp. 1–44. [Google Scholar]
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Vo, A.H.; Van Vleet, T.R.; Gupta, R.R.; Liguori, M.J.; Rao, M.S. An overview of machine learning and big data for drug toxicity evaluation. Chem. Res. Toxicol. 2019, 33, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lengeling, B.; Aspuru-Guzik, A. Inverse molecular design using machine learning: Generative models for matter engineering. Science 2018, 361, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Xiao, K.; Latino, D.A.; Wu, C.; Zhang, Q.; Aires-de-Sousa, J. Machine learning methods to predict density functional theory B3LYP energies of HOMO and LUMO orbitals. J. Chem. Inf. Model. 2017, 57, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Sui, M.-Y.; Yang, Z.-R.; Geng, Y.; Sun, G.-Y.; Hu, L.; Su, Z.-M. Nonfullerene acceptors for organic photovoltaics: From conformation effect to power conversion efficiencies prediction. Sol. RRL 2019, 3, 1900258. [Google Scholar] [CrossRef]
- Chattopadhyay, J.; Srivastava, N. Application of Nanomaterials in Chemical Sensors and Biosensors; CRC Press: Boca Raton, FL, USA, 2021. [Google Scholar]
- Omar, Ö.H.; Del Cueto, M.; Nematiaram, T.; Troisi, A. High-throughput virtual screening for organic electronics: A comparative study of alternative strategies. J. Mater. Chem. C 2021, 9, 13557–13583. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Pattanaik, L.; Coley, C.W. Molecular representation: Going long on fingerprints. Chem 2020, 6, 1204–1207. [Google Scholar] [CrossRef]
- Mahmood, A.; Hu, J.-Y.; Xiao, B.; Tang, A.; Wang, X.; Zhou, E. Recent progress in porphyrin-based materials for organic solar cells. J. Mater. Chem. A 2018, 6, 16769–16797. [Google Scholar] [CrossRef]
- Zhang, C.; Song, X.; Liu, K.K.; Zhang, M.; Qu, J.; Yang, C.; Yuan, G.Z.; Mahmood, A.; Liu, F.; He, F. Electron-Deficient and Quinoid Central Unit Engineering for Unfused Ring-Based A1–D–A2–D–A1-Type Acceptor Enables High Performance Nonfullerene Polymer Solar Cells with High Voc and PCE Simultaneously. Small 2020, 16, 1907681. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A.; Tang, A.; Wang, X.; Zhou, E. First-principles theoretical designing of planar non-fullerene small molecular acceptors for organic solar cells: Manipulation of noncovalent interactions. Phys. Chem. Chem. Phys. 2019, 21, 2128–2139. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.-K.; Xu, X.; Wang, J.-L.; Zhang, C.; Ge, G.-Y.; Zhuang, F.-D.; Zhang, H.-J.; Yang, C.; Peng, Q.; Pei, J. Achieving high-performance non-halogenated nonfullerene acceptor-based organic solar cells with 13.7% efficiency via a synergistic strategy of an indacenodithieno [3, 2-b] selenophene core unit and non-halogenated thiophene-based terminal group. J. Mater. Chem. A 2019, 7, 24389–24399. [Google Scholar] [CrossRef]
- Lopez, S.A.; Sanchez-Lengeling, B.; de Goes Soares, J.; Aspuru-Guzik, A. Design principles and top non-fullerene acceptor candidates for organic photovoltaics. Joule 2017, 1, 857–870. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, W.; Huang, W.; Lin, F.; Li, T.; Liu, S.; Zhan, X.; Liang, Y.; Gao, C.; Wu, H. Assessing the energy offset at the electron donor/acceptor interface in organic solar cells through radiative efficiency measurements. Energy Environ. Sci. 2019, 12, 3556–3566. [Google Scholar] [CrossRef]
- Linderl, T.; Zechel, T.; Brendel, M.; Moseguí González, D.; Müller-Buschbaum, P.; Pflaum, J.; Brütting, W. Energy Losses in Small-Molecule Organic Photovoltaics. Adv. Energy Mater. 2017, 7, 1700237. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, W.; Zhang, M.; Liu, Y.; Zhou, G.; Xu, S.; Zhang, F.; Zhu, H.; Liu, F.; Zhu, X. Revealing the critical role of the HOMO alignment on maximizing current extraction and suppressing energy loss in organic solar cells. IScience 2019, 19, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, P.B.; Mesta, M.; Shil, S.; García Lastra, J.M.; Jacobsen, K.W.; Thygesen, K.S.; Schmidt, M.N. Machine learning-based screening of complex molecules for polymer solar cells. J. Chem. Phys. 2018, 148, 241735. [Google Scholar] [CrossRef]
- Peng, S.-P.; Zhao, Y. Convolutional neural networks for the design and analysis of non-fullerene acceptors. J. Chem. Inf. Model. 2019, 59, 4993–5001. [Google Scholar] [CrossRef] [PubMed]
- Padula, D.; Troisi, A. Concurrent optimization of organic donor–acceptor pairs through machine learning. Adv. Energy Mater. 2019, 9, 1902463. [Google Scholar] [CrossRef]
- Wu, Y.; Guo, J.; Sun, R.; Min, J. Machine learning for accelerating the discovery of high-performance donor/acceptor pairs in non-fullerene organic solar cells. npj Comput. Mater. 2020, 6, 120. [Google Scholar] [CrossRef]
- Li, Y.; Cai, Y.; Xie, Y.; Song, J.; Wu, H.; Tang, Z.; Zhang, J.; Huang, F.; Sun, Y. A facile strategy for third-component selection in non-fullerene acceptor-based ternary organic solar cells. Energy Environ. Sci. 2021, 14, 5009–5016. [Google Scholar] [CrossRef]
- Sun, W.; Li, M.; Li, Y.; Wu, Z.; Sun, Y.; Lu, S.; Xiao, Z.; Zhao, B.; Sun, K. The use of deep learning to fast evaluate organic photovoltaic materials. Adv. Theory Simul. 2019, 2, 1800116. [Google Scholar] [CrossRef]
- Sahu, H.; Rao, W.; Troisi, A.; Ma, H. Toward predicting efficiency of organic solar cells via machine learning and improved descriptors. Adv. Energy Mater. 2018, 8, 1801032. [Google Scholar] [CrossRef]
- Zhao, Z.-W.; del Cueto, M.; Geng, Y.; Troisi, A. Effect of increasing the descriptor set on machine learning prediction of small molecule-based organic solar cells. Chem. Mater. 2020, 32, 7777–7787. [Google Scholar] [CrossRef]
- Ye, L.; Zhao, W.; Li, S.; Mukherjee, S.; Carpenter, J.H.; Awartani, O.; Jiao, X.; Hou, J.; Ade, H. High-efficiency nonfullerene organic solar cells: Critical factors that affect complex multi-length scale morphology and device performance. Adv. Energy Mater. 2017, 7, 1602000. [Google Scholar] [CrossRef]
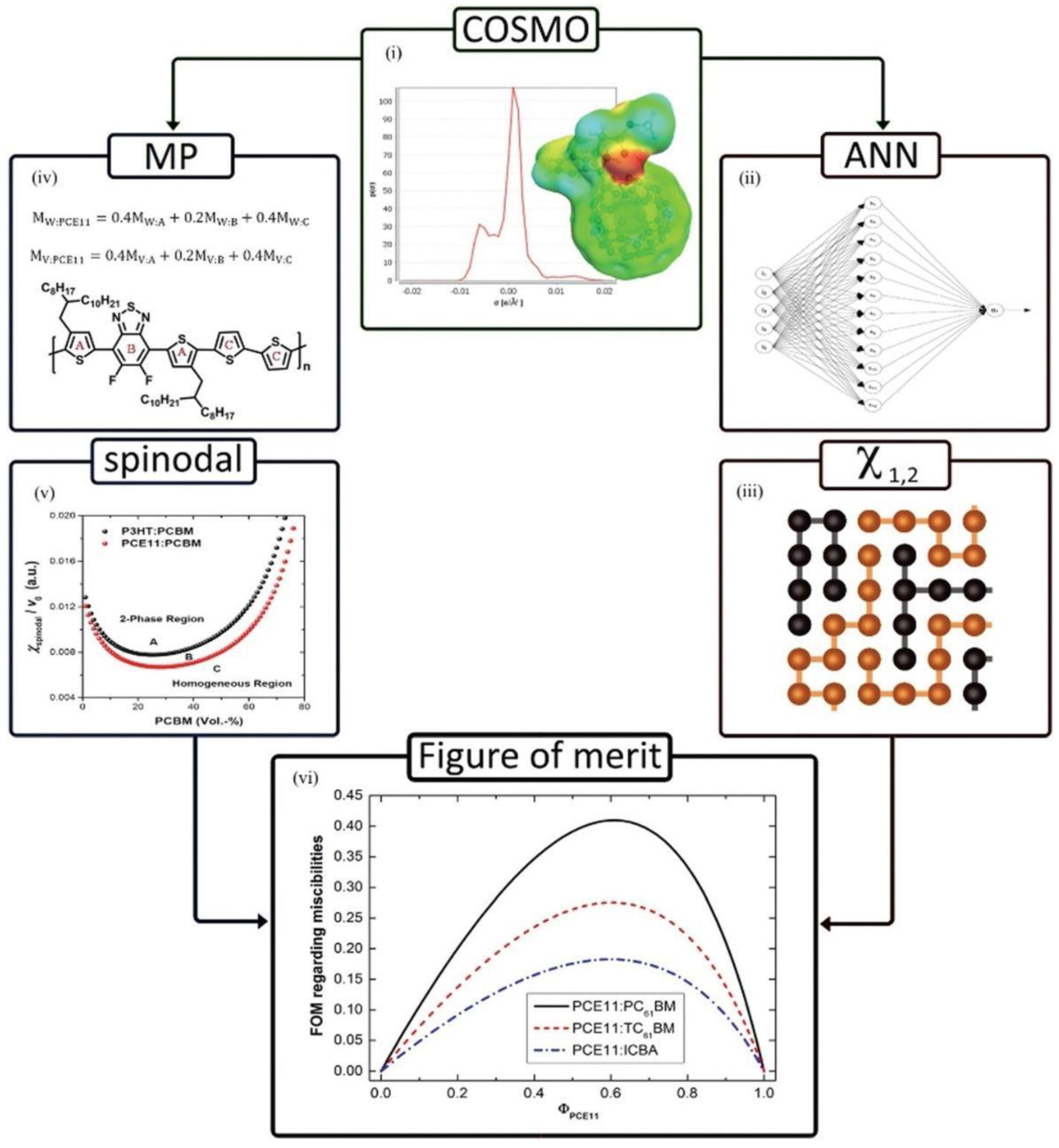
- Duong, D.T.; Walker, B.; Lin, J.; Kim, C.; Love, J.; Purushothaman, B.; Anthony, J.E.; Nguyen, T.Q. Molecular solubility and hansen solubility parameters for the analysis of phase separation in bulk heterojunctions. J. Polym. Sci. Part B Polym. Phys. 2012, 50, 1405–1413. [Google Scholar] [CrossRef]
- Perea, J.D.; Langner, S.; Salvador, M.; Sanchez-Lengeling, B.; Li, N.; Zhang, C.; Jarvas, G.; Kontos, J.; Dallos, A.; Aspuru-Guzik, A. Introducing a new potential figure of merit for evaluating microstructure stability in photovoltaic polymer-fullerene blends. J. Phys. Chem. C 2017, 121, 18153–18161. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, H.; Zhang, R.; Wang, Y.; Hou, J.; Leclerc, M.; Zhan, X.; Huang, F.; Gao, F.; Zou, Y. Reducing voltage losses in the A-DA′ DA acceptor-based organic solar cells. Chem 2020, 6, 2147–2161. [Google Scholar] [CrossRef]
- Hachmann, J.; Olivares-Amaya, R.; Jinich, A.; Appleton, A.L.; Blood-Forsythe, M.A.; Seress, L.R.; Román-Salgado, C.; Trepte, K.; Atahan-Evrenk, S.; Er, S. Lead candidates for high-performance organic photovoltaics from high-throughput quantum chemistry–the Harvard Clean Energy Project. Energy Environ. Sci. 2014, 7, 698–704. [Google Scholar] [CrossRef]
- Imamura, Y.; Tashiro, M.; Katouda, M.; Hada, M. Automatic high-throughput screening scheme for organic photovoltaics: Estimating the orbital energies of polymers from oligomers and evaluating the photovoltaic characteristics. J. Phys. Chem. C 2017, 121, 28275–28286. [Google Scholar] [CrossRef]
- Pyzer-Knapp, E.O.; Simm, G.N.; Guzik, A.A. A Bayesian approach to calibrating high-throughput virtual screening results and application to organic photovoltaic materials. Mater. Horiz. 2016, 3, 226–233. [Google Scholar] [CrossRef]
- Lee, M.-H. Robust random forest based non-fullerene organic solar cells efficiency prediction. Org. Electron. 2020, 76, 105465. [Google Scholar] [CrossRef]
- Yue, Q.; Liu, W.; Zhu, X. n-Type molecular photovoltaic materials: Design strategies and device applications. J. Am. Chem. Soc. 2020, 142, 11613–11628. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Gao, W.; Ma, X.; Hu, Z.; Xu, C.; Wang, X.; An, Q.; Yang, C.; Zhang, X.; Zhang, F. Over 14.5% efficiency and 71.6% fill factor of ternary organic solar cells with 300 nm thick active layers. Energy Environ. Sci. 2020, 13, 958–967. [Google Scholar] [CrossRef]
- Liu, T.; Ma, R.; Luo, Z.; Guo, Y.; Zhang, G.; Xiao, Y.; Yang, T.; Chen, Y.; Li, G.; Yi, Y. Concurrent improvement in J sc and V oc in high-efficiency ternary organic solar cells enabled by a red-absorbing small-molecule acceptor with a high LUMO level. Energy Environ. Sci. 2020, 13, 2115–2123. [Google Scholar] [CrossRef]
- Lee, M.H. Insights from machine learning techniques for predicting the efficiency of fullerene derivatives-based ternary organic solar cells at ternary blend design. Adv. Energy Mater. 2019, 9, 1900891. [Google Scholar] [CrossRef]
- Lee, M.-H. A Machine Learning–Based Design Rule for Improved Open-Circuit Voltage in Ternary Organic Solar Cells. Adv. Intell. Syst. 2020, 2, 1900108. [Google Scholar] [CrossRef]
- Zhou, Z.; Xu, S.; Song, J.; Jin, Y.; Yue, Q.; Qian, Y.; Liu, F.; Zhang, F.; Zhu, X. High-efficiency small-molecule ternary solar cells with a hierarchical morphology enabled by synergizing fullerene and non-fullerene acceptors. Nat. Energy 2018, 3, 952–959. [Google Scholar] [CrossRef]
- Liu, G.; Jia, J.; Zhang, K.; Jia, X.e.; Yin, Q.; Zhong, W.; Li, L.; Huang, F.; Cao, Y. 15% efficiency tandem organic solar cell based on a novel highly efficient wide-bandgap nonfullerene acceptor with low energy loss. Adv. Energy Mater. 2019, 9, 1803657. [Google Scholar] [CrossRef]
- Lee, M.-H. Performance and matching band structure analysis of tandem organic solar cells using machine learning approaches. Energy Technol. 2020, 8, 1900974. [Google Scholar] [CrossRef]
- Du, P.; Zebrowski, A.; Zola, J.; Ganapathysubramanian, B.; Wodo, O. Microstructure design using graphs. npj Comput. Mater. 2018, 4, 50. [Google Scholar] [CrossRef]
- Pfeifer, S.; Pokuri, B.S.S.; Du, P.; Ganapathysubramanian, B. Process optimization for microstructure-dependent properties in thin film organic electronics. Mater. Discov. 2018, 11, 6–13. [Google Scholar] [CrossRef]
- Noruzi, R.; Ghadai, S.; Bingol, O.R.; Krishnamurthy, A.; Ganapathysubramanian, B. NURBS-based microstructure design for organic photovoltaics. Comput. -Aided Des. 2020, 118, 102771. [Google Scholar] [CrossRef]
- Wodo, O.; Tirthapura, S.; Chaudhary, S.; Ganapathysubramanian, B. A graph-based formulation for computational characterization of bulk heterojunction morphology. Org. Electron. 2012, 13, 1105–1113. [Google Scholar] [CrossRef]
- Pokuri, B.S.S.; Ghosal, S.; Kokate, A.; Sarkar, S.; Ganapathysubramanian, B. Interpretable deep learning for guided microstructure-property explorations in photovoltaics. npj Comput. Mater. 2019, 5, 95. [Google Scholar] [CrossRef]
- Wodo, O.; Ganapathysubramanian, B. Modeling morphology evolution during solvent-based fabrication of organic solar cells. Comput. Mater. Sci. 2012, 55, 113–126. [Google Scholar] [CrossRef]
- Kodali, H.K.; Ganapathysubramanian, B. Computer simulation of heterogeneous polymer photovoltaic devices. Model. Simul. Mater. Sci. Eng. 2012, 20, 035015. [Google Scholar] [CrossRef]
- Majeed, N.; Saladina, M.; Krompiec, M.; Greedy, S.; Deibel, C.; MacKenzie, R.C. Using deep machine learning to understand the physical performance bottlenecks in novel thin-film solar cells. Adv. Funct. Mater. 2020, 30, 1907259. [Google Scholar] [CrossRef]
- Zhang, Y.; Ling, C. A strategy to apply machine learning to small datasets in materials science. npj Comput. Mater. 2018, 4, 25. [Google Scholar] [CrossRef]
- Mahmood, A.; Wang, J.L. A review of grazing incidence small-and wide-angle x-ray scattering techniques for exploring the film morphology of organic solar cells. Sol. RRL 2020, 4, 2000337. [Google Scholar] [CrossRef]
- Jones, L.; Nellist, P.D. Identifying and correcting scan noise and drift in the scanning transmission electron microscope. Microsc. Microanal. 2013, 19, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Zawodzki, M.; Resel, R.; Sferrazza, M.; Kettner, O.; Friedel, B. Interfacial morphology and effects on device performance of organic bilayer heterojunction solar cells. ACS Appl. Mater. Interfaces 2015, 7, 16161–16168. [Google Scholar] [CrossRef] [PubMed]
- Pokuri, B.S.S.; Stimes, J.; O’Hara, K.; Chabinyc, M.L.; Ganapathysubramanian, B. GRATE: A framework and software for GRaph based Analysis of Transmission Electron Microscopy images of polymer films. Comput. Mater. Sci. 2019, 163, 1–10. [Google Scholar] [CrossRef]
- Paul, A.; Jha, D.; Al-Bahrani, R.; Liao, W.-k.; Choudhary, A.; Agrawal, A. Transfer learning using ensemble neural networks for organic solar cell screening. In Proceedings of the 2019 International Joint Conference on Neural Networks (IJCNN), Budapest, Hungary, 14–19 July 2019; pp. 1–8. [Google Scholar]
- Osterrieder, T.; Schmitt, F.; Lüer, L.; Wagner, J.; Heumüller, T.; Hauch, J.; Brabec, C.J. Autonomous optimization of an organic solar cell in a 4-dimensional parameter space. Energy Environ. Sci. 2023, 16, 3984–3993. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, D.R.; Muhammadsharif, F.F. A Review of Machine Learning in Organic Solar Cells. Processes 2025, 13, 393. https://doi.org/10.3390/pr13020393
Ahmed DR, Muhammadsharif FF. A Review of Machine Learning in Organic Solar Cells. Processes. 2025; 13(2):393. https://doi.org/10.3390/pr13020393
Chicago/Turabian StyleAhmed, Darya Rasul, and Fahmi F. Muhammadsharif. 2025. "A Review of Machine Learning in Organic Solar Cells" Processes 13, no. 2: 393. https://doi.org/10.3390/pr13020393
APA StyleAhmed, D. R., & Muhammadsharif, F. F. (2025). A Review of Machine Learning in Organic Solar Cells. Processes, 13(2), 393. https://doi.org/10.3390/pr13020393