
Metagenomic Analysis of Bacterial Community Structure and Dynamics of a Digestate and a More Stabilized Digestate-Derived Compost from Agricultural Waste





Abstract
1. Introduction
2. Materials and Methods
2.1. Digestate and Stabilized Digestate-Derived Compost Sources
2.2. Molecular Analysis of Microbiota from Digestate and Stabilized Digestate-Derived Compost
2.2.1. DNA Extraction
2.2.2. PCR Amplification, Sequencing, and Bioinformatics Analyses
2.3. Statistical Analyses
3. Results
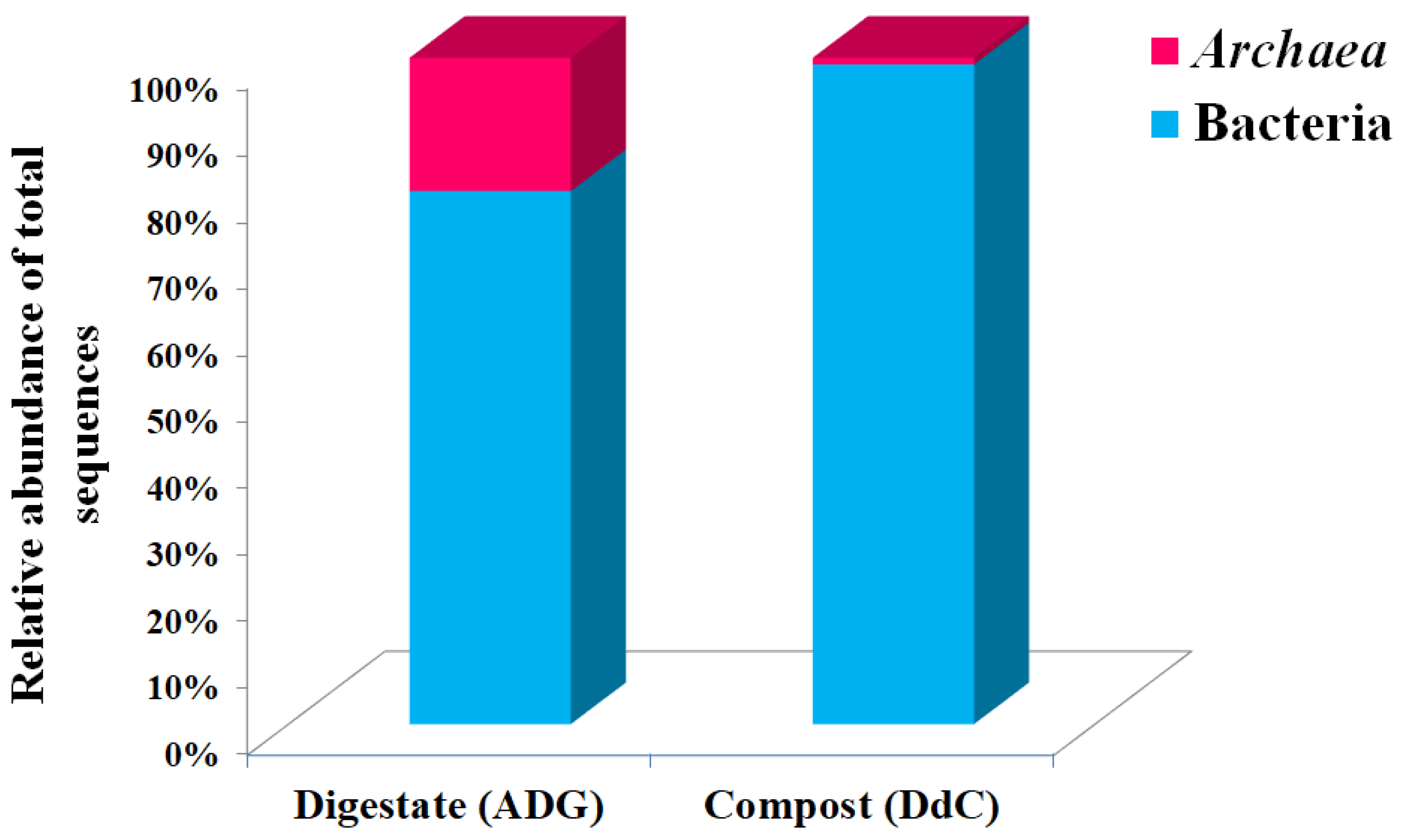
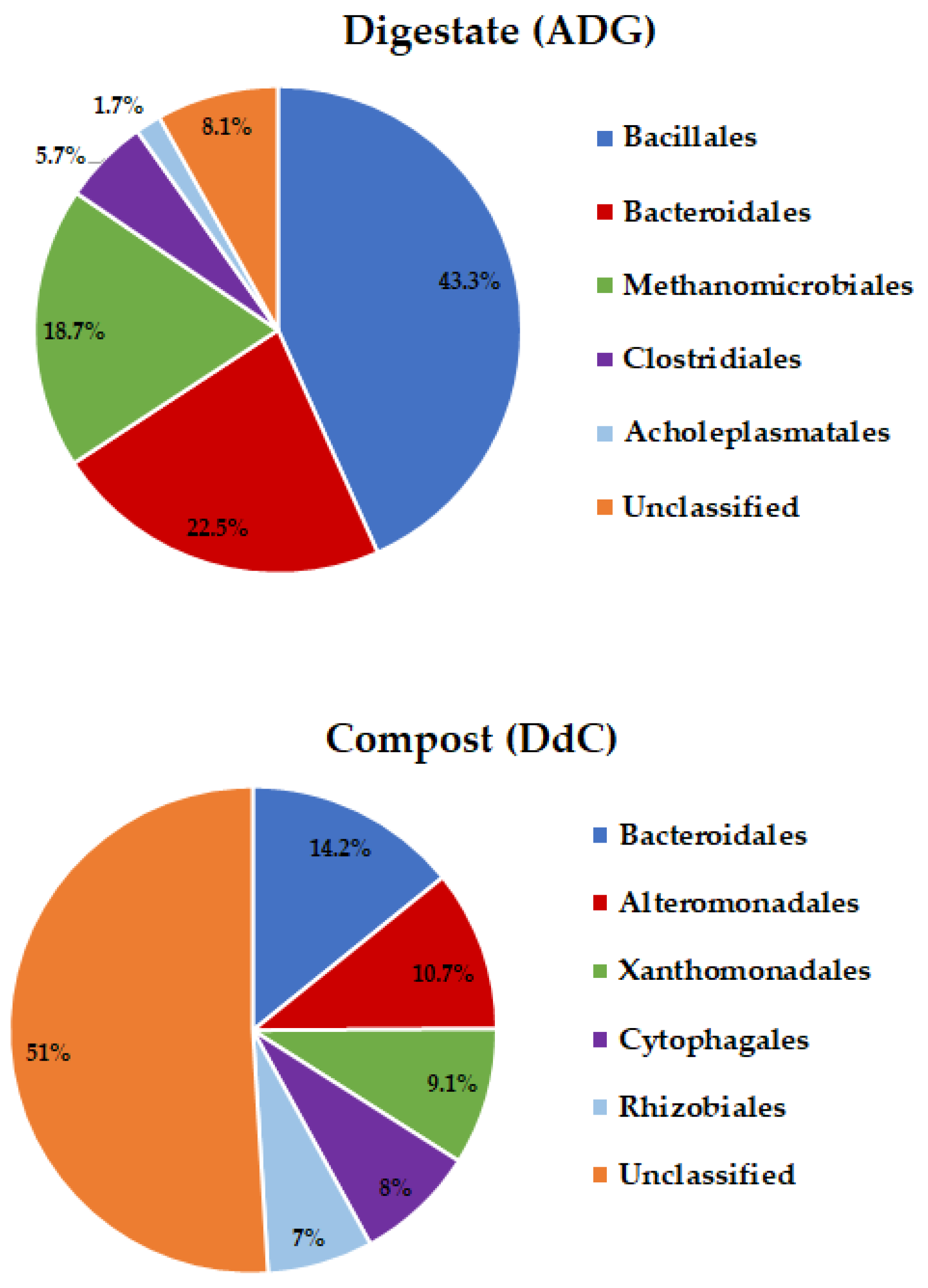
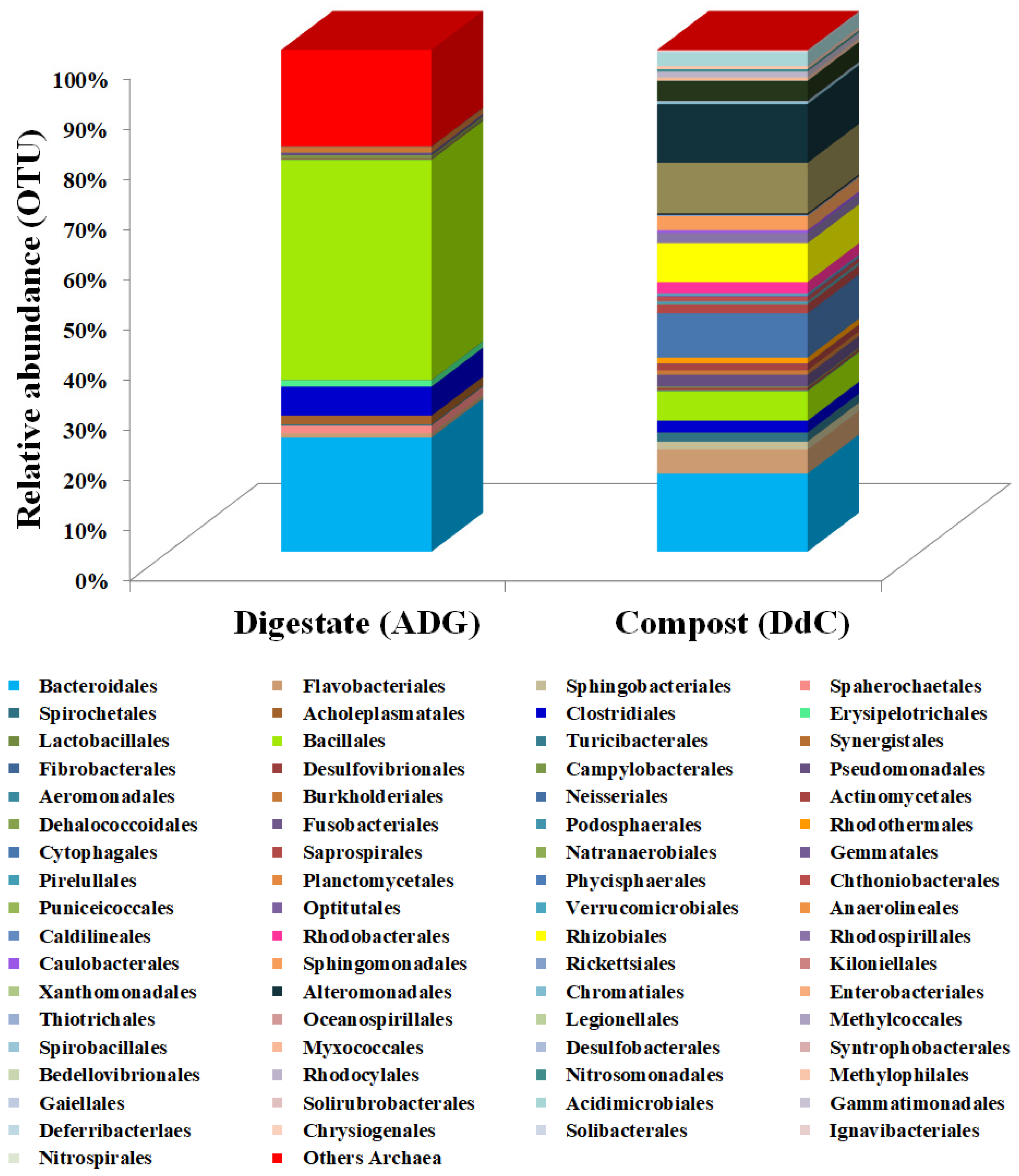
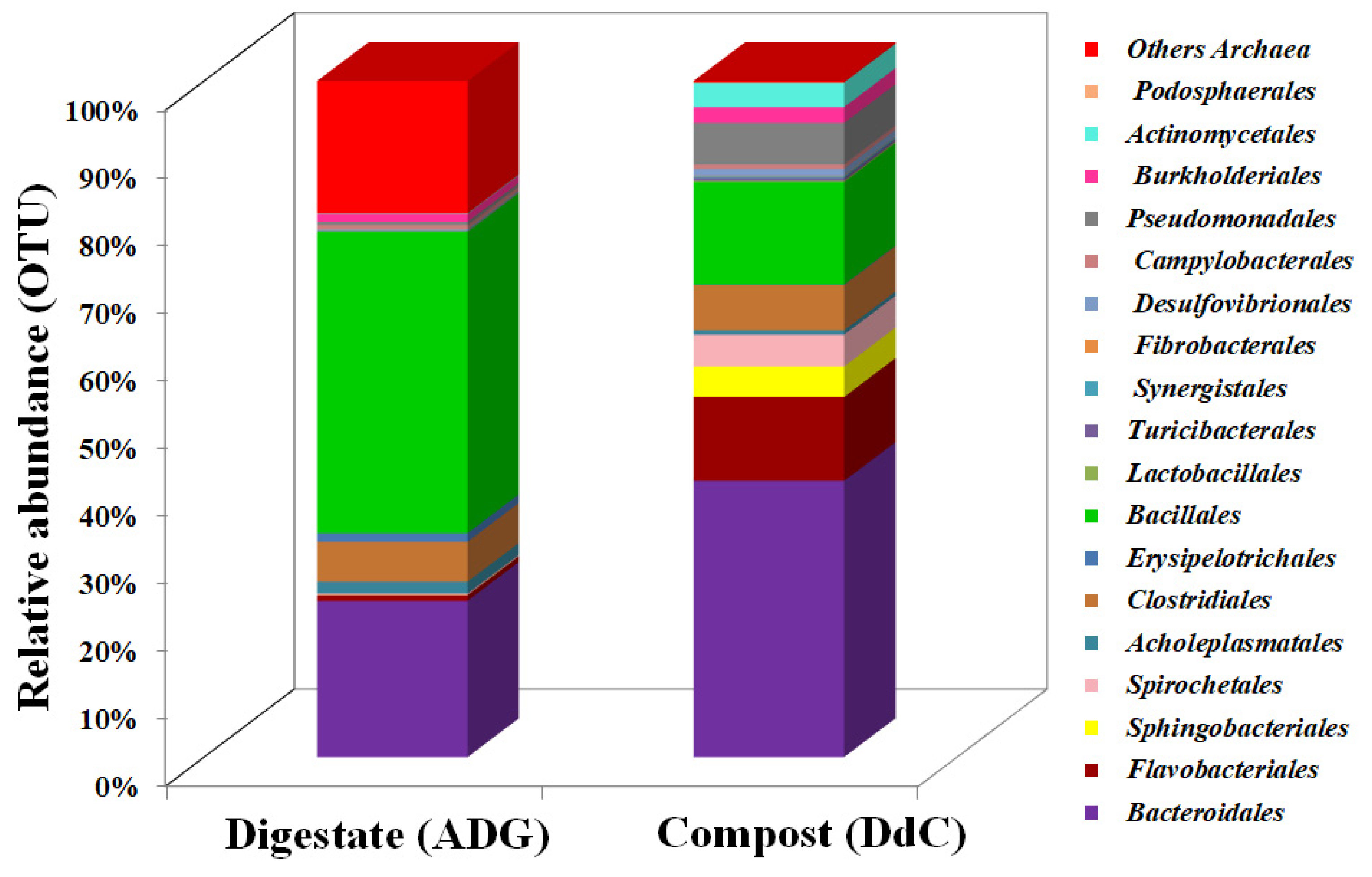
3.1. Taxonomic Classification of Digestate and Stabilized Digestate-Derived Compost Microbiome
3.2. Species Diversity and Richness
3.3. Shared Microbioma Composition
4. Discussion
4.1. Microbiota Diversity and Characterization of the Bacterial Community
4.2. Environmental Factors Influencing Micobioma, the ADG, and the DdC
4.3. Microbial Community Changes over Time
4.4. Role of the Microbial Composition in Controlling Phytopathogens
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salminen, E.; Rintala, J. Anaerobic digestion of organics solid poultry slaughterhouse waste—A review. Bioresour. Technol. 2002, 83, 13–26. [Google Scholar] [CrossRef]
- Zhang, C.; Su, H.; Baeyens, J.; Tan, T. Reviewing the anaerobic digestion of food waste for biogas production. Renew. Sustain. Energy Rev. 2014, 38, 383–392. [Google Scholar] [CrossRef]
- Obileke, K.; Nuwokolo, N.; Makaka, G.; Mukumba, P.; Onyeaka, H. Anaerobic digestion: Technology for biogas production as a source of renewable energy—A review. Energy Environ. 2021, 32, 191–225. [Google Scholar] [CrossRef]
- Pavlostathis, S.G.; Giraldo-Gomez, E. Kinetics of anaerobic treatment: A critical review. Crit. Rev. Environ. Control 1991, 21, 411–490. [Google Scholar] [CrossRef]
- Maeda, K.; Hanajima, D.; Toyoda, S.; Yoshida, N.; Morioka, R.; Osada, T. Microbiology of nitrogen cycle in animal compost. Microb. Biotechnol. 2011, 4, 700–709. [Google Scholar] [CrossRef]
- Bernal, M.; Alburquerque, J.; Moral, R. Composting of animal manures and chemical criteria for compost maturity assessment. A review. Bioresour. Technol. 2009, 100, 5444–5545. [Google Scholar] [CrossRef]
- Rudolf, M.; Kroneck, P. The nitrogen cycle: Its biology. Met. Ions. Biol. Syst. 2005, 43, 75–104. [Google Scholar]
- Batstone, D.J.; Keller, J.; Angelidaki, I.; Kalyuzhnyi, S.V.; Pavlostathis, S.G.; Rozzi, A.; Sanders, W.T.M.; Siegrist, H.; Vavilin, V.A. Anaerobic Digestion Model No. 1. IWA Task Group on Mathematical Modelling of Anaerobic Digestion Processes; IWA Publishing: London, UK, 2002; 80p. [Google Scholar]
- Gilbert, J.; Jürgensen-Ricci, M.; Ramola, A. Benefits of Compost and Anaerobic Digestate When Applied to Soil; The International Solid Waste Association (ISWA): Rotterdam, The Netherlands, 2020. [Google Scholar]
- Ezemagu, I.G.; Ejimofor, M.I.; Menkiti, M.C. Biofertilizer production via composting of digestate obtained from anaerobic digestion of postbiocoagulation sludge blended with saw dust: Physiochemical characterization and kinetic study. Environ. Chall. 2021, 5, 100288. [Google Scholar] [CrossRef]
- European Biogas Association (EBA). Contribution of Anaerobic Digestion to Circular Economy. Available online: www.european.biogas.eu (accessed on 14 January 2021).
- Directive 2008/98/EC of the European Parliament and of the Council of 19 November 2008 on Waste and Repealing Certain Directives (Text with EEA Relevance). An Official Website of the European Union. 2008. Available online: https://data.europa.eu/eli/dir/2008/98/oj (accessed on 10 June 2020).
- European Environment Agency. Bio-Waste in Europe—Turning Challenges into Opportunities; EEA Report, No. 4; Publications Office of the European Union: Luxembourg, 2020; Available online: https://www.eea.europa.eu/publications/bio-waste-in-europe/ (accessed on 1 September 2021).
- Cioabla, A.E.; Ionel, I.; Dumitrel, G.-A.; Popescu, F. Comparative study on factors affecting anaerobic digestion of agricultural vegetal residues. Biotechnol. Biofuels 2012, 5, 39–48. [Google Scholar] [CrossRef]
- Gholamreza, M.; Samaneh, G.; Anoshiravan, M.B. The biodegradation and COD removal of 2-chlorophenolina granular anoxic baffled reactor. J. Biotechnol. 2014, 184, 111–117. [Google Scholar] [CrossRef]
- Ward, A.J.; Hobbs, P.J.; Holliman, P.J.; Jones, D.L. Optimization of the anaerobic digestion of agricultural resources. Bioresour. Technol. 2008, 99, 7928–7940. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Dong, F.; Chen, M.; Zhu, J.; Tan, J.; Fu, X.; Wang, Y.; Chen, S. Advances in recycling and utilization of agricultural wastes in China: Based on environmental risk, crucial pathways, influencing factors, policy mechanism. Procedia Environ. Sci. 2016, 31, 12–17. [Google Scholar] [CrossRef]
- Maina, S.; Kachrimanidou, V.; Koutinas, A. A roadmap towards a circular and sustainable bioeconomy through waste valorization. Curr. Oppin. Green Sustain. Chem. 2017, 8, 18–23. [Google Scholar] [CrossRef]
- De Corato, U. Agricultural waste recycling in horticultural intensive farming systems by on-farm composing and compost-based tea application improve soil quality and plant health: A review under the perspective of a circular economy. Sci. Total Environ. 2020, 738, 139840. [Google Scholar] [CrossRef] [PubMed]
- Zarbá, C.; Chinnici, G.; La Via, G.; Bracco, S.; Pecorino, B.; D’Amico, M. Regulatory Elements on the Circular Economy: Driving into the Agri-Food System. Sustainability 2021, 13, 8350. [Google Scholar] [CrossRef]
- Smidt, E.; Tintner, J.; Böhm, K.; Binner, E. Transforming of Biogenic Waste Materials through Anaerobic Digestion and Subsequent Composting of the Residues—A Case Study. Dyn. Soil Dyn. Plant 2011, 5, 63–69. [Google Scholar]
- Weiland, P. Biomass Digestion in Agriculture: A Successful Pathway for the Energy Production and Waste Treatment in Germany. Review. Eng. Life Sci. 2003, 6, 302–309. [Google Scholar] [CrossRef]
- Sedlmeier, R.; Rombach, M.; Bitsch, V. Making Food Rescue Your Business: Cases Studies in Germany. Sustainability 2019, 11, 5101. [Google Scholar] [CrossRef]
- Bywater, A. A Review of Anaerobic Digestion Plants on UK Farms. Barriers, Benefits and Case Studies; Royal Agricultural Society of England: Stoneleigh Park, UK, 2011; 104p. [Google Scholar]
- Demichelis, F.; Piovano, F.; Fiore, S. Biowaste management in Italy: Challenges and Perspectives. Sustainability 2019, 11, 4213. [Google Scholar] [CrossRef]
- Diacono, M.; Montemurro, F. Long-term effects of organic amendments on soil fertility. A review. Agron. Sustain. Dev. 2010, 30, 401–422. [Google Scholar] [CrossRef]
- Larney, F.J.; Angers, D.A. The role of organic amendments in soil reclamation: A review. Can. J. Soil Sci. 2012, 92, 19–38. [Google Scholar] [CrossRef]
- Ramos, T.M.; Jay-Rusell, M.T.; Milner, P.D.; Shade, J.; Misiewicz, T.; Sorge, U.S.; Hutchinson, M.; Lilley, J.; Pires, A.F.A. Assessment of Biological Soil Amendments of Animal Origin use, Research Needs, and Extension Opportunities in Organic Production. Front. Sustain. Food Syst. 2019, 3, 73. [Google Scholar] [CrossRef]
- Lim, Y.L.; Bong, C.P.C.; Lee, C.T.; Kleměs, J.J.; Sarmidi, M.J.; Lim, J.S. Review on the Current Composting Practices and the Potential of Improvement Using Two-Stage Composting. Chem. Eng. Trans. 2017, 61, 1051–1056. [Google Scholar] [CrossRef]
- Bian, B.; Hu, X.; Zhang, S.; Lv, C.; Yang, Z.; Yang, W.; Zang, L. Pilot-scale composting of typical multiple agricultural wastes: Parameter optimization and mechanisms. Bioresour. Technol. 2019, 287, 121482. [Google Scholar] [CrossRef]
- Sayara, T.; Salimia-Basheer, R.; Hawamnde, F.; Sánchez, A. Recycling of Organic Wastes through Composting: Process Performance and Compost Application in Agriculture. Agronomy 2020, 10, 1838. [Google Scholar] [CrossRef]
- Palese, A.M.; Persiani, A.; D’Adamo, C.; Pergola, M.; Pastore, V.; Sileo, R.; Ippolito, G.; Lombardi, M.A.; Celano, G. Composting as Manure Disposal Strategy in Small/Medium-Size Livestock Farms: Some Demonstrations with Operative Indications. Sustainability 2020, 12, 3315. [Google Scholar] [CrossRef]
- Ahmad, R.; Jilani, G.; Arshad, M.; Zahir, Z.A.; Khalid, Z. Bio-conversion of organic wastes for their recycling in agriculture: An overview of perspectives and prospects. Ann. Microbiol. 2007, 57, 471–479. [Google Scholar] [CrossRef]
- Martinéz-Balnco, J.; Lazcano, C.; Christensen, T.H.; Muňoz, P.; Rieradevall, J.; Møller, J.; Antón, A.; Boldrin, A. Compost benefits for agriculture evaluated by life cycle assessment. A review. Agron. Sustain. Dev. 2013, 33, 721–732. [Google Scholar] [CrossRef]
- Tully, K.L.; McAskill, C. Promoting soil health in organically managed systems: A review. Org. Agric. 2020, 10, 339–358. [Google Scholar] [CrossRef]
- Cozzolino, V.; Di Meo, V.; Monda, H.; Spaccini, R.; Piccolo, A. The molecular characteristics of compost affect plant growth, arbuscular mycorrhizal fungi, and soil microbial composition. Biol. Fertil. Soils 2016, 52, 15–29. [Google Scholar] [CrossRef]
- Sánchez, Ó.J.; Ospina, D.A.; Montoya, S. Compost supplementation with nutrients and microorganisms in composting process. Waste Manag. 2017, 69, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, Y.; Xia, Y.; Wu, T.; Zhu, J.; Wang, Z.; Yang, J. The succession pattern of bacterial diversity in compost using pig manure mixed with wood chips analyzed by 16S rRNA gene analysis. bioRxiv 2019, 674069. [Google Scholar] [CrossRef]
- Ryckeboer, J.; Mergaert, J.; Vaes, K.; Klammer, S.; De Clercq, D.; Coosemans, J.; Insam, H.; Swings, J. A survey of bacteria and fungi occurring during composting and self-heating processes. Ann. Microbiol. 2003, 53, 349–410. [Google Scholar]
- Hultman, J. Microbial Diversity in the Municipal Composting Process and Development of Detection Methods. Ph.D. Thesis, Department of Ecological and Environmental Sciences, Faculty of Biosciences, Institute of Biotechnology, Viikki Graduate School in Biosciences, University of Helsinki, Helsinki, Finland, 2009; 47p. [Google Scholar]
- Liu, A.-C.; Chou, C.-Y.; Chen, L.-L.; Kuo, C.-H. Bacterial community dynamics in a swine wastewater anaerobic reactor revealed by 16S rDNA sequence analysis. J. Biotechnol. 2015, 194, 124–131. [Google Scholar] [CrossRef]
- Bhatia, A.; Rajpal, A.; Madan, S.; Kazmi, A.A. Techniques to analyze microbial diversity during composting—A mini review. Indian J. Biotechnol. 2015, 14, 19–25. [Google Scholar]
- Palaniveloo, K.; Amran, M.A.; Norhashim, N.A.; Mohamad-Fauzi, N.; Peng-Hui, F.; Hui-Wen, L.; Kai-Lin, Y.; Jiale, L.; Chian-Yee, M.G.; Jing-Yi, L.; et al. Food Waste Composting and Microbial Community Profiling. Review. Processes 2020, 8, 723. [Google Scholar] [CrossRef]
- Martins, L.F.; Antunes, L.P.; Pascon, R.C.; de Oliveira, J.C.F.; Digiampietri, L.A.; Barbosa, D.; Peixoto, B.M.; Vallim, M.A.; Viana-Niero, C.; Ostroski, E.H. Metagenomic analysis of a tropical composting operation at the São Paulo Zoo Park reveals diversity of biomass degradation functions and organisms. PLoS ONE 2013, 8, e61928. [Google Scholar] [CrossRef]
- Mahir, B.; Çağrı, A.; Orhan, I.; Sevcan, A.; Bahar, I. Application of next-generation sequencing methods for microbial monitoring of anaerobic digestion of lignocellulosic biomass. Appl. Microbiol. Biotechnol. 2017, 101, 6849–6864. [Google Scholar] [CrossRef]
- Meneghine, A.K.; Nielsen, S.; Varani, A.M.; Thomas, T.; Alves, L.M.C. Metagenomic analysis of soil and freshwater from zoo agricultural area with organic fertilization. PLoS ONE 2017, 12, e0190178. [Google Scholar] [CrossRef]
- De Corato, U. Use of omic approaches for characterizing microbiota from suppressive compost to control soil-borne plant pathogens. Arch. Phytopathol. Plant Prot. 2019, 52, 757–775. [Google Scholar] [CrossRef]
- Enebe, M.C.; Babalola, O.O. Effects of inorganic and organic treatments on the microbial community of maize rhizosphere by a shotgun metagenomics approach. Ann. Microbiol. 2020, 70, 49. [Google Scholar] [CrossRef]
- Young, J.P.W.; Downer, H.L.; Eardly, B.D. Phylogeny of the phototropic Rhizobium strain BTAi1by polymerase chain reaction-based sequencing of a 16S rRNA gene segment. J. Bacteriol. 1991, 173, 2271–2277. [Google Scholar] [CrossRef]
- Dong, B.; Yi, J.; Dai, L.; Dai, X. Evaluation of Several DNA Extraction Methods for Obtaining Total Community DNA from Anaerobic Digestion Sludge. Procedia Environ. Sci. 2013, 18, 856–863. [Google Scholar] [CrossRef][Green Version]
- Vitti, A.; Elshafie, H.S.; Logozzo, G.; Marzario, S.; Scopa, A.; Camele, I.; Nuzzaci, M. Physico-Chemical Characterization and Biological Activities of a Digesate and a more Stabilized Digestate-Derived Compost from Agro-Waste. Plants 2021, 10, 386. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a Prokaryotic Universal Primer for Simultaneous Analysis of Bacteria and Archaea Using Next-Generation Sequencing. PLoS ONE 2014, 9, e105592. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Caporaso, G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 5 May 2019).
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological Statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Thummes, K.; Schäfer, J.; Kämpfer, P.; Jäckel, U. Thermophilic methanogenic Archaea in compost material: Occurrence, persistence and possible mechanisms for their distribution to other environments. Syst. Appl. Microbiol. 2007, 30, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Oshi, R.; Tada, C.; Asano, R.; Yamamoto, N.; Suyama, Y.; Nakai, Y. Growth of ammonia-oxidizing archaea and bacteria in cattle manure compost under various temperatures and ammonia concentrations. Microb. Ecol. 2012, 63, 787–793. [Google Scholar] [CrossRef]
- Antunes, L.P.; Martins, L.F.; Pereira, L.V.; Thomas, A.M.; Barbosa, D.; Lemos, L.N.; Machado Silva, G.M.; Moura, L.M.S.; Epamino Condomiti, G.W.; Digiampietri, L.A.; et al. Microbial community structure and dymamics in thermophilic composting viewed through metagenomics and metatranscriptomics. Sci. Rep. 2016, 6, 38915. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Massé, D.; McAllister, T.A.; Kong, Y.; Seviour, R.; Beaulieu, C. Identity and diversity of archaeal communities during anaerobis co-digestion of chicken feathers and other animal wastes. Bioresour. Technol. 2012, 110, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Kundu, K.; Bergmann, I.; Kloche, M.; Sharma, S.; Sreekrishnan, T.R. Impact of abrupt temperature increase on the performance of an anaerobic hybrid bioreactor and its intrinsic microbial community. Bioresour. Technol. 2014, 168, 72–79. [Google Scholar] [CrossRef] [PubMed]
- De Vrieze, J.; Saunders, A.M.; He, Y.; Fang, J.; Nielsen, P.H.; Vestraete, W.; Boon, N. Ammonia and temperature determine potential clustering in the anaerobic digestion microbiome. Water Res. 2015, 75, 312–323. [Google Scholar] [CrossRef]
- Chang, R.; Li, Y.; Li, N.; Wu, X.; Chen, Q. Effect of microbial transformation induced by metallic compound additives and temperature variations during composting on suppression of soil-borne pathogens. J. Environ. Manag. 2021, 279, 111816. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, H.; Wang, Z.; Chen, G.; Wang, L. Dynamic changes of the dominant functioning microbial community in the compost of a 90-m3 aerobic solid state fermentor revealed by integrated meta-omics. Bioresour. Technol. 2016, 203, 1–10. [Google Scholar] [CrossRef]
- Cardinali-Rezende, J.; Colturato, L.; Colturato, T.D.B.; Chartone-Souza, E.; Nascimento, A.M.A.; Sanz, J.L. Prokaryotic divesity and dynamics in a full-scale municipal solid waste anaerobic reactor from start-up to steady-state conditions. Bioresour. Technol. 2012, 119, 373–383. [Google Scholar] [CrossRef]
- Werner, J.J.; Knights, D.; Garcia, M.L.; Scalfone, N.B.; Smith, S.; Yarasheski, K.; Cummings, T.A.; Beers, A.R.; Knight, R.; Angenent, L.T. Bacterial community structures are unique and resilient in full-scale bioenergy systems. Proc. Natl. Acad. Sci. USA 2011, 108, 4158–4163. [Google Scholar] [CrossRef]
- Nelson, M.C.; Morrison, M.; Yu, Z.T. A meta-analysis of the microbial diversity observed in anaerobic digesters. Bioresour. Technol. 2011, 102, 3730–3739. [Google Scholar] [CrossRef]
- Zhang, L.; Li, L.; Pan, X.; Shi, Z.; Feng, X.; Gong, B.; Li, J.; Wang, L. Enhanced Growth and Activities of the Dominant Functional Microbiota of Chicken Manure Composts in the Presence of Maize Straw. Front. Microbiol. 2018, 9, 1131. [Google Scholar] [CrossRef]
- Jurado, M.; Lopéz, M.; Suárez-Estrella, F.; Vargas-Garcia, M.C.; López-Gonzáles, J.A.; Moreno, J. Exploiting compost biodiversity: Study of the persistent biotechnologically relevant microorganisms from lignocellulose-based composting. Bioresour. Technol. 2014, 162, 283–293. [Google Scholar] [CrossRef]
- Partanen, P.; Hultman, J.; Paulin, L.; Auvinen, P.; Romantschuk, M. Bacterial diversity at different stages of composting process. BMC Microbiol. 2010, 10, 94. [Google Scholar] [CrossRef]
- Holman, D.B.; Hao, X.; Topp, E.; Yang, H.E.; Alexander, T.W. Effect of Co-Composting Cattle Manure with Construction and Demolition Waste on the Archaeal, Bacterial, and Fungal Microbiota, and on Antimicrobial Resistance Determinants. PLoS ONE 2016, 11, e0157539. [Google Scholar] [CrossRef]
- Franke-Whittle, I.H.; Confaloneri, A.; Insam, H.; Schlegelmilch, M.; Körner, I. Changes in the microbial communities during co-composting of digestates. Waste Manag. 2014, 34, 632–641. [Google Scholar] [CrossRef]
- Ryckeboer, J.; Mergaert, J.; Coosemans, J.; Deprins, K.; Swings, J. Microbiological aspects of biowaste during composting in a monitored compost bin. J. Appl. Microbiol. 2003, 94, 127–137. [Google Scholar] [CrossRef]
- Varma, V.S.; Dhamodharan, K.; Kalamdhad, A.S. Characterization of bacterial community structure during in-vessel composting of agricultural waste by 16S rRNA sequencing. Biotech 2018, 8, 301. [Google Scholar] [CrossRef]
- Chandna, P.; Nain, L.; Singh, S.; Kuhad, R.C. Assessment of Bacterial Diversity during Composting of Agricultural Byproducts. BMC Microbiol. 2013, 13, 99. Available online: https//www.bmcmicrobiol.biomedcentral.com/1471.2180/13/99 (accessed on 1 September 2021). [CrossRef]
- Meng, Q.; Wang, W.; Men, M.; Bello, A.; Xu, X.; Xu, B.; Deng, L.; Jiang, X.; Sheng, S.; Wu, X.; et al. Microbial Community Succession and Response to Environmental Variables during Cow Manure and Corn Straw Composting. Front. Microbiol. 2019, 10, 529. [Google Scholar] [CrossRef]
- Ishii, K.; Takii, S. Comparison of microbial communities in four different composting processes as evaluated by denaturing gradient gel electrophoresis analysis. J. Appl. Micobiol. 2003, 95, 109–119. [Google Scholar] [CrossRef]
- Fracchia, L.; Dohrmann, A.B.; Martinotti, M.G.; Tebbe, C.C. Bacterial diversity in a finished compost and vermicompost: Differences revealed by cultivation independent analyses of PCR-amplified 16S rRNA genes. Appl. Microbiol. Biotechnol. 2006, 71, 942–952. [Google Scholar] [CrossRef]
- Hadar, Y.; Mandelbaum, R. Suppressive compost for biocontrol of soil-borne plant pathogens. Phytoparasitica 1992, 20, S113–S116. [Google Scholar] [CrossRef]
- Tubeileh, A.M.; Stephenson, G.T. Soil amendment by composted plant wastes reduces the Verticillium dahliae abundance and changes soil chemical properties in a bell pepper cropping system. Curr. Plant Biol. 2020, 22, 100148. [Google Scholar] [CrossRef]
- Rady, M.M.; Semida, W.M.; Hemida, K.A.; Abdelhamid, M.T. The effect of compost on growth and yield of Phaseolus vulgaris plants grown under saline soil. Int. J. Recycl. Org. Waste Agric. 2016, 5, 311–321. [Google Scholar] [CrossRef]
- Ibukunoluwa Moyin-Jesu, E. Use of different organic fertilizers on soil fertility improvement, growth and head yield parameters of cabbage (Brassica oleraceae L.). Int. J. Recycl. Org. Waste Agric. 2015, 4, 291–298. [Google Scholar] [CrossRef]
- Postma, J.; Montanari, M.; van den Boorget, P.H.J.F. Microbial enrichment to enhance the disease suppressive activity of compost. Eur. J. Soil Biol. 2003, 39, 157–163. [Google Scholar] [CrossRef]
- Zaccardelli, M.; de Nicola, F.; Villecco, D.; Scotti, R. The development and suppressive activity of soil microbial communities under compost amendment. J. Soil Sci. Plant Nutr. 2013, 13, 740–742. [Google Scholar] [CrossRef]
- Bellini, A.; Ferrocino, I.; Cucu, M.A.; Pugliese, M.; Garibaldi, A.; Gullino, M.L. A Compost Treatment Acts as a Suppressive Agent in Phytophthora capsici—Cucurbita pepo Pathosystem by Modifying the Rhizosphere Microbiota. Front. Plant Sci. 2020, 11, 885. [Google Scholar] [CrossRef]
- Lutz, S.; Thuerig, B.; Oberhaensli, T.; Mayerhofer, J.; Fuchs, J.G.; Widmer, F.; Freimoser, F.M.; Ahrens, C.H. Harnessing the Microbiomes of Suppressive Composts for Plant Protection: From Metagenomes to Beneficial Microorganisms and Reliable Diagnostics. Front. Microbiol. 2020, 11, 1810. [Google Scholar] [CrossRef]
- Piciarello, E.; Pucci, L.; Carotenuto, M.; Libralato, G.; Lofrano, G.; Baldantoni, D. Compost and Sewage Sludge for the Improvement of Soil Chemical and Biological Quality of Mediterranean Agroecosystems. Sustainability 2021, 13, 26. [Google Scholar] [CrossRef]
- Baldantoni, D.; Saviello, G.; Alfani, A. Nutrients and non-essential elements in edible crops following long-term mineral and compost fertilization of a Mediterranean agricultural soil. Environ. Sci. Pollut. Res. 2018, 26, 35353–35364. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total No./Taxonomic Classification | Digestate (ADG) | Compost (DdC) |
|---|---|---|
| Sequences obtained | 55,616 | 11,229 |
| Unclassified sequences (%) | 1.34 | 9.09 |
| Classified sequences * (%) | 98.66 | 90.91 |
| Orders identified | 27 | 74 |
| Orders included (shared by the two types of matrices and including Archaea) | 23 | 23 |
| Orders excluded | 4 | 51 |
| Families identified | 54 | 125 |
| Families included (shared by the two types of matrices) | 37 | 37 |
| Families excluded | 17 | 88 |
| Order | Digestate (ADG) | Compost (DdC) | p (t) |
|---|---|---|---|
| Acholeplasmatales | 1.7 ± 0.02 | 0.02 ± 0.0058 | 8.27 × 10−5 |
| Acidimicrobiales | 0 ± 0 | 2.4 ± 0.1155 | 0.0023 |
| Actinomycetales | 0.08 ± 0.0058 | 1.25 ± 0.01 | 2.43 × 10−5 |
| Aeromonadales | 0.02 ± 0.0058 | 0.01 ± 0 | 0.225 |
| Alteromonadales | 0 ± 0 | 10.66 ± 0.0346 | 1.06 × 10−5 |
| Anaerolineales | 0 ± 0 | 0.03 ± 0.0058 | 0.035 |
| Bacillales | 43.27 ± 0.0519 | 5.27 ± 0.0230 | 5.778 × 10−5 |
| Bacteroidales | 22.47 ± 0.4798 | 14.21 ± 0.060 | 0.004 |
| Bedellovibrionales | 0 ± 0 | 0.08 ± 0.0058 | 0.0041 |
| Burkholderiales | 1.11 ± 0.0058 | 0.82 ± 0.0058 | 0.00013 |
| Caldilineales | 0 ± 0 | 0.13 ± 0.0058 | 0.0019 |
| Campylobacterales | 0.6 ± 0.0288 | 0.23 ± 0.01 | 0.00315 |
| Caulobacterales | 0 ± 0 | 0.5 ± 0.0152 | 0.0009 |
| Chromatiales | 0 ± 0 | 0.32 ± 0.0058 | 0.00032 |
| Chrysiogenales | 0 ± 0 | 0.03 ± 0.0058 | 0.035 |
| Chthoniobacterales | 0 ± 0 | 0.7 ± 0.0058 | 0.0067 |
| Clostridiales | 5.72 ± 0.0723 | 2.13 ± 0.0115 | 0.00028 |
| Cytophagales | 0 ± 0 | 8.04 ± 0.0115 | 2.06 × 10−6 |
| Deferribacterales | 0.01± 0 | 0 ± 0 | 0.0078 |
| Dehalococcoidales | 0.01 ± 0 | 0 ± 0 | NA * |
| Desulfobacterales | 0 ± 0 | 0.05 ± 0.0058 | 0.013 |
| Desulfovibrionales | 0.11 ± 0.0058 | 0.34 ± 0.0058 | 0.00063 |
| Enterobacteriales | 0 ± 0 | 0.03 ± 0.01 | 0.09 |
| Erysipelotrichales | 1.25 ± 0.0173 | 0.03 ± 0.0058 | 8.96 × 10−5 |
| Fibrobacterales | 0.04 ± 0.0058 | 0.03 ± 0.01 | 0.22 |
| Flavobacteriales | 0.8 ± 0.0208 | 4.32 ± 0.01 | 1.08 × 10−5 |
| Fusobacteriales | 0.01 ± 0 | 0 ± 0 | NA * |
| Gaiellales | 0 ± 0 | 0.01 ± 0 | NA * |
| Gammatimonadales | 0 ± 0 | 0.04 ± 0.0058 | 0.02 |
| Gemmatales | 0 ± 0 | 0.01 ± 0 | NA * |
| Ignavibacteriales | 0 ± 0 | 0.15 ± 0.0058 | 0.0015 |
| Kiloniellales | 0 ± 0 | 0.33 ± 0.0058 | 0.00148 |
| Lactobacillales | 0.08 ± 0.0058 | 0.07 ± 0.0058 | 0.225 |
| Legionellales | 0 ± 0 | 0.02 ± 0 | NA * |
| Methylcoccales | 0 ± 0 | 0.02 ± 0.0058 | 0.074 |
| Methylophilales | 0 ± 0 | 0.51 ± 0.01 | 0.00038 |
| Myxococcales | 0 ± 0 | 0.44 ± 0.02 | 0.0021 |
| Natranaerobiales | 0 ± 0 | 0.04 ± 0.0058 | 0.02 |
| Neiseriales | 0.01 | 0 | NA * |
| Nitrosomonadales | 0 ± 0 | 0.41 ± 0.0152 | 0.0014 |
| Nitrospirales | 0 ± 0 | 0.01 ± 0 | NA * |
| Oceanospirillales | 0 ± 0 | 3.64 ± 0.0230 | 4.03 × 10−5 |
| Optitutales | 0 ± 0 | 0.24 ± 0.0058 | 0.00058 |
| Phycisphaerales | 0 ± 0 | 0.12 ± 0.0058 | 0.0023 |
| Pirelullales | 0 ± 0 | 0.37 ± 0.0058 | 0.00024 |
| Planctomycetales | 0 ± 0 | 0.08 ± 0.0058 | 0.0052 |
| Podosphaerales | 0.02 ± 0.01 | 0.02 ± 0 | 1 |
| Pseudomonadales | 0.5 ± 0.0058 | 2.13 ± 0.01 | 1.25 × 10−5 |
| Puniceicoccales | 0 ± 0 | 0.01 ± 0 | NA * |
| Rhizobiales | 0 ± 0 | 7.05 ± 0.0058 | 6.71 × 10−7 |
| Rhodobacterales | 0 ± 0 | 2.05 ± 0.01 | 2.38 × 10−5 |
| Rhodocylales | 0 ± 0 | 0.95 ± 0.0153 | 0.000258 |
| Rhodospirillales | 0 ± 0 | 1.88 ± 0.0251 | 0.000179 |
| Rhodothermales | 0 ± 0 | 1.04 ± 0.0058 | 3.08 × 10−5 |
| Rickettsiales | 0 ± 0 | 0.22 ± 0.0058 | 0.000688 |
| Saprospirales | 0 ± 0 | 1.64 ± 0.1001 | 0.0037 |
| Solibacterales | 0 ± 0 | 0.09 ± 0.0058 | 0.0041 |
| Solirubrobacterales | 0 ± 0 | 0.07 ± 0.01 | 0.019 |
| Spaherochaetales | 1.55 ± 0.0058 | 0 ± 0 | 1.39 × 10−5 |
| Sphingobacteriales | 0.02 ± 0.0058 | 1.47 ± 0.0115 | 0.00011 |
| Sphingomonadales | 0 ± 0 | 2.53 ± 0.0404 | 0.00025 |
| Spirobacillales | 0 ± 0 | 0.04 ± 0.0058 | 0.02 |
| Spirochetales | 0.22 ± 0.0058 | 1.64 ± 0.0058 | 1.97 × 10−6 |
| Synergistales | 0.09 ± 0.0058 | 0.09 ± 0.01 | 1 |
| Syntrophobacterales | 0 ± 0 | 0.11 ± 0.0058 | 0.0027 |
| Thiotrichales | 0 ± 0 | 0.24 ± 0.0115 | 0.0023 |
| Turicibacterales | 0.06 ± 0.0058 | 0.15 ± 0.0153 | 0.012 |
| Verrucomicrobiales | 0 ± 0 | 0.27 ± 0.01 | 0.00137 |
| Xanthomonadales | 0 ± 0 | 9.16 ± 0.0058 | 3.97 × 10−7 |
| Others Archaea | 19 ± 0.0305 | 0.08 ± 0.0058 | 1.77 × 10−5 |
| Diversity Index | ADG | DdC |
|---|---|---|
| Shannon | 1.573 | 3.014 |
| Chao-1 | 24.75 | 68 |
| Order | Digestate (ADG) | Compost (DdC) | p (t) |
|---|---|---|---|
| Acholeplasmatales | 1.7 ± 0.0577 | 0.22 ± 0.0058 | 0.0013 |
| Actinomycetales | 0.08 ± 0.0058 | 1.25 ± 0.0058 | 8.094 × 10−8 |
| Bacillales | 43.27 ± 0.5832 | 5.27 ± 0.0115 | 0.00024 |
| Bacteroidales | 22.47 ± 0.4798 | 14.21 ± 0.0682 | 0.0041 |
| Burkholderiales | 1.11 ± 0.0058 | 0.82 ± 0.0058 | 0.00119 |
| Campylobacterales | 0.6 ± 0.0577 | 0.23 ± 0.0058 | 0.0214 |
| Clostridiales | 5.72 ± 0.0321 | 2.33 ± 0.0058 | 0.00012 |
| Desulfovibrionales | 0.11 ± 0.0058 | 0.34 ± 0.0058 | 0.00188 |
| Erysipelotrichales | 1.25 ± 0.0058 | 0.03 ± 0.0058 | 6.72 × 10−5 |
| Fibrobacterales | 0.04 ± 0.0058 | 0.03 ± 0.0058 | 0.225 |
| Flavobacteriales | 0.8 ± 0.0208 | 4.32 ± 0.01 | 1.07 × 10−5 |
| Lactobacillales | 0.08 ± 0.0058 | 0.07 ± 0.0058 | 0.422 |
| Podosphaerales | 0.02 ± 0.0058 | 0.02 ± 0 | 1 |
| Pseudomonadales | 0.5 ± 0.0577 | 2.13 ± 0.0058 | 0.00139 |
| Sphingobacteriales | 0.02 ± 0.0058 | 1.47 ± 0.0115 | 0.00011 |
| Spirochetales | 0.22 ± 0.0058 | 1.64 ± 0.0058 | 1.04 × 10−6 |
| Synergistales | 0.09 ± 0.005 | 0.09 ± 0 | 1 |
| Turicibacterales | 0.06 ± 0.0058 | 0.15 ± 0.0058 | 0.004 |
| Others Archaea | 19 ± 0.0577 | 0.07 ± 0.0058 | 0.00092 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mang, S.M.; Trotta, V.; Scopa, A.; Camele, I. Metagenomic Analysis of Bacterial Community Structure and Dynamics of a Digestate and a More Stabilized Digestate-Derived Compost from Agricultural Waste. Processes 2022, 10, 379. https://doi.org/10.3390/pr10020379
Mang SM, Trotta V, Scopa A, Camele I. Metagenomic Analysis of Bacterial Community Structure and Dynamics of a Digestate and a More Stabilized Digestate-Derived Compost from Agricultural Waste. Processes. 2022; 10(2):379. https://doi.org/10.3390/pr10020379
Chicago/Turabian StyleMang, Stefania Mirela, Vincenzo Trotta, Antonio Scopa, and Ippolito Camele. 2022. "Metagenomic Analysis of Bacterial Community Structure and Dynamics of a Digestate and a More Stabilized Digestate-Derived Compost from Agricultural Waste" Processes 10, no. 2: 379. https://doi.org/10.3390/pr10020379
APA StyleMang, S. M., Trotta, V., Scopa, A., & Camele, I. (2022). Metagenomic Analysis of Bacterial Community Structure and Dynamics of a Digestate and a More Stabilized Digestate-Derived Compost from Agricultural Waste. Processes, 10(2), 379. https://doi.org/10.3390/pr10020379