

Analogues of Oxamate, Pyruvate, and Lactate as Potential Inhibitors of Plasmodium knowlesi Lactate Dehydrogenase Identified Using Virtual Screening and Verified via Inhibition Assays

Abstract
1. Introduction
2. Materials and Methods
2.1. Ligand-Based, Structure-Based Drug Design and Toxicity Tests
2.2. Compounds for Experimental Tests
2.3. Pk-LDH Enzyme
2.4. Steady-State Enzymatic Assays Monitored with Pk-LDH Enzyme Reaction
2.5. Inhibition of Pk-LDH Activity by Oxalic Acid, Glycolamide, 3-Hydroxytetrahydrofuran, and 3,3-Difluoropyrrolidine Hydrochloride
3. Results and Discussion
3.1. Identification of Small Molecule Analogues of Pyruvate and Lactate by USRCAT and Toxicity Test
3.2. Molecular Docking for Structure-Based Drug Design
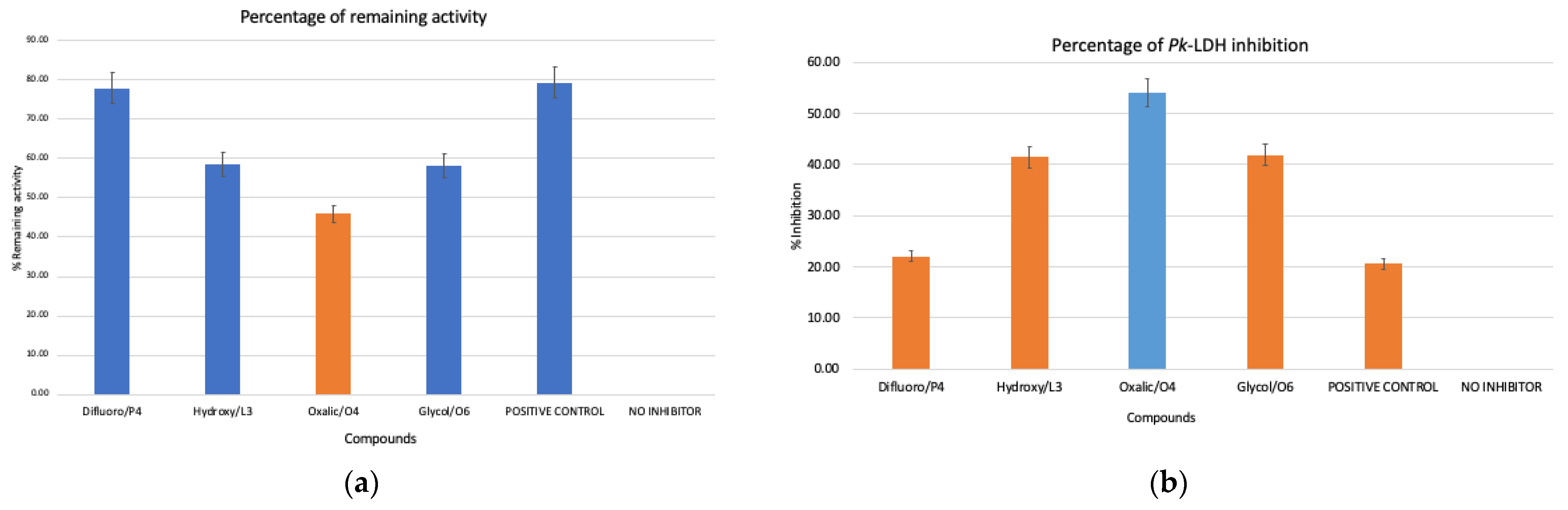
3.3. Inhibition of Pk-LDH Activity by Oxalic Acid, Glycolamide, 3-Hydroxytetrahydrofuran, and 3,3-Difluoropyrrolidine Hydrochloride
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- WHO. The E-2020 Initiative of 21 Malaria-Eliminating Countries: 2019 Progress Report; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Cox, F.E. History of the Discovery of the Malaria Parasites and Their Vectors. Parasit Vectors 2010, 3, 5. [Google Scholar] [CrossRef]
- Sabbatani, S.; Fiorino, S.; Manfredi, R. The Emerging of the Fifth Malaria Parasite (Plasmodium Knowlesi). A Public Health Concern? Braz. J. Infect. Dis. 2010, 14, 299–309. [Google Scholar] [CrossRef][Green Version]
- Amir, A.; Cheong, F.W.; de Silva, J.R.; Liew, J.W.K.; Lau, Y.L. Plasmodium Knowlesi Malaria: Current Research Perspectives. Infect. Drug Resist. 2018, 11, 1145–1155. [Google Scholar] [CrossRef]
- Cooper, D.J.; Rajahram, G.S.; William, T.; Jelip, J.; Mohammad, R.; Benedict, J.; Alaza, D.A.; Malacova, E.; Yeo, T.W.; Grigg, M.J.; et al. Plasmodium Knowlesi Malaria in Sabah, Malaysia, 2015–2017: Ongoing Increase in Incidence Despite Near-Elimination of the Human-Only Plasmodium Species. Clin. Infect. Dis. 2019, 70, 361–367. [Google Scholar] [CrossRef]
- Imwong, M.; Madmanee, W.; Suwannasin, K.; Kunasol, C.; Peto, T.J.; Tripura, R.; von Seidlein, L.; Nguon, C.; Davoeung, C.; Day, N.P.J.; et al. Asymptomatic Natural Human Infections with the Simian Malaria Parasites Plasmodium Cynomolgi and Plasmodium Knowlesi. J. Infect. Dis. 2019, 219, 695–702. [Google Scholar] [CrossRef]
- Singh, B.; Daneshvar, C.; Siner, A.; Ahmed, M.A.; Woon, L.C.; Pasini, E.M.; Kocken, C.H.; Singh, B.; Cox-Singh, J.; Krishna, S.; et al. Human Infections and Detection of Plasmodium Knowlesi. Clin. Microbiol. Rev. 2013, 26, 165–184. [Google Scholar] [CrossRef]
- Parija, S.; Janagond, A.; Jeremiah, S. Challenges in Diagnosis of Plasmodium Knowlesi Infections. Trop. Parasitol. 2014, 4, 25. [Google Scholar] [CrossRef]
- Antinori, S.; Galimberti, L.; Milazzo, L.; Corbellino, M. Plasmodium Knowlesi: The Emerging Zoonotic Malaria Parasite. Acta Trop. 2013, 125, 191–201. [Google Scholar] [CrossRef]
- Mahittikorn, A.; Masangkay, F.R.; Kotepui, K.U.; Milanez, G.D.J.; Kotepui, M. Quantification of the Misidentification of Plasmodium Knowlesi as Plasmodium Malariae by Microscopy: An Analysis of 1569 P. Knowlesi Cases. Malar. J. 2021, 20, 1–11. [Google Scholar] [CrossRef]
- Ashley, E.A.; Dhorda, M.; Fairhurst, R.M.; Amaratunga, C.; Lim, P.; Suon, S.; Sreng, S.; Anderson, J.M.; Mao, S.; Sam, B.; et al. Spread of Artemisinin Resistance in Plasmodium Falciparum Malaria. N. Engl. J. Med. 2014, 371, 411–423. [Google Scholar] [CrossRef]
- Andrews, K.T.; Fisher, G.; Skinner-Adams, T.S. Drug Repurposing and Human Parasitic Protozoan Diseases. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Kaushal, D.C.; Rathaur, S.; Kumar, N.; Kaushal, N.A. Cloning, Overexpression, Purification and Characterization of Plasmodium Knowlesi Lactate Dehydrogenase. Protein. Expr. Purif. 2012, 84, 195–203. [Google Scholar] [CrossRef] [PubMed]
- de Souza, N.B.; Carmo, A.M.L.; da Silva, A.D.; França, T.C.C.; Krettli, A.U. Antiplasmodial Activity of Chloroquine Analogs against Chloroquine-Resistant Parasites, Docking Studies and Mechanisms of Drug Action. Malar. J. 2014, 13, 469. [Google Scholar] [CrossRef][Green Version]
- Penna-Coutinho, J.; Cortopassi, W.A.; Oliveira, A.A.; França, T.C.C.; Krettli, A.U. Antimalarial Activity of Potential Inhibitors of Plasmodium Falciparum Lactate Dehydrogenase Enzyme Selected by Docking Studies. PLoS ONE 2011, 6, e21237. [Google Scholar] [CrossRef] [PubMed]
- Nurhainis, O.S.; Fuad, F.A.A. Screening Potential Inhibitors of Lactate Dehydrogenase from Plasmodium Knowlesi via Virtual Screening Approaches. Trop. Biomed. 2017, 34, 841–854. [Google Scholar] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Salim, N.O.; Fuad, F.A.A.; Khairuddin, F.; Seman, W.M.K.W.; Jonet, M.A. Purifying and Characterizing Bacterially Expressed Soluble Lactate Dehydrogenase from Plasmodium Knowlesi for the Development of Anti-Malarial Drugs. Molecules 2021, 26, 6625. [Google Scholar] [CrossRef]
- Saxena, N.; Pandey, V.; Dutta, G.; Ghatak, S. Characterization of Lactate Dehydrogenase of Plasmodium Knowlesi. Mol. Biochem. Parasitol. 1986, 21, 199–202. [Google Scholar] [CrossRef]
- Goldberg, E. Lactate Dehydrogenases in Spermatozoa: Subunit Interactions in Vitro. Arch. Biochem. Biophys. 1965, 109, 134–141. [Google Scholar] [CrossRef]
- Khanna, S.; Madan, M.; Vangoori, A.; Banerjee, R.; Thaimattam, R.; Jafar Sadik Basha, S.K.; Ramesh, M.; Casturi, S.R.; Pal, M. Evaluation of Glycolamide Esters of Indomethacin as Potential Cyclooxygenase-2 (COX-2) Inhibitors. Bioorg. Med. Chem. 2006, 14, 4820–4833. [Google Scholar] [CrossRef]
- Quaytman, S.L.; Schwartz, S.D. Comparison Studies of the Human Heart and Bacillus Stearothermophilus Lactate Dehydrogreanse by Transition Path Sampling. J. Phys. Chem. A 2009, 113, 1892–1897. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, S.; Pradhan, A.; Hammond, N.L.; Chittiboyina, A.G.; Tekwani, B.L.; Avery, M.A. Design, Synthesis, and Biological Evaluation of Plasmodium Falciparum Lactate Dehydrogenase Inhibitors. J. Med. Chem. 2007, 50, 3841–3850. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Compound Similar to Pyruvate (P) | Compound Name | LBDD Similarity Score (sqc) | SBDD Mın. Binding Energy (kcal/mol) |
|---|---|---|---|
| Compound P1 | 2-Amino-1-methyl-azetidine dihydrochloride | 0.882 | −4.43 |
| Compound P2 | Methyl acetate | 0.874 | −3.19 |
| Compound P3 | 6-Oxa-3-azabicyclo[3.1.1]heptane | 0.872 | −5.25 |
| Compound P4 | 3,3-Difluoropyrrolidine hydrochloride | 0.868 | −3.25 |
| Compound P5 | 1,2,2-Trifluoropropane 97% | 0.861 | −1.99 |
| Compound P6 | 1,2-Difluoropropane (FC-272ea) 97% | 0.859 | −4.78 |
| Pyruvate | - | - | −3.66 |
| Compounds Similar to Lactate (L) | Compound Name | LBDD Similarity Score (sqc) | SBDD Min. Binding Energy (kcal/mol) |
|---|---|---|---|
| Compound L1 | 3-Methyloxetan-3-ol | 0.849 | −3.9 |
| Compound L2 | 2-Fluoropropan-1-ol | 0.848 | −2.81 |
| Compound L3 | 3-Hydroxytetrahydrofuran | 0.830 | −3.63 |
| Compound L4 | Azetidin-2-one | 0.827 | −2.99 |
| Compound L5 | 2-Methyloxetan-3-ol | 0.824 | −3.6 |
| Compound L6 | 3-Hydroxytetrahydrofuran | 0.822 | −3.74 |
| Lactate | - | - | −4.29 |
| Toxicity Test | Compound Similar to Pyruvate | Predicted Test Value | Compound Similar to Lactate | Predicted Test Value |
|---|---|---|---|---|
| Rat Oral LD50 (mg/kg) | P2 | 3133.46 | L5 | 3755.16 |
| Fathead minnow LC50 (mg/L)—96 h | P2 | 838.37 | L6 | 3581.10 |
| D. magna LC50 (mg/L)—48 h | P4 | 490.09 | L6 | 2085.12 |
| T. pyriformis IGC50 (mg/L)—48 h | P4 | 3946.89 | L1 | 5657.5 |
| Developmental Toxicity | P5 | Non-toxicant | L5 | Non-toxicant |
| Mutagenicity | P2, P4, P5 | Negative | All except L4 | Negative |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad Fuad, F.A.; Ogu Salim, N. Analogues of Oxamate, Pyruvate, and Lactate as Potential Inhibitors of Plasmodium knowlesi Lactate Dehydrogenase Identified Using Virtual Screening and Verified via Inhibition Assays. Processes 2022, 10, 2443. https://doi.org/10.3390/pr10112443
Ahmad Fuad FA, Ogu Salim N. Analogues of Oxamate, Pyruvate, and Lactate as Potential Inhibitors of Plasmodium knowlesi Lactate Dehydrogenase Identified Using Virtual Screening and Verified via Inhibition Assays. Processes. 2022; 10(11):2443. https://doi.org/10.3390/pr10112443
Chicago/Turabian StyleAhmad Fuad, Fazia Adyani, and Nurhainis Ogu Salim. 2022. "Analogues of Oxamate, Pyruvate, and Lactate as Potential Inhibitors of Plasmodium knowlesi Lactate Dehydrogenase Identified Using Virtual Screening and Verified via Inhibition Assays" Processes 10, no. 11: 2443. https://doi.org/10.3390/pr10112443
APA StyleAhmad Fuad, F. A., & Ogu Salim, N. (2022). Analogues of Oxamate, Pyruvate, and Lactate as Potential Inhibitors of Plasmodium knowlesi Lactate Dehydrogenase Identified Using Virtual Screening and Verified via Inhibition Assays. Processes, 10(11), 2443. https://doi.org/10.3390/pr10112443