
Web Apps Come of Age for Molecular Sciences


Abstract
1. Introduction
2. Timeline of JavaScript in Science
3. JavaScript Libraries for Online, Interactive Data Display
JavaScript Molecular Viewers, Editors, and Calculators, from Chemistry to Cheminformatics and Structural Biology
4. JavaScript Tools for Numerical Calculations in Simulations, Data Processing, and Analysis
5. JavaScript Tools for Handling Strings, Text Mining, and Linguistics
6. On-the-Fly Data Retrieval and Utilization within JavaScript Web Apps
7. State-of-the-Art JavaScript Web Apps to Inspire Advanced Applications in Molecular Biosciences
8. Modularity, Integrability, and Open Nature of JavaScript Libraries
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stein, L. Web applets: Java, JavaScript and ActiveX. Trends Genet. TIG 1996, 12, 484–485. [Google Scholar] [CrossRef]
- Sedova, M.; Jaroszewski, L.; Godzik, A. Protael: Protein data visualization library for the web. Bioinform. Oxf. Engl. 2016, 32, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.C.R. Viewing multiple sequence alignments with the JavaScript Sequence Alignment Viewer (JSAV). F1000Research 2014, 3, 249. [Google Scholar] [CrossRef] [PubMed]
- Yachdav, G.; Wilzbach, S.; Rauscher, B.; Sheridan, R.; Sillitoe, I.; Procter, J.; Lewis, S.E.; Rost, B.; Goldberg, T. MSAViewer: Interactive JavaScript visualization of multiple sequence alignments. Bioinform. Oxf. Engl. 2016, 32, 3501–3503. [Google Scholar] [CrossRef] [PubMed]
- Deu-Pons, J.; Schroeder, M.P.; Lopez-Bigas, N. jHeatmap: An interactive heatmap viewer for the web. Bioinform. Oxf. Engl. 2014, 30, 1757–1758. [Google Scholar] [CrossRef] [PubMed]
- Gómez, J.; García, L.J.; Salazar, G.A.; Villaveces, J.; Gore, S.; García, A.; Martín, M.J.; Launay, G.; Alcántara, R.; Del-Toro, N.; et al. BioJS: An open source JavaScript framework for biological data visualization. Bioinform. Oxf. Engl. 2013, 29, 1103–1104. [Google Scholar] [CrossRef] [PubMed]
- Corpas, M. The BioJS article collection of open source components for biological data visualisation. F1000Research 2014, 3, 56. [Google Scholar] [CrossRef] [PubMed]
- Kreft, L.; Botzki, A.; Coppens, F.; Vandepoele, K.; Van Bel, M. PhyD3: A phylogenetic tree viewer with extended phyloXML support for functional genomics data visualization. Bioinform. Oxf. Engl. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hanson, R.M.; Prilusky, J.; Renjian, Z.; Nakane, T.; Sussman, J.L. JSmol and the Next-Generation Web-Based Representation of 3D Molecular Structure as Applied to Proteopedia. Isr. J. Chem. 2013, 53, 207–216. [Google Scholar] [CrossRef]
- Rose, A.S.; Hildebrand, P.W. NGL Viewer: A web application for molecular visualization. Nucleic Acids Res. 2015, 43, W576–W579. [Google Scholar] [CrossRef] [PubMed]
- Rego, N.; Koes, D. 3Dmol.js: Molecular visualization with WebGL. Bioinform. Oxf. Engl. 2015, 31, 1322–1324. [Google Scholar] [CrossRef] [PubMed]
- Bekker, G.-J.; Nakamura, H.; Kinjo, A.R. Molmil: A molecular viewer for the PDB and beyond. J. Cheminform. 2016, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Bienfait, B.; Ertl, P. JSME: A free molecule editor in JavaScript. J. Cheminform. 2013, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Earley, C.W. CH5M3D: An HTML5 program for creating 3D molecular structures. J. Cheminform. 2013, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Mohebifar, M.; Sajadi, F. Chemozart: A web-based 3D molecular structure editor and visualizer platform. J. Cheminform. 2015, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Abriata, L.A.; Bovigny, C.; Dal Peraro, M. Detection and sequence/structure mapping of biophysical constraints to protein variation in saturated mutational libraries and protein sequence alignments with a dedicated server. BMC Bioinform. 2016, 17, 242. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques 2000, 28, 1102–1104. [Google Scholar] [PubMed]
- Jin, X.; Awale, M.; Zasso, M.; Kostro, D.; Patiny, L.; Reymond, J.-L. PDB-Explorer: A web-based interactive map of the protein data bank in shape space. BMC Bioinform. 2015, 16, 339. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Patiny, L.; Sander, T.; Rufener, C.; Zasso, M. Wikipedia Chemical Structure Explorer: Substructure and similarity searching of molecules from Wikipedia. J. Cheminform. 2015, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Patiny, L.; Borel, A. ChemCalc: A building block for tomorrow’s chemical infrastructure. J. Chem. Inf. Model. 2013, 53, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, S.; Park, H.; Varghese, N.; Huang, P.-S.; Pavlopoulos, G.A.; Kim, D.E.; Kamisetty, H.; Kyrpides, N.C.; Baker, D. Protein structure determination using metagenome sequence data. Science 2017, 355, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.S.; Hopf, T.A.; Sander, C. Protein structure prediction from sequence variation. Nat. Biotechnol. 2012, 30, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, S.; Li, Z.; Zhang, R.; Xu, J. Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model. PLoS Comput. Biol. 2017, 13, e1005324. [Google Scholar] [CrossRef] [PubMed]
- Papoutsaki, A.; Sangkloy, P.; Laskey, J.; Daskalova, N.; Huang, J.; Hays, J. WebGazer: Scalable Webcam Eye Tracking Using User Interactions. In Proceedings of the 25th International Joint Conference on Artificial Intelligence, New York, NY, USA, 9–15 July 2016; pp. 3839–3845. [Google Scholar]
- Ince, D.C.; Hatton, L.; Graham-Cumming, J. The case for open computer programs. Nature 2012, 482, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.A.; Reimer, J. Open access and global participation in science. Science 2009, 323, 1025. [Google Scholar] [CrossRef] [PubMed]
- Hanson, B.; Sugden, A.; Alberts, B. Making data maximally available. Science 2011, 331, 649. [Google Scholar] [CrossRef] [PubMed]
- Beisken, S.; Conesa, P.; Haug, K.; Salek, R.M.; Steinbeck, C. SpeckTackle: JavaScript charts for spectroscopy. J. Cheminform. 2015, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Lancashire, R.J. The JSpecView Project: An Open Source Java viewer and converter for JCAMP-DX, and XML spectral data files. Chem. Cent. J. 2007, 1, 31. [Google Scholar] [CrossRef] [PubMed]
- Vosegaard, T. jsNMR: An embedded platform-independent NMR spectrum viewer. Magn. Reson. Chem. MRC 2015, 53, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Mukhyala, K.; Masselot, A. Visualization of protein sequence features using JavaScript and SVG with pViz.js. Bioinform. Oxf. Engl. 2014, 30, 3408–3409. [Google Scholar] [CrossRef] [PubMed]
- Jaschob, D.; Davis, T.N.; Riffle, M. SnipViz: A compact and lightweight web site widget for display and dissemination of multiple versions of gene and protein sequences. BMC Res. Notes 2014, 7, 468. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Chen, X.; Luo, H.; Fan, Z.; Luo, J.; He, S.; Yue, H.; Zhang, P.; Chen, R. BioCircos.js: An interactive Circos JavaScript library for biological data visualization on web applications. Bioinform. Oxf. Engl. 2016, 32, 1740–1742. [Google Scholar] [CrossRef] [PubMed]
- Laird, M.R.; Langille, M.G.I.; Brinkman, F.S.L. GenomeD3Plot: A library for rich, interactive visualizations of genomic data in web applications. Bioinform. Oxf. Engl. 2015, 31, 3348–3349. [Google Scholar] [CrossRef] [PubMed]
- Vanderkam, D.; Aksoy, B.A.; Hodes, I.; Perrone, J.; Hammerbacher, J. pileup.js: A JavaScript library for interactive and in-browser visualization of genomic data. Bioinform. Oxf. Engl. 2016, 32, 2378–2379. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Rusiel, J.L.; Rannou, N.; Gollub, R.L.; Pieper, S.; Murphy, S.; Robertson, R.; Grant, P.E.; Pienaar, R. Reusable Client-Side JavaScript Modules for Immersive Web-Based Real-Time Collaborative Neuroimage Visualization. Front. Neuroinform. 2017, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Virag, I.; Stoicu-Tivadar, L.; Crişan-Vida, M. Gesture Interaction Browser-Based 3D Molecular Viewer. Stud. Health Technol. Inform. 2016, 226, 17–20. [Google Scholar] [PubMed]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlić, A.; Rose, P.W. Web-based Molecular Graphics for Large Complexes. In Proceedings of the 21st International Conference on Web3D Technology, Anaheim, CA, USA, 22–24 July 2016; pp. 185–186. [Google Scholar]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Implementing WebGL and HTML5 in Macromolecular Visualization and Modern Computer-Aided Drug Design. Trends Biotechnol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Saita, E.; Abriata, L.A.; Tsai, Y.T.; Trajtenberg, F.; Lemmin, T.; Buschiazzo, A.; Dal Peraro, M.; de Mendoza, D.; Albanesi, D. A coiled coil switch mediates cold sensing by the thermosensory protein DesK. Mol. Microbiol. 2015, 98, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Abriata, L.A.; Albanesi, D.; Dal Peraro, M.; de Mendoza, D. Signal Sensing and Transduction by Histidine Kinases as Unveiled through Studies on a Temperature Sensor. Acc. Chem. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Mwalongo, F.; Krone, M.; Becher, M.; Reina, G.; Ertl, T. GPU-based remote visualization of dynamic molecular data on the web. Graph. Model. 2016, 88, 57–65. [Google Scholar] [CrossRef]
- Mwalongo, F.; Krone, M.; Becher, M.; Reina, G.; Ertl, T. Remote Visualization of Dynamic Molecular Data Using WebGL. In Proceedings of the 20th International Conference on 3D Web Technology, Heraklion, Greece, 18–21 June 2015; pp. 115–122. [Google Scholar]
- Abriata, L.A.; Palzkill, T.; Dal Peraro, M. How structural and physicochemical determinants shape sequence constraints in a functional enzyme. PLoS ONE 2015, 10, e0118684. [Google Scholar] [CrossRef] [PubMed]
- Hanson, R.M.; Lu, X.-J. DSSR-enhanced visualization of nucleic acid structures in Jmol. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Rothenbreger, O.; Newton, T.; Hanson, R.; Sitzmann, M. The Jmol Virtual Molecular Model Kit: A Resource for Teaching and Learning Chemistry. In CHED Committee on Computers in Chemical Education; American Chemical Society: Washington, DC, USA, 2011. [Google Scholar]
- Abriata, L.A. Structural database resources for biological macromolecules. Brief. Bioinform. 2016. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.H.; Kromann, J.C. The Molecule Calculator: A Web Application for Fast Quantum Mechanics-Based Estimation of Molecular Properties. J. Chem. Educ. 2013, 90, 1093–1095. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Scalfani, V.F.; Williams, A.J.; Tkachenko, V.; Karapetyan, K.; Pshenichnov, A.; Hanson, R.M.; Liddie, J.M.; Bara, J.E. Programmatic conversion of crystal structures into 3D printable files using Jmol. J. Cheminform. 2016, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Jin, X.; Dong, Y.; Chen, M. Kekule.js: An Open Source JavaScript Chemoinformatics Toolkit. J. Chem. Inf. Model. 2016, 56, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.C. ChemDoodle Web Components: HTML5 toolkit for chemical graphics, interfaces, and informatics. J. Cheminform. 2015, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Przybyła, P.; Shardlow, M.; Aubin, S.; Bossy, R.; Eckart de Castilho, R.; Piperidis, S.; McNaught, J.; Ananiadou, S. Text mining resources for the life sciences. Database 2016, 2016. [Google Scholar] [CrossRef]
- Chen, H.; Sharp, B.M. Content-rich biological network constructed by mining PubMed abstracts. BMC Bioinform. 2004, 5, 147. [Google Scholar] [CrossRef] [PubMed]
- Krallinger, M.; Rabal, O.; Leitner, F.; Vazquez, M.; Salgado, D.; Lu, Z.; Leaman, R.; Lu, Y.; Ji, D.; Lowe, D.M.; et al. The CHEMDNER corpus of chemicals and drugs and its annotation principles. J. Cheminform. 2015, 7, S2. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Kim, B.; Cho, H.; Lee, D.; Lee, H. A corpus for plant-chemical relationships in the biomedical domain. BMC Bioinform. 2016, 17, 386. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ohta, T.; Tateisi, Y.; Tsujii, J. GENIA corpus—Semantically annotated corpus for bio-textmining. Bioinformatics 2003, 19, i180–i182. [Google Scholar] [CrossRef] [PubMed]
- Kass-Hout, T.A.; Xu, Z.; Mohebbi, M.; Nelsen, H.; Baker, A.; Levine, J.; Johanson, E.; Bright, R.A. OpenFDA: An innovative platform providing access to a wealth of FDA’s publicly available data. J. Am. Med. Inform. Assoc. 2016, 23, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.; Coll, J.; Haimel, M.; Kandasamy, S.; Tarraga, J.; Furio-Tari, P.; Bari, W.; Bleda, M.; Rueda, A.; Gräf, S.; et al. HGVA: The Human Genome Variation Archive. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chastine, J.W.; Brooks, J.C.; Zhu, Y.; Owen, G.S.; Harrison, R.W.; Weber, I.T. AMMP-Vis: A Collaborative Virtual Environment for Molecular Modeling. In Proceedings of the ACM Symposium on Virtual Reality Software and Technology, Monterey, CA, USA, 7–9 November 2005; pp. 8–15. [Google Scholar]
- Pence, H.E.; Williams, A.J.; Belford, R.E. New Tools and Challenges for Chemical Education: Mobile Learning, Augmented Reality, and Distributed Cognition in the Dawn of the Social and Semantic Web. In Chemistry Education; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; pp. 693–734. ISBN 978-3-527-67930-0. [Google Scholar]
- Gillet, A.; Sanner, M.; Stoffler, D.; Goodsell, D.; Olson, A. Augmented reality with tangible auto-fabricated models for molecular biology applications. IEEE Vis. 2004. [Google Scholar] [CrossRef]
- Gillet, A.; Sanner, M.; Stoffler, D.; Olson, A. Tangible interfaces for structural molecular biology. Structure 2005, 13, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Vega Garzón, J.C.; Magrini, M.L.; Galembeck, E. Using augmented reality to teach and learn biochemistry. Biochem. Mol. Biol. Educ. 2017. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.; Board, J. A Protein in the palm of your hand through augmented reality. Biochem. Mol. Biol. Educ. 2014, 42, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.R.; Almeida, J.S. QMachine: Commodity supercomputing in web browsers. BMC Bioinform. 2014, 15, 176. [Google Scholar] [CrossRef] [PubMed]
- Abriata, L.A. A Simple Spreadsheet Program To Simulate and Analyze the Far-UV Circular Dichroism Spectra of Proteins. J. Chem. Educ. 2011, 88, 1268–1273. [Google Scholar] [CrossRef]
- Stein, L.D. Towards a cyberinfrastructure for the biological sciences: Progress, visions and challenges. Nat. Rev. Genet. 2008, 9, 678–688. [Google Scholar] [CrossRef] [PubMed]




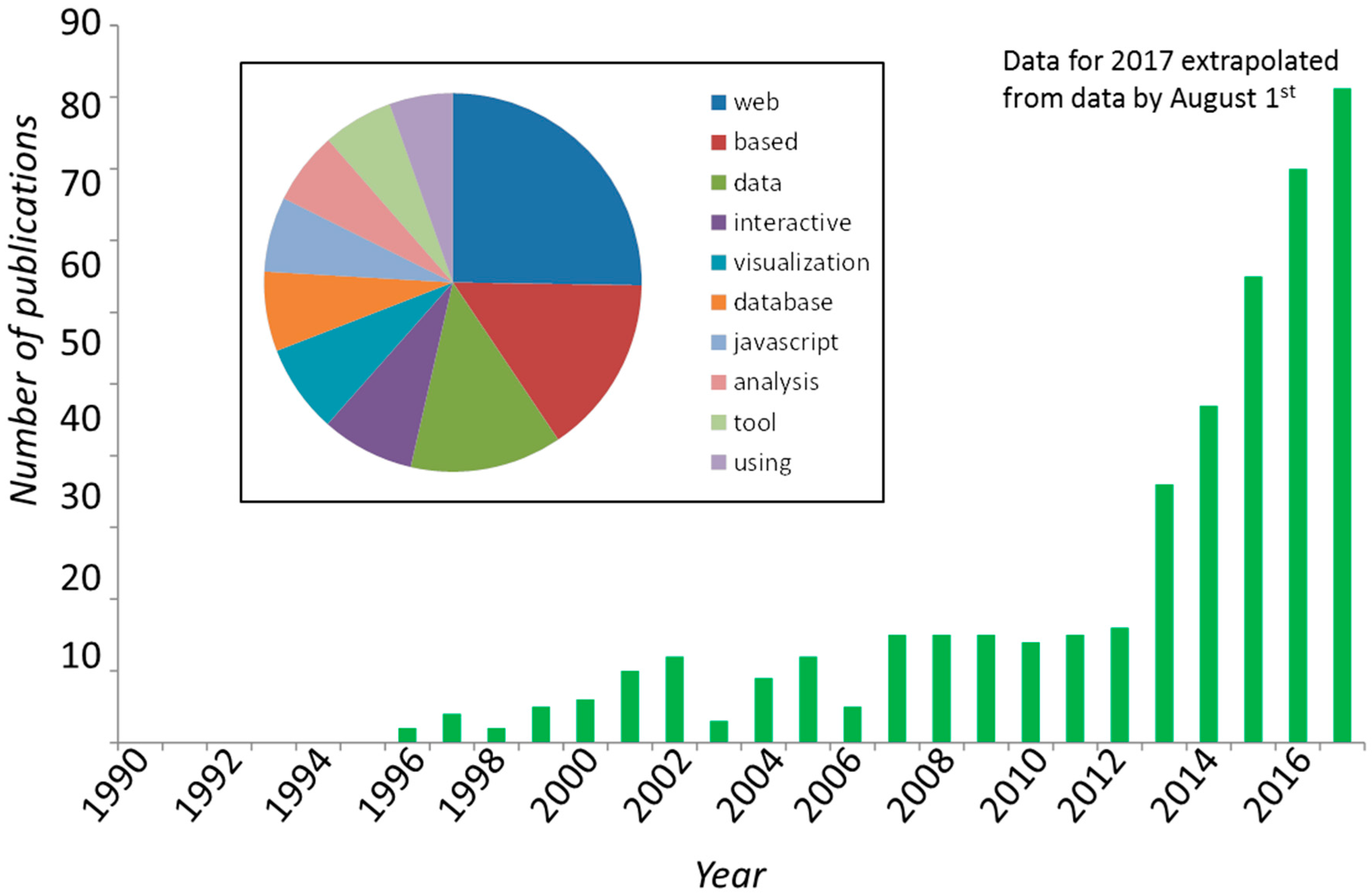{kind=link}
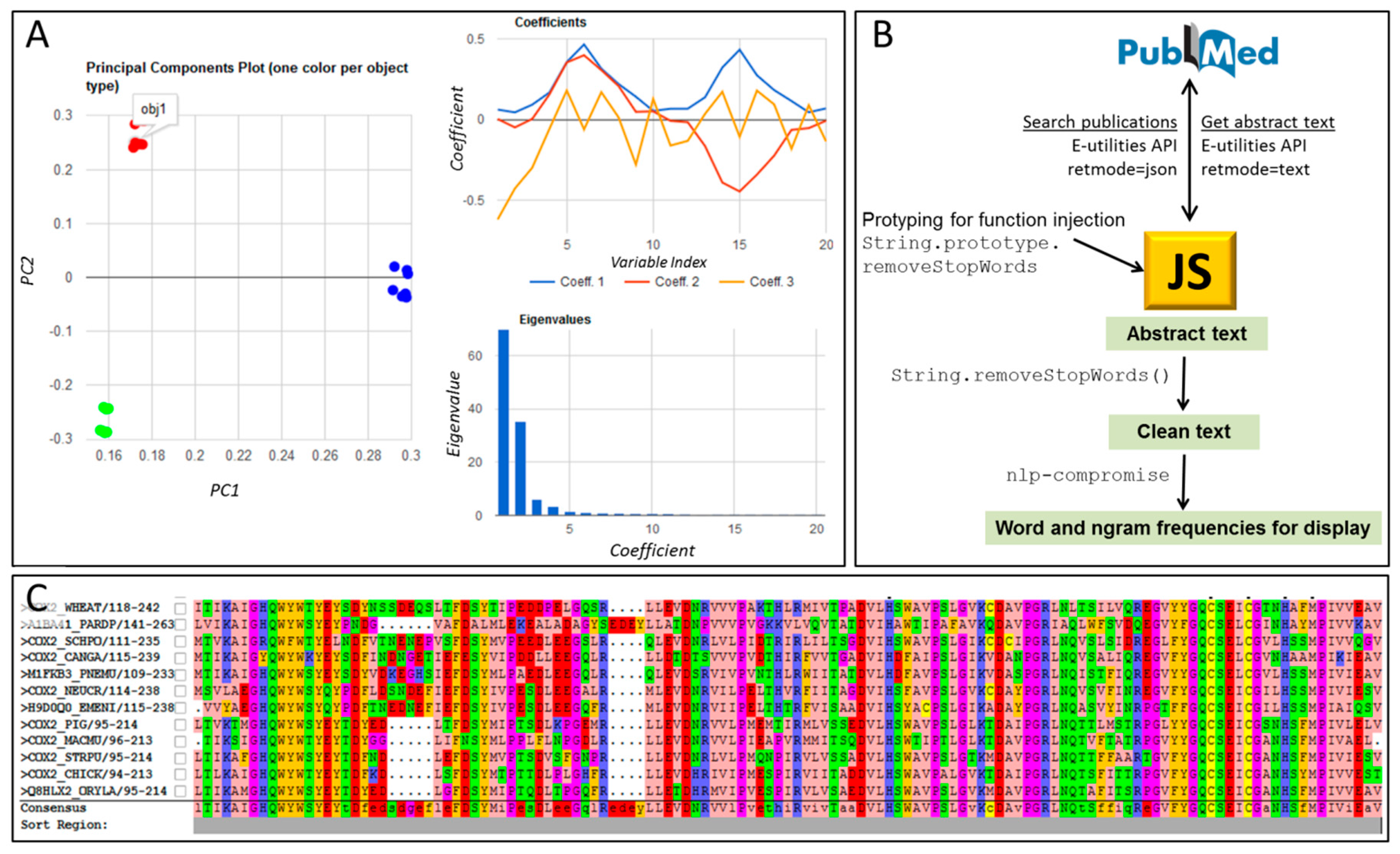{kind=link}
{kind=link}
| Tool and Reference | Brief Description |
|---|---|
| Data visualization | |
| Protael [2] | Protein data visualization library for the web. |
| JSAV [3] | The first JavaScript-based interactive sequence alignment viewer. |
| MSAViewer [4] | Interactive JavaScript visualization of multiple sequence alignments. |
| jHeatmap [5] | Library to display HeatMaps in the browser. |
| BioJS [6] | Framework for biological data visualization, on which several libraries have been built (a collection featuring some of them is available [7]). |
| PhyD3 [8] | Library for phylogenetic tree visualization in the browser. |
| Molecular visualization and editing | |
| JSmol [9] | A full molecular viewer and editor program and library. See text for details. |
| NGL Viewer [10], 3Dmol.js [11], PV, Molmil [12], LiteMol | Various molecular viewers which profit from WebGL technologies better than JSmol providing nicer and smoother rendering, but which are much more limited in capabilities beyond visualization. Most work as embeddable libraries but are also available as full web apps. |
| JSME [13] | A library to create and edit small molecules graphically, often used as the starting point for subsequent calculations or database queries. |
| CH5M3D [14], Chemozart [15] | Libraries to edit molecules in 3D. |
| Full web applications | |
| PsychoProt [16] | A tool to analyze amino acid variation in proteins in terms of protein physical chemistry, to aid in the analysis of sequence alignments and deep-sequencing-based tolerance to substitutions maps. |
| Sequence Manipulation Suite [17] | A collection of tools to handle, build, and analyze DNA and protein sequences. |
| PDB-Explorer [18] | An interactive map to browse the Protein Data Bank. |
| Wikipedia Chemical Structure Explorer [19] | An interactive 2D structure and substructure search engine for small molecules inside Wikipedia. |
| ChemCalc [20] | Calculates molecular formulas, molecular weights, elemental compositions, and isotopic distribution. Works as full web app or as an API for JavaScript calls. |
| ChemInfo.org | A portal to a variety of web-based tools for cheminformatics and related fields, including the PDBExplorer and ChemCalc listed above. |
| Tool | Brief Description and URL |
|---|---|
| Data visualization | |
| Comparing contact predictions and contact maps | Web apps to compare protein contact maps from structures or models to residue-residue contact predictions from Gremlin [21], EVFold [22], and RaptorX [23] contact predictions at http://lucianoabriata.altervista.org/pdbms/ |
| Examples of interactive open data from research articles by this author | Interactive visualization of data from articles: http://lucianoabriata.altervista.org/papersdata/ Interactive 3D visualization of published biomolecular models: http://lucianoabriata.altervista.org/modelshome.html |
| Molecular visualization and editing | |
| Hack-a-mol | Interconvert 2D and 3D molecular structures, SMILES and other codes, and coordinate files in several formats: https://chemapps.stolaf.edu/jmol/jsmol/hackamol.htm |
| Virtual Molecular Model Kit | A web app that provides easy access to JSmol’s model editing ability, also using JSME and API calls to external webservices and databases to provide a full online molecular modeling experience: http://chemagic.org/molecules/mini.html |
| PDB manipulation suite | A growing suite of functions for manipulating PDB files online, for example shifting residue numbers, getting amino acid sequences, and mapping data to B-factors: http://lucianoabriata.altervista.org/pdbms/ |
| Miscellaneous | |
| MultiProtScale | Client-side version of Expasy’s ProtScale server, which handles multiple sequences simultaneously: http://lucianoabriata.altervista.org/multiprotscale/multiprotscale.html |
| CD fitter and simulator | Fit and simulate far-UV protein circular dichroism spectra: http://lucianoabriata.altervista.org/tests/cd2.html |
| Tool | Brief Description, Possible Uses, and URL |
|---|---|
| Charts | |
| Google Charts, uvCharts, Chartist.js | They offer very complete sets of chart types, and are very customizable: https://developers.google.com/chart/ http://imaginea.github.io/uvCharts/ http://gionkunz.github.io/chartist-js/ |
| ChartJS, Highcharts JS, Flot | Also very complete, but here grouped together because they all provide support for older web browsers, yet deliver high-quality graphics, interactivity, and animations in modern browsers. http://www.chartjs.org/ |
| Smoothie Charts | Especially suited for plotting streamed data in real time. http://smoothiecharts.org/ |
| D3.js, n3-charts, Ember charts | D3.js has very broad applications, which allows charting in multiple formats not covered by the libraries mentioned above. Libraries like n3-charts and Ember charts make plotting through D3.js easier. https://github.com/d3/d3/wiki/Gallery http://addepar.github.io/#/ember-charts/overview http://n3-charts.github.io/line-chart |
| Mathematics and statistics | |
| NumCalc.com | A very complete scientific calculator at http://numcalc.com/ |
| LALOLib and Mlweb | Very complete libraries for mathematics in the browser, with tools for advanced matrix algebra, statistics, optimization, and machine learning. Can be used as libraries for web apps or through a complete environment (LALOLab) similar to that of stand-alone math programs, with its own plotting capabilities. See http://mlweb.loria.fr/ |
| Numeric.js | Another powerful library for mathematics in the browser, also usable through an environment similar to that of stand-alone math programs. http://www.numericjs.com/ |
| ConvNetJS, synaptic.js, brain, mind, DN2A | Libraries for neural networks and deep learning, covering from simpler utilities like data and function approximation and regression, to self-organizing maps, image regression and object identification, deep learning, and linguistics. Most can be trained and used online, or trained offline and used online. http://cs.stanford.edu/people/karpathy/convnetjs/ http://caza.la/synaptic/#/ https://github.com/harthur/brain https://github.com/stevenmiller888/mind https://github.com/dn2a/dn2a-javascript |
| jsfft, FFT.js, FFT in many languages, DSP.js, timbre.js | Libraries for Fast Fourier Transforms; together cover direct and inverse transforms, real and complex convolutions, discrete transforms, and other functions (plus, some are actually full packages for signal processing): https://github.com/dntj/jsfft https://www.nayuki.io/page/free-small-fft-in-multiple-languages https://github.com/corbanbrook/dsp.js http://mohayonao.github.io/timbre.js/ |
| Math.js, Sushi, numbers.js, jstat, rift, science, glMatrix | Other math libraries, some with unique features or tailored for specific applications (for example, rift is intended for games, glMatrix, and Sushi for efficient matrix calculations) |
| Strings | |
| String.js | Functions that extend those of standard JavaScript strings http://stringjs.com/ |
| textmining and text-miner | Two similar packages for text mining: https://www.npmjs.com/package/textmining https://www.npmjs.com/package/text-miner/ Potentially useful for mining contents in large corpuses of scientific text; Figure 1B of this article was built using such techniques. Text-miner includes lists of stopwords in four languages. |
| RiTa.js and nlp-compromise | Two libraries for natural language processing: https://rednoise.org/rita/index.php https://nlp-expo.firebaseapp.com/ |
| User interfaces | |
| WebGazer [24] | Eye tracking library that uses common webcams to infer eye-gaze locations of the user on the web page in real time. Could be used to facilitate molecular visualization as shown in the proof-of-concept example at https://lucianoabriata.altervista.org/jsinscience/jsmolwebgazer/jsmolwebgazer.html |
| Tangle | Library to create reactive documents, i.e., which interact with the user in a contextual manner and virtually immediate response. |
| MathJax | Library to display formatted maths in the browser. |
| Emulators and programming | |
| Linux emulators | Two complete emulators of a linux system, the second including internet connectivity and a graphical interface: http://bellard.org/jslinux/, http://s-macke.github.io/jor1k/ Could be used for learning linux in a safe and simple environment for example at the beginning of a tutorial course that requires linux knowledge; also for executing shell commands and scripts as well as programs written for Perl, Python, or even compiling C code, etc. on non-linux computers without the need for complex installations, as exemplified with the NESmapper Perl script. |
| Online programming environments | The two linux emulators listed above include C compilers and allow running Perl, Python, etc. Moreover, there are JavaScript-based web applications specifically tailored for writing, compiling, and running programs. Some notable links: site for learning and running C at http://cs-education.github.io/sys/#VM; a Perl interpreter at https://gfx.github.io/perl.js/; see also Emscripten at http://emscripten.org/ |
| Vi emulator | A working online version of the linux vi editor, could be used to quickly process text files in non-linux computers: http://gpl.internetconnection.net/vi/ |
| 3D graphics, computer vision, and augmented and virtual reality | |
| Three.js, A-Frame | Three.js is probably the most used library for animated 3D graphics, using WebGL. Applications are in molecular visualization and in more generic data visualization, but beyond these uses, it includes multiple numerical algorithms that could be recycled for other purposes: https://threejs.org/ A-Frame is an entity component system framework for Three.js, that makes it much easier to use in virtual and augmented reality applications, the latter especially through AR.js: https://aframe.io/ |
| Jsfeat, tracking.js, js-aruco, AR.js, awe.js, and argon.js | Computer vision and image processing libraries which can identify and track objects or markers. Just like Three.js, they include numerical algorithms that could be used for other purposes. Put together with graphical libraries like Three.js leads directly to augmented reality web apps. https://inspirit.github.io/jsfeat/ https://trackingjs.com/ https://github.com/jcmellado/js-aruco/ https://github.com/jeromeetienne/AR.js/ https://github.com/buildar/awe.js/ https://www.argonjs.io/ |
| Tesseract.js, Ocrad.js | Libraries for optical character recognition from images, useful for retrieving information from article figures. http://antimatter15.com/ocrad.js/demo.html http://tesseract.projectnaptha.com/ |
| Hardware feeds and communication | |
| WebRTC | Not a library but a collection of standards, protocols, and APIs for real-time server-less, plugin-free communication and data exchange directly between web browsers: https://webrtc.org/ See also the example at https://apprtc.appspot.com/ which implements a web app for server-less video conferencing. |
| HTML5 guitar tuner | A web app that guides guitar tuning; exemplifies how to read microphone data and analyze the signal through Fourier transforms to decompose the frequency spectrum: https://jbergknoff.github.io/guitar-tuner/ |
| gyro.js | Simplified access to gyroscope and accelerometer information, expanding the information provided by the built-in Geolocation API (which only locates the user’s position based on network and/or GPS data but gives no orientation information): http://tomg.co/gyrojs/ |
| GPS.js | Access to primary GPS data: https://github.com/infusion/GPS.js |
| Hardware Capabilities | Programming for Non-Experts | Accessibility |
|---|---|---|
|
|
|
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abriata, L.A. Web Apps Come of Age for Molecular Sciences. Informatics 2017, 4, 28. https://doi.org/10.3390/informatics4030028
Abriata LA. Web Apps Come of Age for Molecular Sciences. Informatics. 2017; 4(3):28. https://doi.org/10.3390/informatics4030028
Chicago/Turabian StyleAbriata, Luciano A. 2017. "Web Apps Come of Age for Molecular Sciences" Informatics 4, no. 3: 28. https://doi.org/10.3390/informatics4030028
APA StyleAbriata, L. A. (2017). Web Apps Come of Age for Molecular Sciences. Informatics, 4(3), 28. https://doi.org/10.3390/informatics4030028