
The Dramatic Modulatory Role of the 2'N Substitution of the Terminal Amino Hexose of Globotetraosylceramide in Determining Binding by Members of the Verotoxin Family

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Synthesis of NMe2, NHisoPr, or NHBz derivatives of Gb4•NH2
2.2.2. Synthesis of Gb4•NHCOCF3
2.2.3. Synthesis of Gb4•NHCOH
2.2.4. Mass Spectroscopic Analyses
TLC Overlay
Receptor ELISA (RELISA)
3. Results

{kind=link}
{kind=link}
{kind=link}
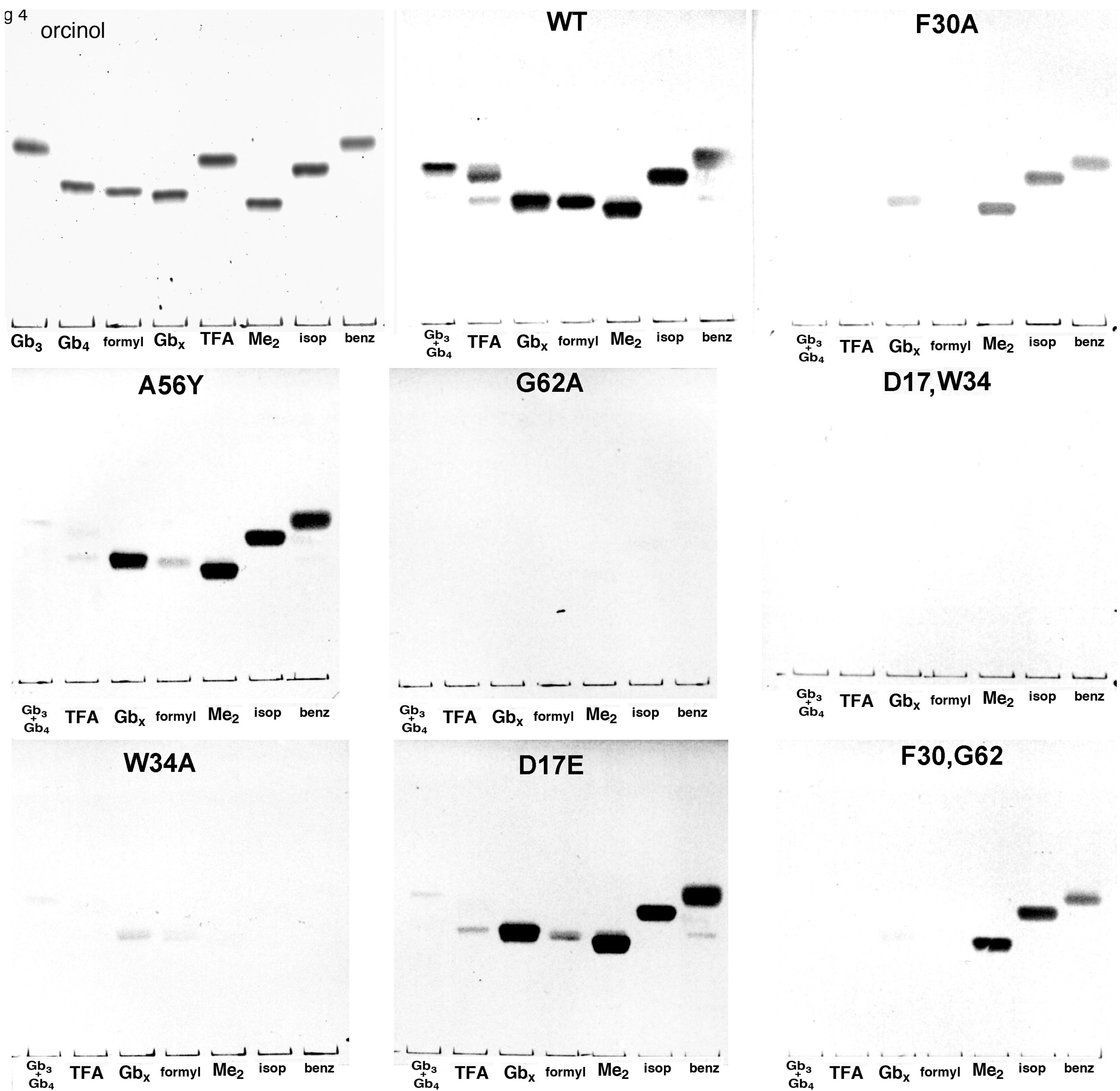{kind=link}
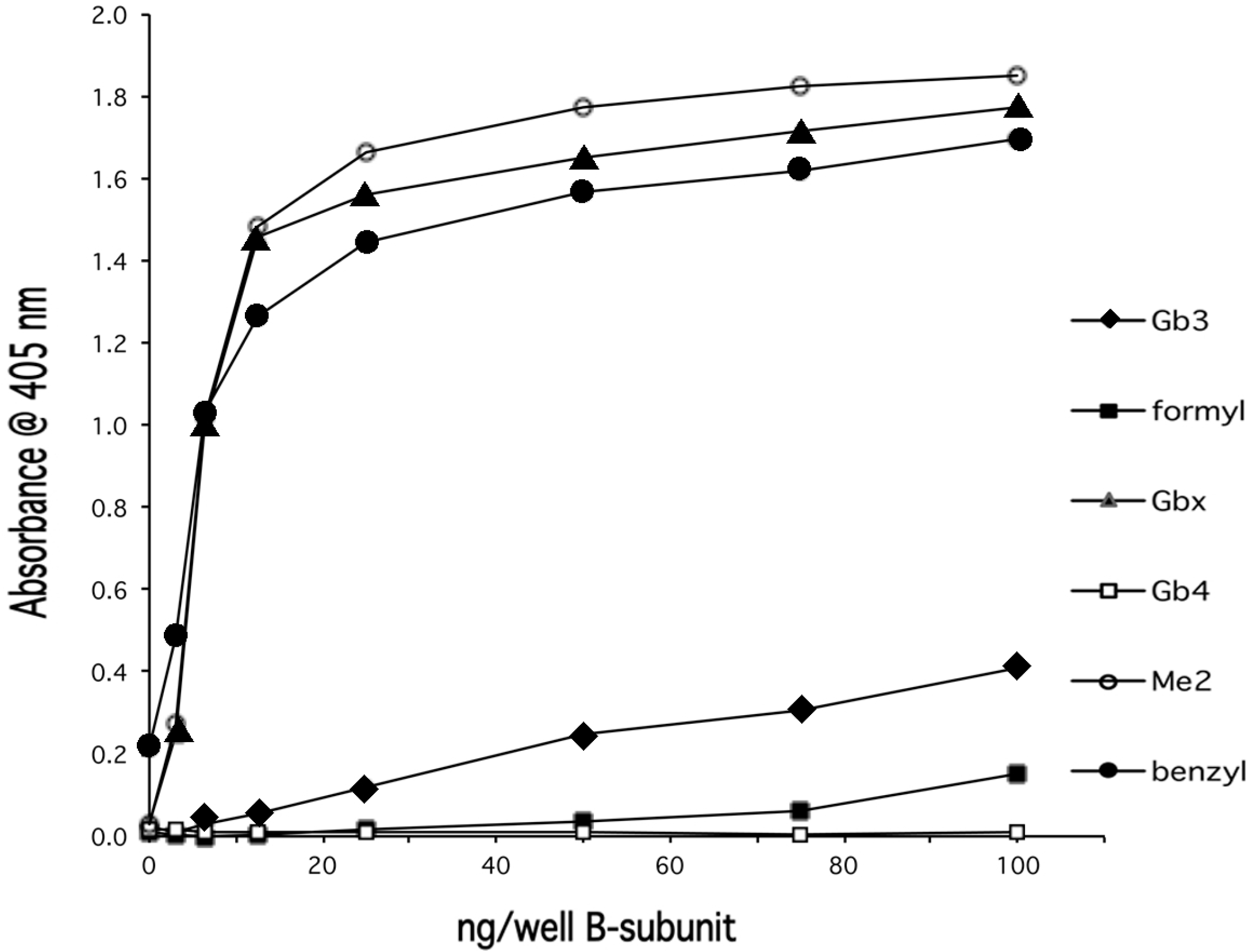{kind=link}
| n | Gb4•NHCOCH3 | Gb4•NHCOH | Gb4•NHCOCF3 | |||
|---|---|---|---|---|---|---|
| M+H (%) | M+Na (%) | M+H (%) | M+Na (%) | M+H (%) | M+Na (%) | |
| 22 | 1312.0 (30) | 1334.0 (74) | ND | 1320.0 (7) | ND | 1388.0 (36) |
| 24 | 1340.0 (39) | 1362.2 (100) | ND | 1348.0 (15) | ND | 1416.2 (43) |
| 24:1 | ND | 1360.0 (78) | ND | 1346.0 (8) | ND | 1414.2 (29) |
| n | Gb4•NH2 | Gb4•NMe2 | Gb4•NHBz- pNMe2 | Gb4•NHisoPr |
|---|---|---|---|---|
| M+H (%) | M+H (%) | M+H (%) | M+H (%) | |
| 22 | 1270.2 (8) | 1298.2 (<2) | 1403.0 (32) | 1340.2 (98) |
| 24 | 1298.0 (16) | 1326.2 (<2) | 1431.0 (39) | 1338.2 (100) |
| 24:1 | 1296.0 (11) | 1324.2 (<2) | 1429.2 (38) | 1312.0 (65) |
| GSL | VT1 | VT2 | VT2e | GT3 |
|---|---|---|---|---|
| Gb3 | + | + | + | + |
| FormylaminoGb4 | + | + | + | + |
| Gb4 | - | - | + | - |
| TFAaminoGb4 | - | - | + | + |
| DimethylaminoGb4 | + | + | - | - |
| isopropylaminoGb4 | + | + | - | - |
| BenzylaminoGb4 | + | + | + | + |
4. Discussion
Acknowledgements
Author Contributions
Conflicts of Interest
Abbreviations
| VT1 | Verotoxin 1 (aka Shiga-like toxin1, Slt1, Shiga toxin1, Stx1), the prototype E. coli derived AB5 subunit toxin with virtual identity to Shiga toxin from S. dysenteriae. Gb3 is the functional receptor |
| VT2 | Verotoxin 2 (Stx2), E. coli toxin B subunit is 66% homologous to VT1B, family member most associated with human disease. Gb3 is the functional receptor |
| VT2e | Less toxic VT2 family member. B subunit 96% homologous to VT2. Gb4 is the functional receptor; RELISA, receptor enzyme linked immunoassay |
| GSL | glycosphingolipid |
References
- Bauwens, A.; Betz, J.; Meisen, I.; Kemper, B.; Karch, H.; Muthing, J. Facing glycosphingolipid-Shiga toxin interaction: Dire straits for endothelial cells of the human vasculature. Cell. Mol. Life Sci. 2012, 70, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Page, A.V.; Liles, W.C. Enterohemorrhagic Escherichia coli Infections and the Hemolytic-Uremic Syndrome. Med. Clin. N. Am. 2013, 97, 681–695. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, D.L.; Gyles, C.L.; Wilcock, B.P. Reproduction of Edema Disease of Swine with Purified Shiga-like Toxin-II Variant. Vet. Pathol. 1991, 28, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, C.A. Verotoxin recognition of its glycolipid receptor, globotriaosylceramide: Role in pathogenesis. In Recent Advances in Verocytotoxin-Producing Escherichia Coli Infections; Karmali, M.A., Goglio, A.G., Eds.; Elsevier Science B.V.: Bergamo, Italy, 1994; pp. 131–137. [Google Scholar]
- DeGrandis, S.; Law, H.; Brunton, J.; Gyles, C.; Lingwood, C.A. Globotetraosyl ceramide is recognized by the pig edema disease toxin. J. Biol. Chem. 1989, 264, 12520–12525. [Google Scholar] [PubMed]
- Head, S.; Karmali, M.; Lingwood, C.A. Preparation of VT1 and VT2 hybrid toxins from their purified dissociated subunits: Evidence for B subunit modulation of A subunit function. J. Biol. Chem. 1991, 266, 3617–3621. [Google Scholar] [PubMed]
- Tyrrell, G.J.; Ramotar, K.; Toye, B.; Boyd, B.; Lingwood, C.A.; Brunton, J.L. Alteration of the carbohydrate binding specificity of verotoxins from Galα1-4Gal to GalNAcß1-3Galα1-4Gal and vice versa by site-directed mutagenesis of the binding subunit. Proc. Natl. Acad. Sci. USA 1992, 89, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Boyd, B.; Tyrrell, G.; Maloney, M.; Gyles, C.; Brunton, J.; Lingwood, C. Alteration of the glycolipid binding specificity of the pig edema toxin from globotetraosyl to globotriaosyl ceramide alters in vivo tissue targetting and results in a VT1-like disease in pigs. J. Exp. Med. 1993, 177, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Nyholm, P.G.; Magnusson, G.; Zheng, Z.; Norel, R.; Binnington-Boyd, B.; Lingwood, C.A. Two distinct binding sites for globotriaosyl ceramide on verotoxins: molecular modelling and confirmation by analogue studies and a new glycolipid receptor for all verotoxins. Chem. Biol. 1996, 3, 263–275. [Google Scholar] [CrossRef]
- Lingwood, C.A.; Nutikka, A. A novel chemical procedure for the selective removal of nonreducing terminal N-acetyl residues from glycolipids. Anal. Biochem. 1994, 217, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Boodhoo, A.; Hazes, B.; Cummings, M.; Armstrong, G.; Brunton, J.; Read, R.J. Structure of the Shiga toxin B-pentamer complexed with an analogue of its receptor Gb3. Biochemistry 1998, 37, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.M.; Evans, P.D.; Homans, S.W.; Donohue-Rolfe, A. Solution structure of the carbohydrate-binding B-subunit homopentamer of verotoxin VT-1 from E. coli. Nat. Struct. Biol. 1996, 4, 190–193. [Google Scholar] [CrossRef]
- Bast, D.J.; Banerjee, L.; Clark, C.; Read, R.J.; Brunton, J.L. The identification of three biologically relevant globotriaosyl ceramide receptor binding sites on the verotoxin 1 B subunit. Mol. Microbiol. 1999, 32, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Soltyk, A.M.; MacKenzie, C.R.; Wolski, V.M.; Hirama, T.; Kitov, P.I.; Bundle, D.R.; Brunton, J. A mutational analysis of the Globotriaosylceramide binding sites of Verotoxin VT1. J. Biol. Chem. 2002, 277, 5351–5359. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Matsuoka, K.; Watanabe, M.; Igai, K.; Hino, K.; Hatano, K.; Yamada, A.; Abe, N.; Terunuma, D.; Kuzuhara, H.; et al. Identification of the optimal structure required for a shiga toxin neutralizer with oriented carbohydrates to function in the circulation. J. Infect. Dis. 2005, 191, 2097–2105. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Igai, K.; Matsuoka, K.; Miyagawa, A.; Watanabe, T.; Yanoshita, R.; Samejima, Y.; Terunuma, D.; Natori, Y.; Nishikawa, K. Structural analysis of the interaction between Shiga toxin B subunits and linear polymers bearing clustered globotriose residues. Infect. Immun. 2006, 74, 1984–1988. [Google Scholar] [CrossRef] [PubMed]
- Kitov, P.I.; Mulvey, G.L.; Griener, T.P.; Lipinski, T.; Solomon, D.; Paszkiewicz, E.; Jacobson, J.; Sadowska, J.; Suzuki, M.; Yamamura, Y.; et al. In vivo supramolecular templating enhances the activity of multivalent ligands: a potential therapeutic against the Escherichia coli O157 AB5 toxins. Proc. Natl. Acad. Sci. USA 2008, 105, 16837–16842. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Field, R.A.; Homans, S.W.; Donohue-Rolfe, A. Solution structure of the complex between the B-subunit homopentamer of verotoxin VT-1 from Escherichia coli and the trisaccharide moiety of globotriaosylceramide. Biochemistry 1998, 37, 11078–11082. [Google Scholar] [CrossRef] [PubMed]
- Thompson, G.; Shimizu, H.; Homans, S.; Donohue-Rolfe, A. Localization of the binding site for the oligosaccharide moiety of Gb3 on verotoxin 1 using NMR residual dipolar coupling measurements. Biochemistry 2000, 39, 13153–13156. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.E.; Fujinaga, M.; Cherney, M.M.; Melton-Celsa, A.R.; Twiddy, E.M.; O'Brien, A.D.; James, M. Structure of shiga toxin type 2 (Stx2) from escherichia coli O157:H7. J. Biol. Chem. 2004, 279, 27511–27517. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, J.M.; Yin, J.; Kitov, P.I.; Mulvey, G.; Griener, T.P.; James, M.N.; Armstrong, G.; Bundle, D. The crystal structure of shiga toxin type 2 with bound disaccharide guides the design of a heterobifunctional toxin inhibitor. J. Biol. Chem. 2014, 289, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Pannu, N.S.; Boodhoo, A.; Armstrong, G.D.; Clark, C.G.; Brunton, J.L.; Read, R. A mutant Shiga-like toxin IIe bound to its receptor Gb3: Structure of a group II Shiga-like toxin with altered binding specificity. Structure 2000, 8, 253–264. [Google Scholar] [CrossRef]
- Nyholm, P.-G.; Pascher, I. Orientation of the saccharide chains of glycolipids at the membrane surface: Conformational analysis of the glucose-ceramide and the glucose-glyceride linkages using molecular mechanics (MM3). Biochemistry 1993, 32, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Watkins, E.B.; Gao, H.; Dennison, A.J.; Chopin, N.; Struth, B.; Arnold, T.; Florent, J.-C.; Johannes, L. Carbohydrate conformation and lipid condensation in monolayers containing glycosphingolipid Gb3: Influence of acyl chain structure. Biophys. J. 2014, 107, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Mahfoud, R.; Manis, A.; Binnington, B.; Ackerley, C.; Lingwood, C.A. A major fraction of glycosphingolipids in model and cellular cholesterol containing membranes are undetectable by their binding proteins. J. Biol. Chem. 2010, 285, 36049–36059. [Google Scholar] [CrossRef] [PubMed]
- Yahi, N.; Aulas, A.; Fantini, J. How cholesterol constrains glycolipid conformation for optimal recognition of Alzheimer's beta amyloid peptide (Abeta1-40). PLoS One 2010, 5, e9079. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Binnington, B.; Róg, T.; Vattulainen, I.; Grzybek, M.; Coskun, U.; Lingwood, C.; Simons, K. Cholesterol modulates glycolipid conformation and receptor activity. Nat. Chem. Biol. 2011, 7, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, C.A. Receptor-related Risk Factors for Verotoxin Pathogenesis. In Bacterial Toxins - Genetics, Molecular Biology and Novel Applications; Proft, T., Ed.; Caister Acedemic Press: Norfolk, UK, 2013; pp. 1–12. [Google Scholar]
- Kale, R.R.; McGannon, C.M.; Fuller-Schaefer, C.; Hatch, D.M.; Flagler, M.J.; Gamage, S.D.; Weiss, A.; Iyer, S. Differentiation between structurally homologous Shiga 1 and Shiga 2 toxins by using synthetic glycoconjugates. Angew. Chem. Int. Ed. Engl. 2008, 47, 1265–1268. [Google Scholar] [CrossRef] [PubMed]
- Flagler, M.J.; Mahajan, S.S.; Kulkarni, A.A.; Iyer, S.S.; Weiss, A.A. Comparison of binding platforms yields insights into receptor binding differences between shiga toxins 1 and 2. Biochemistry 2010, 49, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Kiarash, A.; Boyd, B.; Lingwood, C.A. Glycosphingolipid receptor function is modified by fatty acid content: Verotoxin 1 and Verotoxin 2c preferentially recognize different globotriaosyl ceramide fatty acid homologues. J. Biol. Chem. 1994, 269, 11138–11146. [Google Scholar] [PubMed]
- Gallegos, K.M.; Conrady, D.G.; Karve, S.S.; Gunasekera, T.S.; Herr, A.B.; Weiss, A.A. Shiga toxin binding to glycolipids and glycans. PLoS One 2012, 7, e30368. [Google Scholar] [CrossRef] [PubMed]
- Boyd, B.; Lingwood, C.A. Verotoxin receptor glycolipid in human renal tissue. Nephron 1989, 51, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Nutikka, A.; Binnington-Boyd, B.; Lingwood, C.A. Methods for the purification of Shiga toxin 1. In Methods in Molecular Medicine; Philpot, D., Ebel, F., Eds.; Humana Press: Totowa, NY, USA, 2003; pp. 187–195. [Google Scholar]
- Nutikka, A.; Binnington-Boyd, B.; Lingwood, C. Methods for the Identification of Host Receptors for Shiga toxin. In Methods in Molecular Medicine; Philpot, D., Ebel, F., Eds.; Humana Press: Totowa, NY, USA, 2003; pp. 197–208. [Google Scholar]
- Nakajima, H.; Kiyokawa, N.; Katagiri, Y.U.; Taguchi, T.; Suzuki, T.; Sekino, T.; Mimori, K.; Ebata, T.; Saito, M.; Nakao, H.; et al. Kinetic analysis of binding between Shiga toxin and receptor glycolipid Gb3Cer by surface plasmon resonance. J. Biol. Chem. 2001, 276, 42915–42922. [Google Scholar] [CrossRef] [PubMed]
- Betz, J.; Bielaszewska, M.; Thies, A.; Humpf, H.U.; Dreisewerd, K.; Karch, H.; Kim, K.; Frierich, A.; Muthing, J. Shiga toxin glycosphingolipid receptors in microvascular and macrovascular endothelial cells: association with membrane lipid raft microdomains that differ by their stability to cholesterol depletion. J. Lipid. Res. 2011, 58, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Head, S.; Ramotar, K.; Lingwood, C.A. Modification of the glycolipid-binding specificity of vero cytotoxin by Polymyxin B and other cyclic amphipathic peptides. Infect. Immun. 1990, 58, 1532–1537. [Google Scholar] [PubMed]
- Karve, S.S.; Weiss, A.A. Glycolipid binding preferences of Shiga toxin variants. PLoS One 2014, 9, e101173. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, D.; Jackson, M.; Holmes, R.; O'Brien, A. Cloning and sequencing of a Shiga-like toxin II variant from an Escherichia coli strain responsible for edema disease of swine. J. Bacteriol. 1988, 170, 4223–4230. [Google Scholar] [PubMed]
- Cummings, M.; Ling, H.; Armstrong, G.; Brunton, J.; Read, R. Modeling the carbohydrate-binding specificity of pig edema toxin. Biochemistry 1998, 37, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mylvaganam, M.; Binnington, B.; Budani, M.; Soltyk, A.M.; Lingwood, C.A. The Dramatic Modulatory Role of the 2'N Substitution of the Terminal Amino Hexose of Globotetraosylceramide in Determining Binding by Members of the Verotoxin Family. Chromatography 2015, 2, 529-544. https://doi.org/10.3390/chromatography2030529
Mylvaganam M, Binnington B, Budani M, Soltyk AM, Lingwood CA. The Dramatic Modulatory Role of the 2'N Substitution of the Terminal Amino Hexose of Globotetraosylceramide in Determining Binding by Members of the Verotoxin Family. Chromatography. 2015; 2(3):529-544. https://doi.org/10.3390/chromatography2030529
Chicago/Turabian StyleMylvaganam, Murugesapillai, Beth Binnington, Monique Budani, Anna M. Soltyk, and Clifford A. Lingwood. 2015. "The Dramatic Modulatory Role of the 2'N Substitution of the Terminal Amino Hexose of Globotetraosylceramide in Determining Binding by Members of the Verotoxin Family" Chromatography 2, no. 3: 529-544. https://doi.org/10.3390/chromatography2030529
APA StyleMylvaganam, M., Binnington, B., Budani, M., Soltyk, A. M., & Lingwood, C. A. (2015). The Dramatic Modulatory Role of the 2'N Substitution of the Terminal Amino Hexose of Globotetraosylceramide in Determining Binding by Members of the Verotoxin Family. Chromatography, 2(3), 529-544. https://doi.org/10.3390/chromatography2030529