A Sensitive and Robust Ultra HPLC Assay with Tandem Mass Spectrometric Detection for the Quantitation of the PARP Inhibitor Olaparib (AZD2281) in Human Plasma for Pharmacokinetic Application


Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
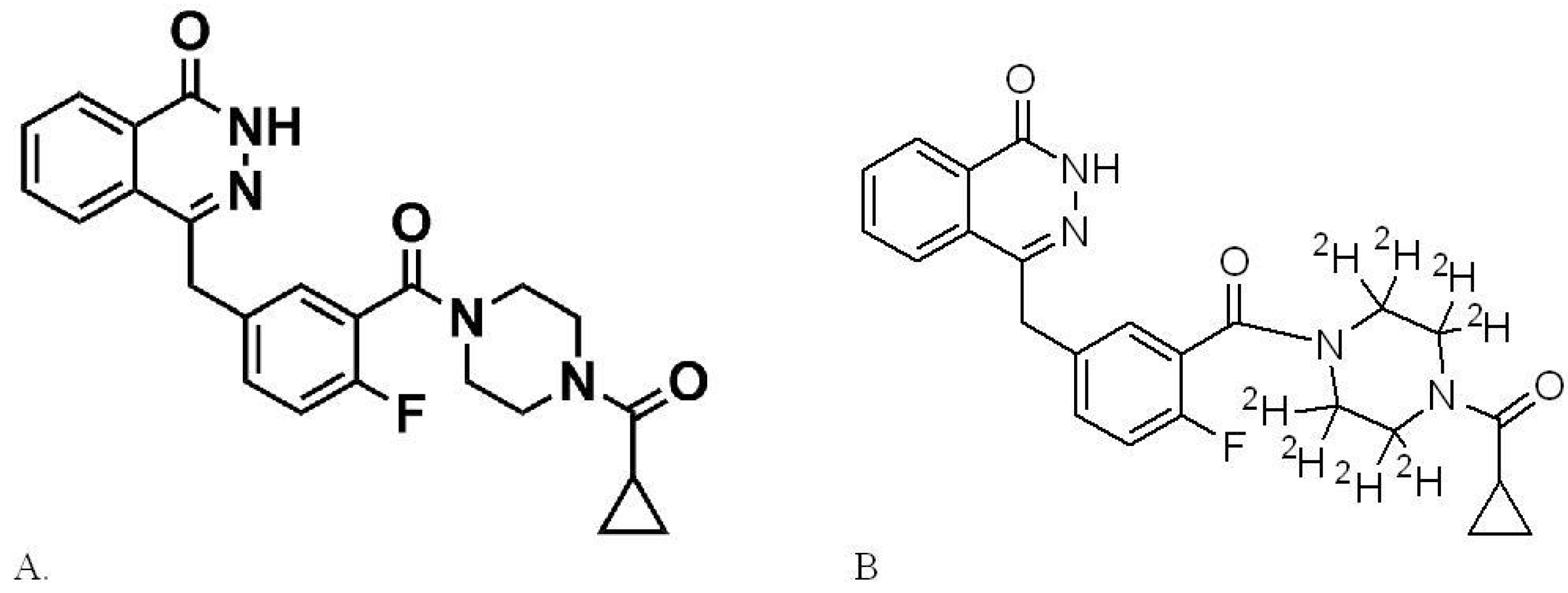
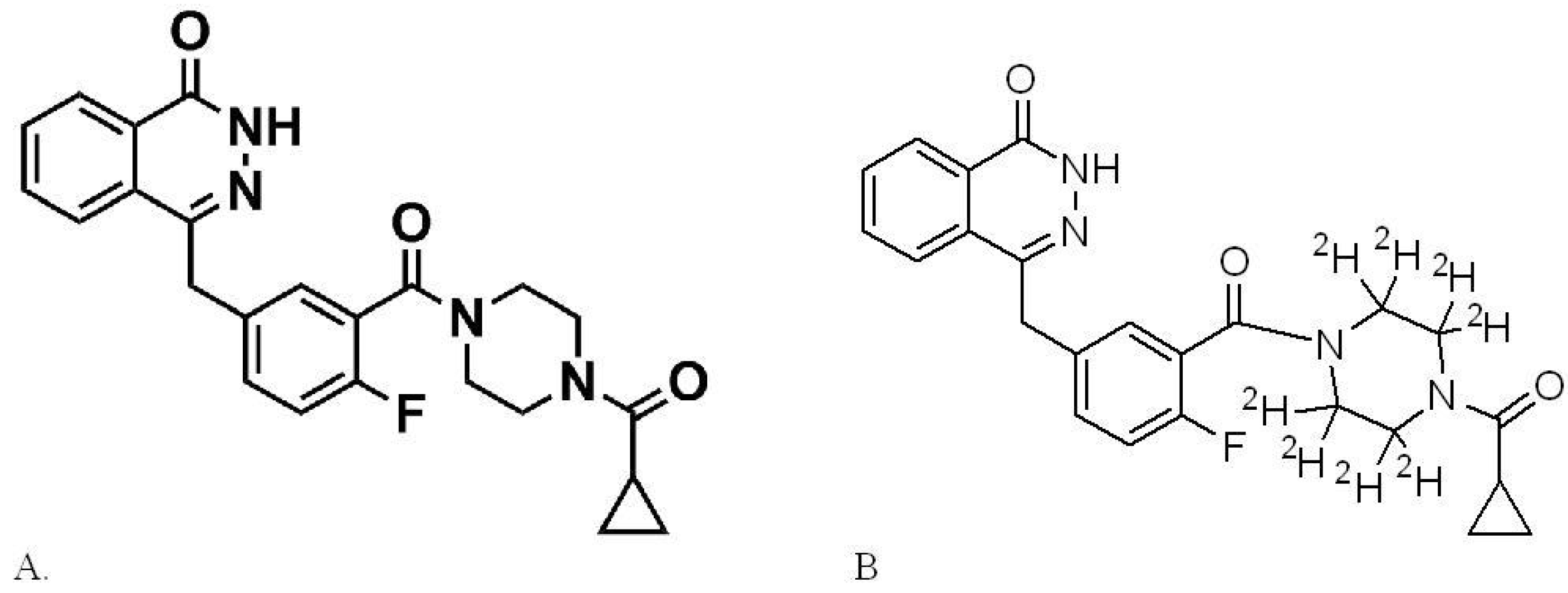
2.2. Preparation of Stock Solutions
2.3. Sample Preparation
2.4. Instrument Conditions
2.5. Validation
2.5.1. Linearity
2.5.2. Accuracy and Precision
2.5.3. Stability
2.5.4. Extraction Recovery and Matrix Effects
2.6. Clinical Application
3. Results and Discussion
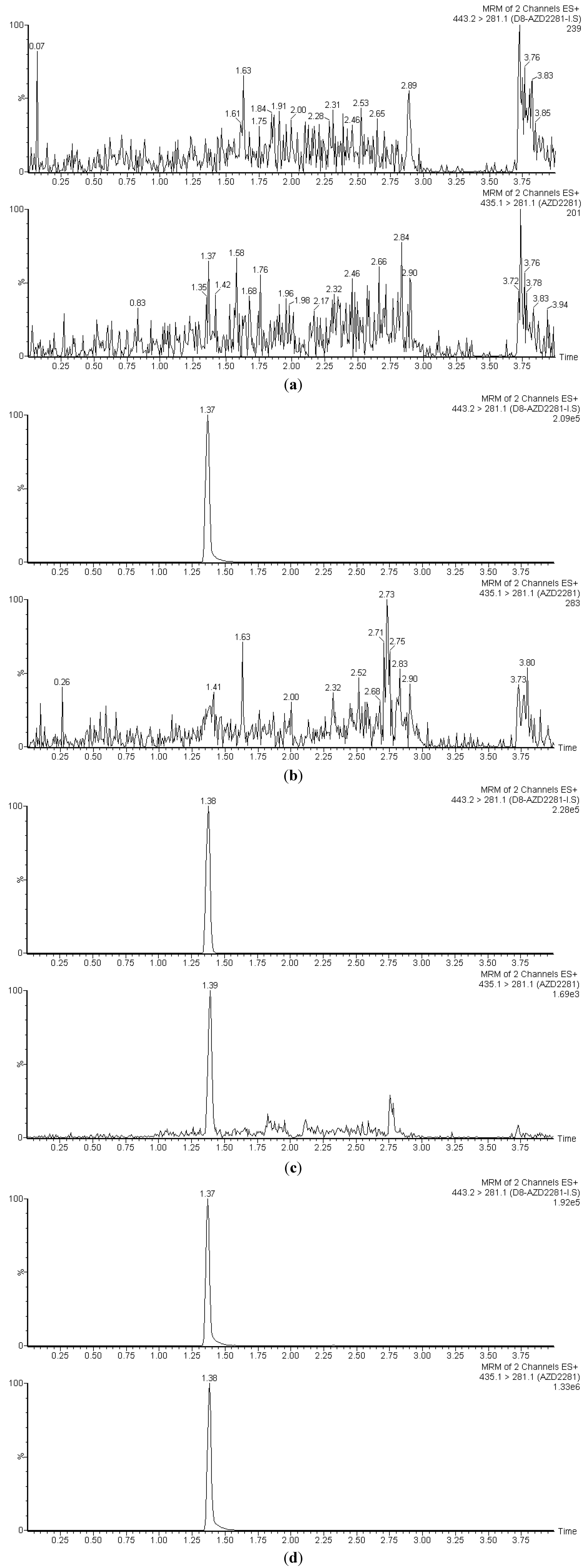
3.1. Selectivity
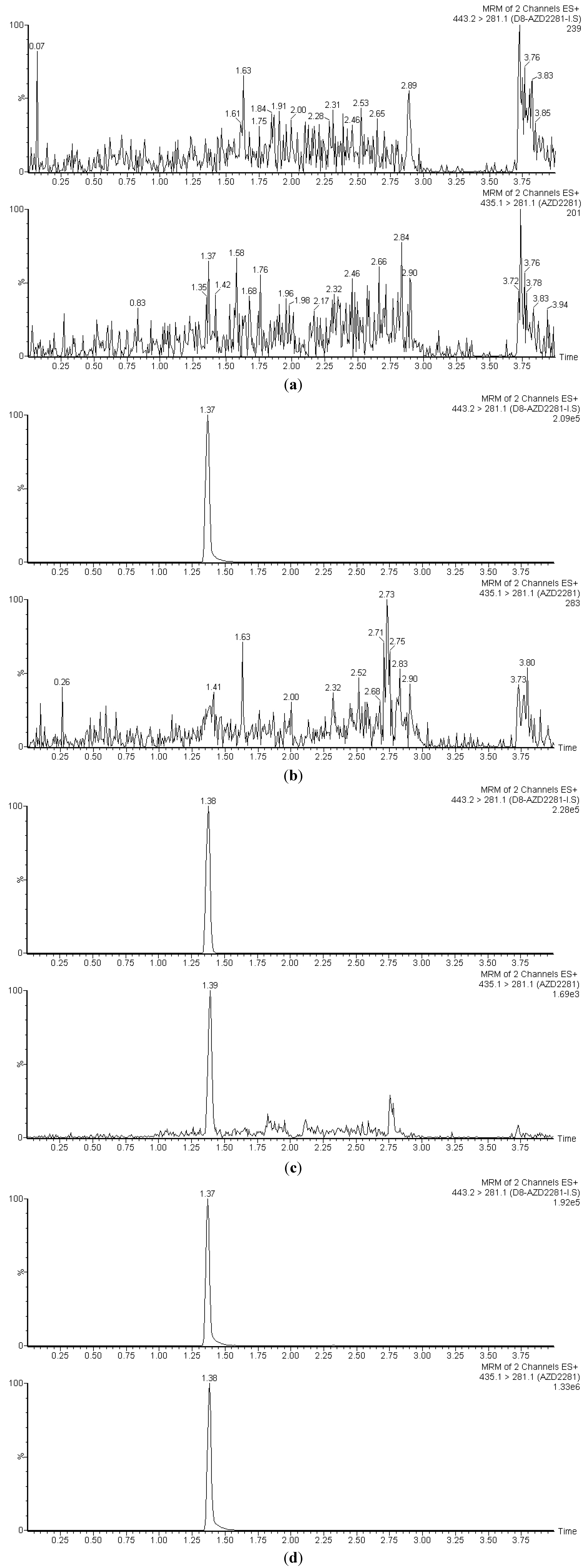
3.2. Linearity, Accuracy and Precision

{kind=link}
{kind=link}
{kind=link}
| Nominal (ng/mL) | GM (ng/mL) | SD (ng/mL) | DEV (%) | CV (%) | n |
|---|---|---|---|---|---|
| 0.50 | 0.52 | 0.06 | 3.00 | 11.88 | 8 |
| 5.0 | 5.38 | 0.31 | 7.50 | 5.78 | 8 |
| 25 | 24.99 | 1.17 | −0.04 | 4.67 | 8 |
| 100 | 104.30 | 2.58 | 4.30 | 2.47 | 8 |
| 500 | 494.13 | 16.09 | −1.17 | 3.26 | 8 |
| 1000 | 1001.39 | 36.39 | 0.14 | 3.63 | 8 |
| 2000 | 1963.61 | 74.09 | −1.82 | 3.77 | 8 |
| 5000 | 4594.08 | 94.75 | −8.12 | 2.06 | 8 |
| Nominal (ng/mL) | GM (ng/mL) | SD (ng/mL) | DEV (%) | WRP (%) | BRP (%) | n |
|---|---|---|---|---|---|---|
| 0.5 (LLOQ) | 0.49 | 0.05 | −2.60 | 9.10 | 1.75 | 20 |
| 1.0 (LQC) | 1.01 | 0.12 | 1.10 | 10.79 | 3.65 | 20 |
| 900 (MQC) | 876.89 | 39.72 | −2.57 | 1.53 | 2.77 | 20 |
| 4500 (HQC) | 4105.04 | 133.88 | −8.78 | 1.59 | 1.85 | 20 |
| 50,000 (DQC) | 48,028.85 | 1397.15 | −3.94 | 2.20 | 1.24 | 20 |
3.3. Stability
| 6.50 ng/mL (low) | 150 ng/mL (mid) | 1600 (high) | ||||
|---|---|---|---|---|---|---|
| Freeze/Thaw Cycles | GM (ng/mL) | DEV from Fresh (%) | GM (ng/mL) | DEV from Fresh (%) | GM (ng/mL) | DEV from Fresh (%) |
| 0 (Fresh) | 5.94 | - | 149.52 | - | 1489.40 | - |
| 1 | 6.10 | 2.69 | 146.70 | −1.89 | 1512.20 | 1.53 |
| 2 | 6.22 | 4.71 | 156.40 | 4.60 | 1606.48 | 7.86 |
| 3 | 6.50 | 9.43 | 153.66 | 2.77 | 1572.46 | 5.58 |
3.4. Extraction Recovery and Matrix Effects
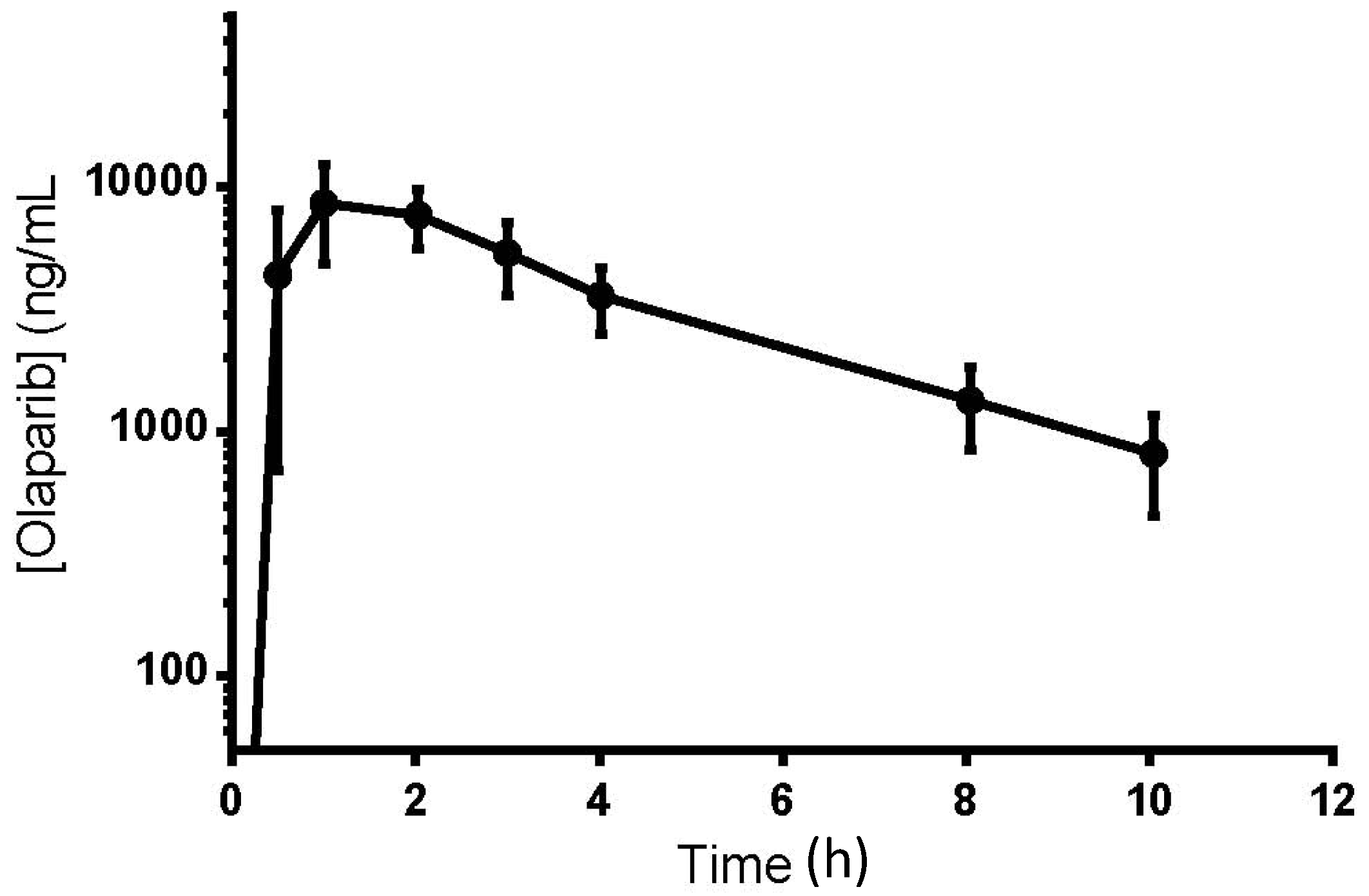
3.5. Clinical Application
4. Conclusions
Acknowledgments
Authors Contributions
Disclaimer
Conflicts of Interest
References
- Dantzer, F.; de La Rubia, G.; Menissier-de Murcia, J.; Hostomsky, Z.; de Murcia, G.; Schreiber, V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry 2000, 39, 7559–7569. [Google Scholar]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 75, 5588–5599. [Google Scholar]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Bundred, N.; Gardovskis, J.; Jaskiewicz, J.; Eglitis, J.; Paramonov, V.; McCormack, P.; Swaisland, H.; Cavallin, M.; Parry, T.; Carmichael, J.; et al. Evaluation of the pharmacodynamics and pharmacokinetics of the PARP inhibitor olaparib: A Phase I multicentre trial in patients scheduled for elective breast cancer surgery. Investig. New Drugs 2013, 31, 949–958. [Google Scholar] [CrossRef]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, L.; Hao, Q. Olaparib: A promising PARP inhibitor in ovarian cancer therapy. Arch. Gynecol. Obstet. 2013, 288, 367–374. [Google Scholar] [CrossRef]
- Kaye, S.B.; Lubinski, J.; Matulonis, U.; Ang, J.E.; Gourley, C.; Karlan, B.Y.; Amnon, A.; Bell-McGuinn, K.M.; Chen, L.M.; Friedlander, M.; et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J. Clin. Oncol. 2012, 30, 372–379. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef]
- Liu, J.F.; Tolaney, S.M.; Birrer, M.; Fleming, G.F.; Buss, M.K.; Dahlberg, S.E.; Lee, H.; Whalen, C.; Tyburski, K.; Winer, E.; et al. A Phase 1 trial of the poly(ADP-ribose) polymerase inhibitor olaparib (AZD2281) in combination with the anti-angiogenic cediranib (AZD2171) in recurrent epithelial ovarian or triple-negative breast cancer. Eur. J. Cancer 2013, 49, 2972–2978. [Google Scholar] [CrossRef]
- Moon, D.H.; Lee, J.M.; Noonan, A.M.; Annunziata, C.M.; Minasian, L.; Houston, N.; Hays, J.L.; Kohn, E.C. Deleterious BRCA1/2 mutation is an independent risk factor for carboplatin hypersensitivity reactions. Br. J. Cancer 2013, 109, 1072–1078. [Google Scholar] [CrossRef]
- Miura, K.; Sakata, K.; Someya, M.; Matsumoto, Y.; Matsumoto, H.; Takahashi, A.; Hareyama, M. The combination of olaparib and camptothecin for effective radiosensitization. Radiat. Oncol. 2012, 7, 62. [Google Scholar] [CrossRef]
- Senra, J.M.; Telfer, B.A.; Cherry, K.E.; McCrudden, C.M.; Hirst, D.G.; O’Connor, M.J.; Wedge, S.R.; Stratford, I.J. Inhibition of PARP-1 by olaparib (AZD2281) increases the radiosensitivity of a lung tumor xenograft. Mol. Cancer Ther. 2011, 10, 1949–1958. [Google Scholar] [CrossRef]
- Yamamoto, N.; Nokihara, H.; Yamada, Y.; Goto, Y.; Tanioka, M.; Shibata, T.; Yamada, K.; Asahina, H.; Kawata, T.; Shi, X.; et al. A Phase I, dose-finding and pharmacokinetic study of olaparib (AZD2281) in Japanese patients with advanced solid tumors. Cancer Sci. 2012, 103, 504–509. [Google Scholar] [CrossRef]
- Khan, O.A.; Gore, M.; Lorigan, P.; Stone, J.; Greystoke, A.; Burke, W.; Carmichael, J.; Watson, A.J.; McGown, G.; Thorncroft, M.; et al. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br. J. Cancer 2011, 104, 750–755. [Google Scholar] [CrossRef]
- Samol, J.; Ranson, M.; Scott, E.; Macpherson, E.; Carmichael, J.; Thomas, A.; Cassidy, J. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: A phase I study. Investig. New Drugs 2012, 30, 1493–1500. [Google Scholar] [CrossRef]
- Dean, E.; Middleton, M.R.; Pwint, T.; Swaisland, H.; Carmichael, J.; Goodege-Kunwar, P.; Ranson, M. Phase I study to assess the safety and tolerability of olaparib in combination with bevacizumab in patients with advanced solid tumours. Br. J Cancer 2012, 106, 468–474. [Google Scholar] [CrossRef]
- Rajan, A.; Carter, C.A.; Kelly, R.J.; Gutierrez, M.; Kummar, S.; Szabo, E.; Yancey, M.A.; Ji, J.; Mannargudi, B.; Woo, S.; et al. A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin. Cancer Res. 2012, 18, 2344–2351. [Google Scholar] [CrossRef]
- Nijenhuis, C.M.; Lucas, L.; Rosing, H.; Schellens, J.H.M.; Beijnen, J.H. Development and validation of high-performance liquid chromatography-tandem mass spectrometry assay quantifying olaparib in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 940, 121–125. [Google Scholar] [CrossRef]
- Sparidans, R.W.; Martens, I.; Valkenburg-van Iersel, L.B.; den Hartigh, J.; Schellens, J.H.; Beijnen, J.H. Liquid chromatography-tandem mass spectrometric assay for the PARP-1 inhibitor olaparib in combination with the nitrogen mustard melphalan in human plasma. J. Chromatogr. B Analy. Technol. Biomed. Life Sci. 2011, 879, 1851–1856. [Google Scholar]
- U.S. Food and Drug Administration. Guidance for Industry: Bioanalytical Method Validation; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2001.
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Roth, J.; Peer, C.J.; Mannargudi, B.; Swaisland, H.; Lee, J.-M.; Kohn, E.C.; Figg, W.D. A Sensitive and Robust Ultra HPLC Assay with Tandem Mass Spectrometric Detection for the Quantitation of the PARP Inhibitor Olaparib (AZD2281) in Human Plasma for Pharmacokinetic Application. Chromatography 2014, 1, 82-95. https://doi.org/10.3390/chromatography1020082
Roth J, Peer CJ, Mannargudi B, Swaisland H, Lee J-M, Kohn EC, Figg WD. A Sensitive and Robust Ultra HPLC Assay with Tandem Mass Spectrometric Detection for the Quantitation of the PARP Inhibitor Olaparib (AZD2281) in Human Plasma for Pharmacokinetic Application. Chromatography. 2014; 1(2):82-95. https://doi.org/10.3390/chromatography1020082
Chicago/Turabian StyleRoth, Jeffrey, Cody J. Peer, Baskar Mannargudi, Helen Swaisland, Jung-Min Lee, Elise C. Kohn, and William D. Figg. 2014. "A Sensitive and Robust Ultra HPLC Assay with Tandem Mass Spectrometric Detection for the Quantitation of the PARP Inhibitor Olaparib (AZD2281) in Human Plasma for Pharmacokinetic Application" Chromatography 1, no. 2: 82-95. https://doi.org/10.3390/chromatography1020082
APA StyleRoth, J., Peer, C. J., Mannargudi, B., Swaisland, H., Lee, J.-M., Kohn, E. C., & Figg, W. D. (2014). A Sensitive and Robust Ultra HPLC Assay with Tandem Mass Spectrometric Detection for the Quantitation of the PARP Inhibitor Olaparib (AZD2281) in Human Plasma for Pharmacokinetic Application. Chromatography, 1(2), 82-95. https://doi.org/10.3390/chromatography1020082