
Long-Term Study on Therapeutic Strategy for Treatment of Eisenmenger Syndrome Patients: A Case Series Study

 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
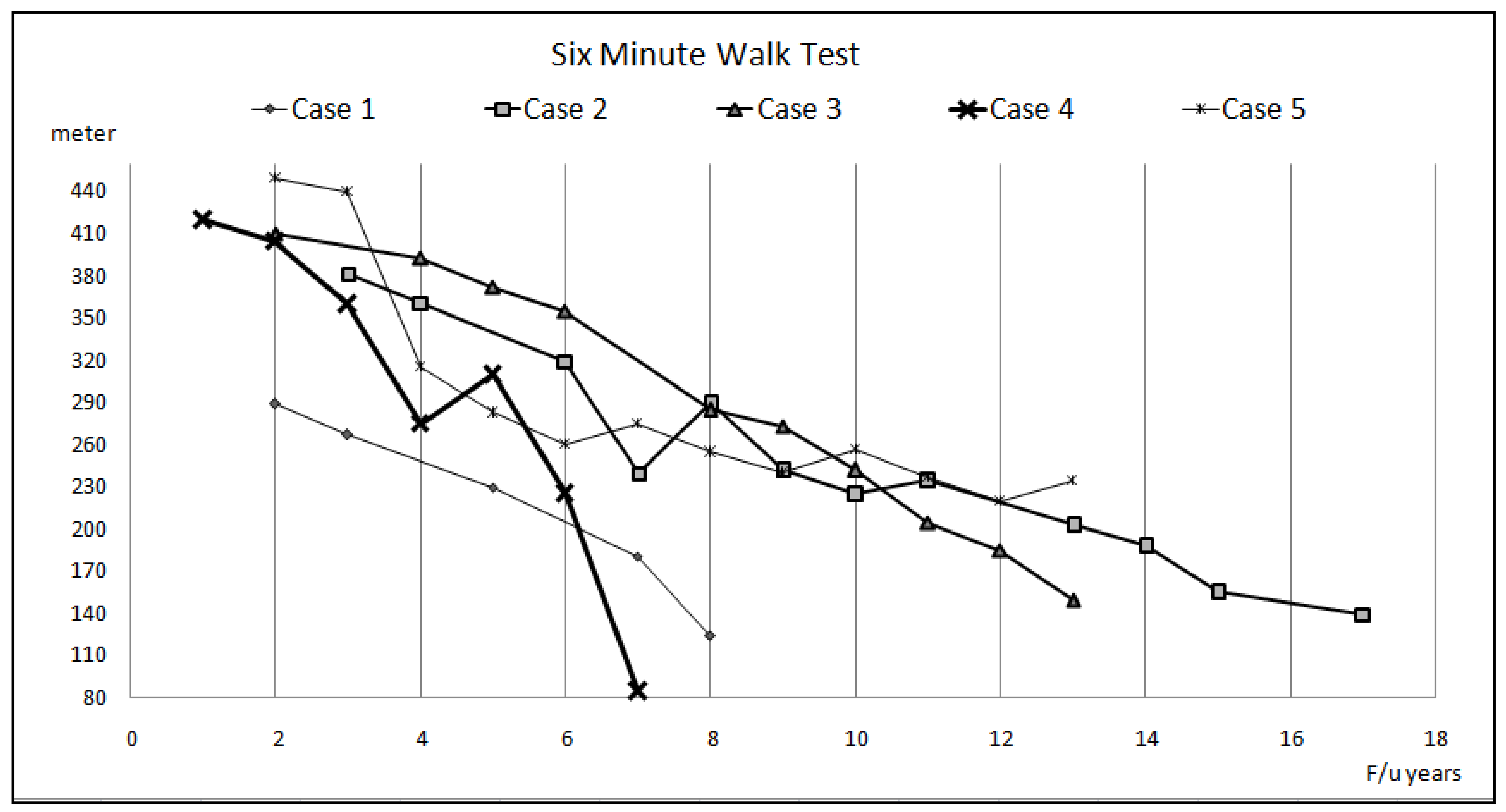
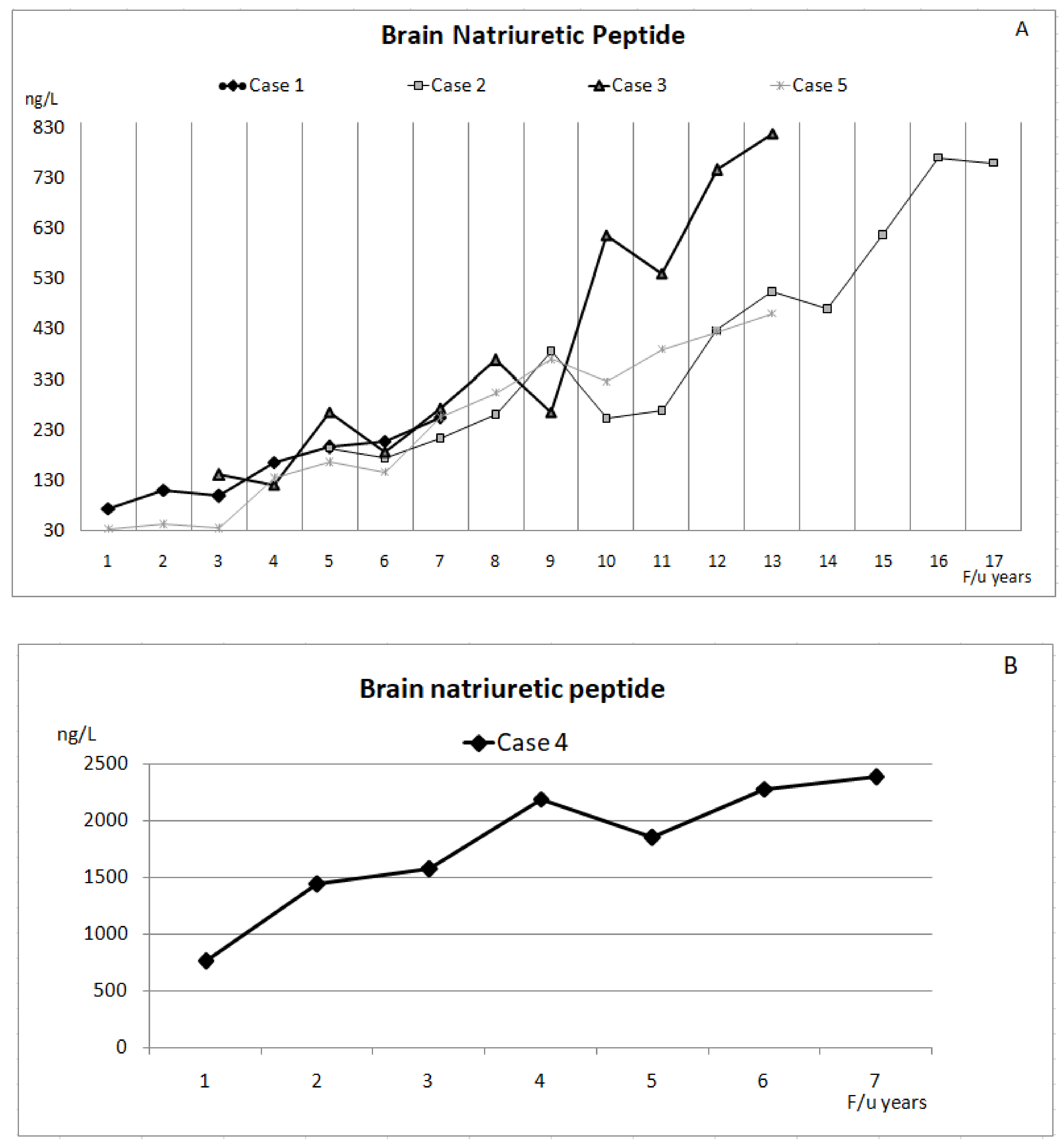
3. Results
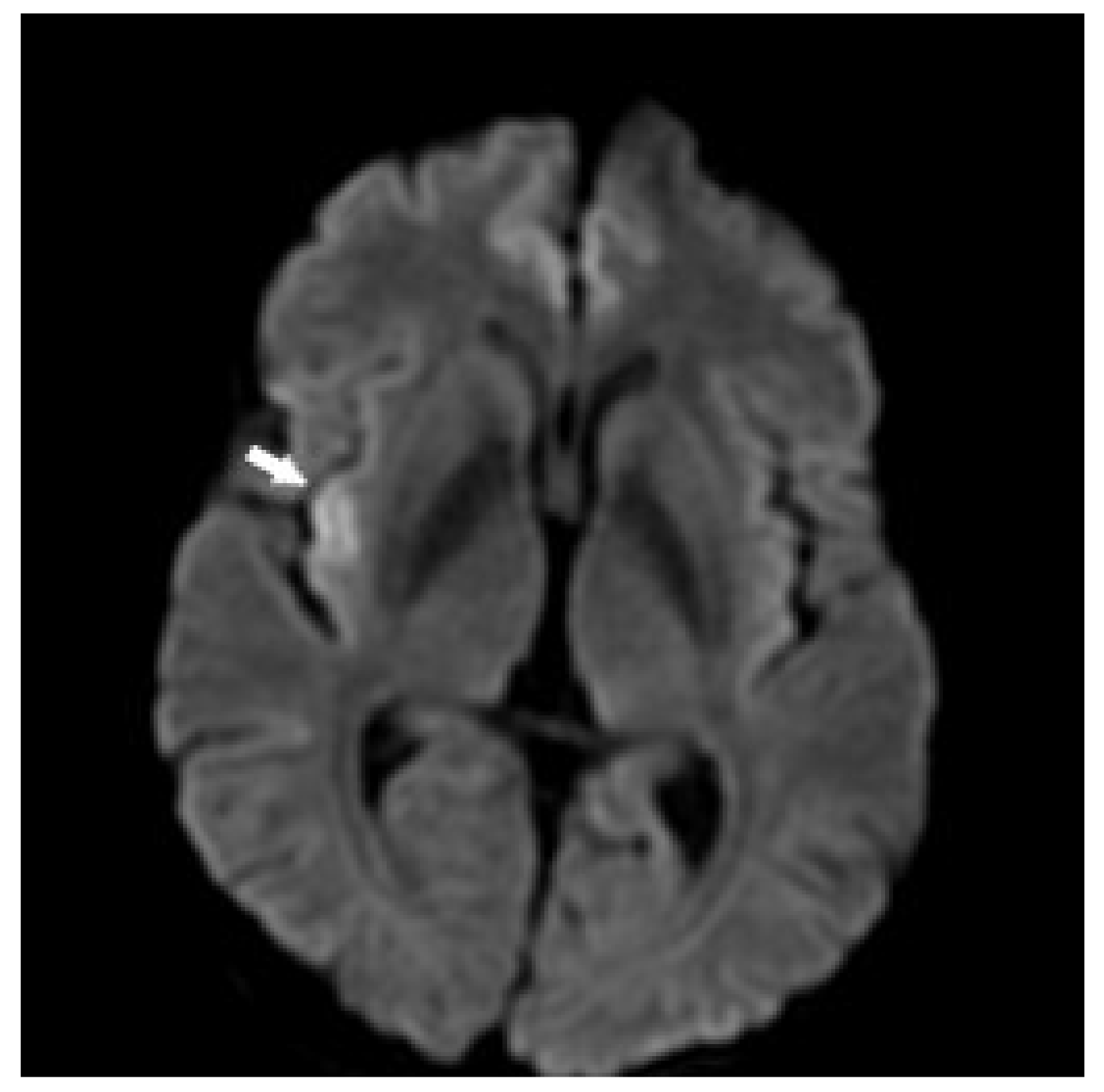
3.1. Case 1
3.2. Case 2
3.3. Case 3
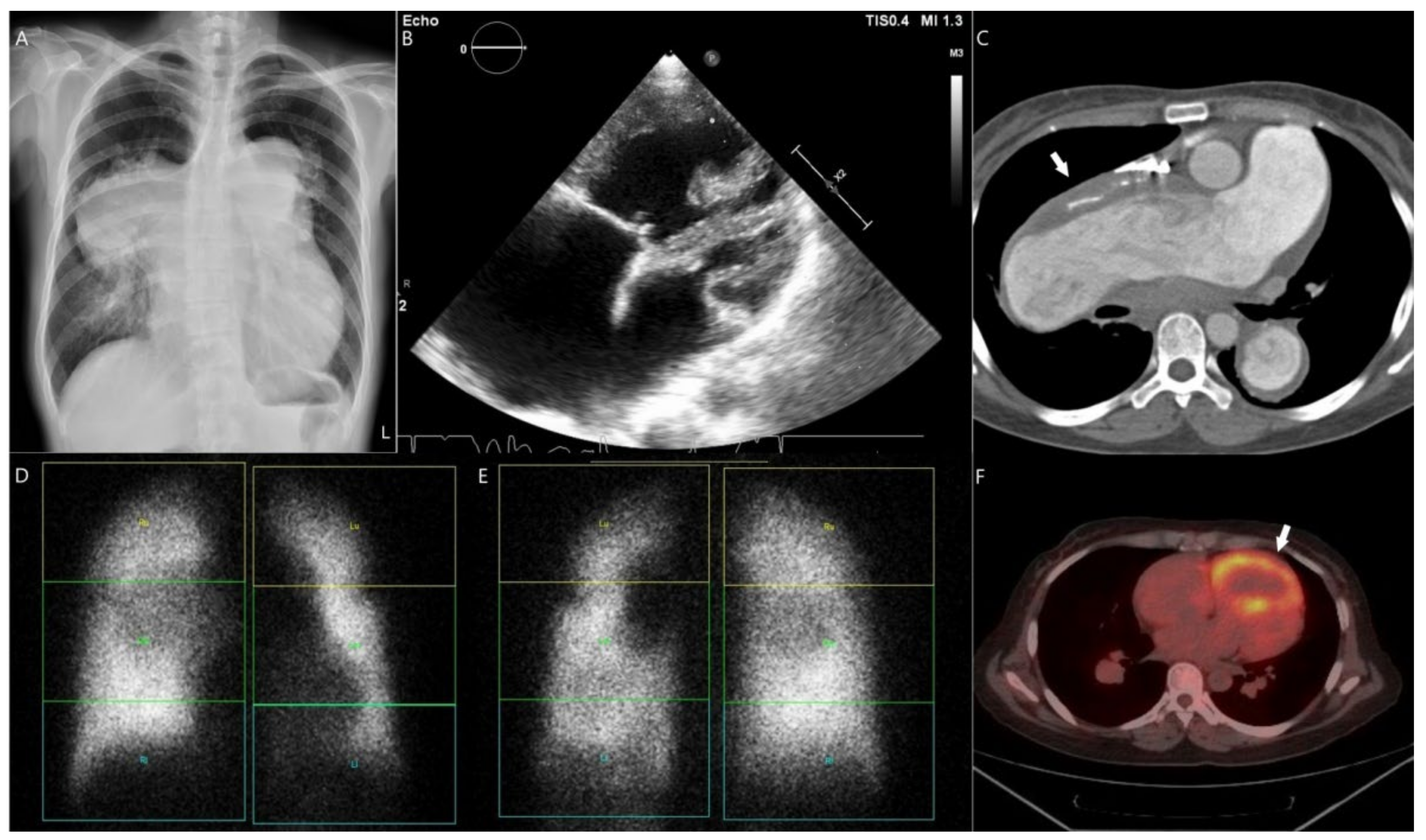
3.4. Case 4
3.5. Case 5
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Arvanitaki, A.; Giannakoulas, G.; Baumgartner, H.; Lammers, A.E. Eisenmenger syndrome: Diagnosis, prognosis and clinical management. Heart 2020, 106, 1638–1645. [Google Scholar] [CrossRef] [PubMed]
- Amaducci, A.; Vitulo, P. Stenting of multiple peripheral pulmonary stenosis with the coronary technique in an adul patient with eisenmenger-like syndrome. G. Ital. Cardiol. 2017, 18, 33S–36S. [Google Scholar]
- Chen, I.C.; Dai, Z.K. Insight into pulmonary arterial hypertension associated with congenital heart disease (pah-chd): Classification and pharmacological management from a pediatric cardiological point of view. Acta Cardiol. Sin. 2015, 31, 507–515. [Google Scholar] [PubMed]
- Diller, G.P.; Gatzoulis, M.A. Pulmonary vascular disease in adults with congenital heart disease. Circulation 2007, 115, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 esc/ers guidelines for the diagnosis and treatment of pulmonary hypertension: The joint task force for the diagnosis and treatment of pulmonary hypertension of the european society of cardiology (esc) and the european respiratory society (ers): Endorsed by: Association for european paediatric and congenital cardiology (aepc), international society for heart and lung transplantation (ishlt). Eur. Heart J. 2016, 37, 67–119. [Google Scholar]
- Hjortshoj, C.S.; Gilljam, T.; Dellgren, G.; Pentikainen, M.O.; Moller, T.; Jensen, A.S.; Turanlahti, M.; Thilen, U.; Gustafsson, F.; Sondergaard, L. Outcome after heart-lung or lung transplantation in patients with eisenmenger syndrome. Heart 2020, 106, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Idrees, J.J.; Pettersson, G.B. State of the art of combined heart-lung transplantation for advanced cardiac and pulmonary dysfunction. Curr. Cardiol. Rep. 2016, 18, 36. [Google Scholar] [CrossRef] [PubMed]
- Iversen, K.; Jensen, A.S.; Jensen, T.V.; Vejlstrup, N.G.; Sondergaard, L. Combination therapy with bosentan and sildenafil in eisenmenger syndrome: A randomized, placebo-controlled, double-blinded trial. Eur. Heart J. 2010, 31, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Ereminiene, E.; Kinderyte, M.; Miliauskas, S. Impact of advanced medical therapy for the outcome of an adult patient with eisenmenger syndrome. Respir. Med. Case Rep. 2017, 21, 16–20. [Google Scholar] [CrossRef]
- Chaix, M.A.; Gatzoulis, M.A.; Diller, G.P.; Khairy, P.; Oechslin, E.N. Eisenmenger syndrome: A multisystem disorder-do not destabilize the balanced but fragile physiology. Can. J. Cardiol. 2019, 35, 1664–1674. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, R.L.; Roofthooft, M.T.; Hillege, H.L.; ten Harkel, A.D.; van Osch-Gevers, M.; Delhaas, T.; Kapusta, L.; Strengers, J.L.; Rammeloo, L.; Clur, S.A.; et al. Pediatric pulmonary hypertension in the netherlands: Epidemiology and characterization during the period 1991 to 2005. Circulation 2011, 124, 1755–1764. [Google Scholar] [CrossRef]
- Haworth, S.G.; Hislop, A.A. Treatment and survival in children with pulmonary arterial hypertension: The uk pulmonary hypertension service for children 2001–2006. Heart 2009, 95, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Diller, G.P.; Korten, M.A.; Bauer, U.M.; Miera, O.; Tutarel, O.; Kaemmerer, H.; Berger, F.; Baumgartner, H.; German Competence Network for Congenital Heart Defects, I. Current therapy and outcome of eisenmenger syndrome: Data of the german national register for congenital heart defects. Eur. Heart J. 2016, 37, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Hjortshoj, C.M.S.; Kempny, A.; Jensen, A.S.; Sorensen, K.; Nagy, E.; Dellborg, M.; Johansson, B.; Rudiene, V.; Hong, G.; Opotowsky, A.R.; et al. Past and current cause-specific mortality in eisenmenger syndrome. Eur. Heart J. 2017, 38, 2060–2067. [Google Scholar] [CrossRef]
- Barst, R.J.; Ivy, D.D.; Foreman, A.J.; McGoon, M.D.; Rosenzweig, E.B. Four- and seven-year outcomes of patients with congenital heart disease-associated pulmonary arterial hypertension (from the reveal registry). Am. J. Cardiol. 2014, 113, 147–155. [Google Scholar] [CrossRef]
- Moceri, P.; Bouvier, P.; Baudouy, D.; Dimopoulos, K.; Cerboni, P.; Wort, S.J.; Doyen, D.; Schouver, E.D.; Gibelin, P.; Senior, R.; et al. Cardiac remodelling amongst adults with various aetiologies of pulmonary arterial hypertension including eisenmenger syndrome-implications on survival and the role of right ventricular transverse strain. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 1262–1270. [Google Scholar] [CrossRef]
- Arvanitaki, A.; Gatzoulis, M.A.; Opotowsky, A.R.; Khairy, P.; Dimopoulos, K.; Diller, G.P.; Giannakoulas, G.; Brida, M.; Griselli, M.; Grunig, E.; et al. Eisenmenger syndrome: Jacc state-of-the-art review. J. Am. Coll. Cardiol. 2022, 79, 1183–1198. [Google Scholar] [CrossRef]
- Perloff, J.K.; Hart, E.M.; Greaves, S.M.; Miner, P.D.; Child, J.S. Proximal pulmonary arterial and intrapulmonary radiologic features of eisenmenger syndrome and primary pulmonary hypertension. Am. J. Cardiol. 2003, 92, 182–187. [Google Scholar] [CrossRef]
- Silversides, C.K.; Granton, J.T.; Konen, E.; Hart, M.A.; Webb, G.D.; Therrien, J. Pulmonary thrombosis in adults with eisenmenger syndrome. J. Am. Coll. Cardiol. 2003, 42, 1982–1987. [Google Scholar] [CrossRef]
- Daliento, L.; Somerville, J.; Presbitero, P.; Menti, L.; Brach-Prever, S.; Rizzoli, G.; Stone, S. Eisenmenger syndrome. Factors relating to deterioration and death. Eur. Heart J. 1998, 19, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Broberg, C.S.; Ujita, M.; Prasad, S.; Li, W.; Rubens, M.; Bax, B.E.; Davidson, S.J.; Bouzas, B.; Gibbs, J.S.; Burman, J.; et al. Pulmonary arterial thrombosis in eisenmenger syndrome is associated with biventricular dysfunction and decreased pulmonary flow velocity. J. Am. Coll. Cardiol. 2007, 50, 634–642. [Google Scholar] [CrossRef]
- Sandoval, J.; Santos, L.E.; Cordova, J.; Pulido, T.; Gutierrez, G.; Bautista, E.; Martinez Guerra, M.L.; Pena, H.; Broberg, C.S. Does anticoagulation in eisenmenger syndrome impact long-term survival? Congenit. Heart Dis. 2012, 7, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, H.; De Backer, J.; Babu-Narayan, S.V.; Budts, W.; Chessa, M.; Diller, G.P.; Lung, B.; Kluin, J.; Lang, I.M.; Meijboom, F.; et al. 2020 esc guidelines for the management of adult congenital heart disease. Eur. Heart J. 2021, 42, 563–645. [Google Scholar] [CrossRef] [PubMed]
- Shen, A.Y.; Yao, J.F.; Brar, S.S.; Jorgensen, M.B.; Chen, W. Racial/ethnic differences in the risk of intracranial hemorrhage among patients with atrial fibrillation. J. Am. Coll. Cardiol. 2007, 50, 309–315. [Google Scholar] [CrossRef]
- Chiang, C.E.; Wu, T.J.; Ueng, K.C.; Chao, T.F.; Chang, K.C.; Wang, C.C.; Lin, Y.J.; Yin, W.H.; Kuo, J.Y.; Lin, W.S.; et al. 2016 guidelines of the taiwan heart rhythm society and the taiwan society of cardiology for the management of atrial fibrillation. J. Formos. Med. Assoc. 2016, 115, 893–952. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.S.; Idorn, L.; Thomsen, C.; von der Recke, P.; Mortensen, J.; Sorensen, K.E.; Thilen, U.; Nagy, E.; Kofoed, K.F.; Ostrowski, S.R.; et al. Prevalence of cerebral and pulmonary thrombosis in patients with cyanotic congenital heart disease. Heart 2015, 101, 1540–1546. [Google Scholar] [CrossRef]
- Hayabuchi, Y.; Matsuoka, S.; Akita, H.; Kuroda, Y. Hyperuricaemia in cyanotic congenital heart disease. Eur. J. Pediatr. 1993, 152, 873–876. [Google Scholar] [CrossRef]
- Niwa, K.; Perloff, J.K.; Kaplan, S.; Child, J.S.; Miner, P.D. Eisenmenger syndrome in adults: Ventricular septal defect, truncus arteriosus, univentricular heart. J. Am. Coll. Cardiol. 1999, 34, 223–232. [Google Scholar] [CrossRef]
- Oya, H.; Nagaya, N.; Satoh, T.; Sakamaki, F.; Kyotani, S.; Fujita, M.; Nakanishi, N.; Miyatake, K. Haemodynamic correlates and prognostic significance of serum uric acid in adult patients with eisenmenger syndrome. Heart 2000, 84, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, A.; Ohira, H.; Mielniczuk, L.M. Fdg pet imaging for identifying pulmonary hypertension and right heart failure. Curr. Cardiol. Rep. 2015, 17, 555. [Google Scholar] [CrossRef] [PubMed]
- Mielniczuk, L.M.; Birnie, D.; Ziadi, M.C.; deKemp, R.A.; DaSilva, J.N.; Burwash, I.; Tang, A.T.; Davies, R.A.; Haddad, H.; Guo, A.; et al. Relation between right ventricular function and increased right ventricular [18f]fluorodeoxyglucose accumulation in patients with heart failure. Circ. Cardiovasc. Imaging 2011, 4, 59–66. [Google Scholar] [CrossRef]
- Oikawa, M.; Kagaya, Y.; Otani, H.; Sakuma, M.; Demachi, J.; Suzuki, J.; Takahashi, T.; Nawata, J.; Ido, T.; Watanabe, J.; et al. Increased [18f]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J. Am. Coll. Cardiol. 2005, 45, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Can, M.M.; Kaymaz, C.; Tanboga, I.H.; Tokgoz, H.C.; Canpolat, N.; Turkyilmaz, E.; Sonmez, K.; Ozdemir, N. Increased right ventricular glucose metabolism in patients with pulmonary arterial hypertension. Clin. Nucl. Med. 2011, 36, 743–748. [Google Scholar] [CrossRef]
- Galie, N.; Hoeper, M.M.; Humbert, M.; Torbicki, A.; Vachiery, J.L.; Barbera, J.A.; Beghetti, M.; Corris, P.; Gaine, S.; Gibbs, J.S.; et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The task force for the diagnosis and treatment of pulmonary hypertension of the european society of cardiology (esc) and the european respiratory society (ers), endorsed by the international society of heart and lung transplantation (ishlt). Eur. Heart J. 2009, 30, 2493–2537. [Google Scholar] [PubMed]
- McLaughlin, V.V.; Archer, S.L.; Badesch, D.B.; Barst, R.J.; Farber, H.W.; Lindner, J.R.; Mathier, M.A.; McGoon, M.D.; Park, M.H.; Rosenson, R.S.; et al. Accf/aha 2009 expert consensus document on pulmonary hypertension: A report of the american college of cardiology foundation task force on expert consensus documents and the american heart association: Developed in collaboration with the american college of chest physicians, american thoracic society, inc., and the pulmonary hypertension association. Circulation 2009, 119, 2250–2294. [Google Scholar]
- Galie, N.; Beghetti, M.; Gatzoulis, M.A.; Granton, J.; Berger, R.M.; Lauer, A.; Chiossi, E.; Landzberg, M.; Bosentan Randomized Trial of Endothelin Antagonist Therapy, I. Bosentan therapy in patients with eisenmenger syndrome: A multicenter, double-blind, randomized, placebo-controlled study. Circulation 2006, 114, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Chen, J.; Chen, C.; Zheng, H.; Chen, Y.; Liu, M.; Zheng, B. Systematic review and cost-effectiveness of bosentan and sildenafil as therapeutic drugs for pediatric pulmonary arterial hypertension. Pediatr. Pulmonol. 2021, 56, 2250–2258. [Google Scholar] [CrossRef]
- Zhu, N.; Welch, C.L.; Wang, J.; Allen, P.M.; Gonzaga-Jauregui, C.; Ma, L.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D.; et al. Rare variants in sox17 are associated with pulmonary arterial hypertension with congenital heart disease. Genome. Med. 2018, 10, 56. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Sex | Dx | PET/CT | Tx | Complication | F/u(Yr) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| RV (SUV) | RV/LV | Thrombus In PA | AF | UA | Others | |||||
| 1 | M | VSDII | S, B, I | Bil | - | I | Stroke | 8 | ||
| 2 | F | ASDII | 12.7 | 1.65 | S, B, I | Bil | + | I | - | 17 |
| 3 | F | ASDI | 6.2 | 1.44 | S, B, I | RPA | + | N | Stroke | 13 |
| 4 | F | ASDII | 5.8 | 2.15 | S, B, I | None | - | I | - | 7 |
| 5 | M | ASDII | 12.7 | 1.31 | S, B | LPA | + | I | - | 13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.-C.; Chen, Y.-W.; Chen, I.-C.; Wu, Y.-H.; Lo, S.-H.; Hsu, J.-S.; Hsu, J.-H.; Wu, B.-N.; Cheng, Y.-F.; Dai, Z.-K. Long-Term Study on Therapeutic Strategy for Treatment of Eisenmenger Syndrome Patients: A Case Series Study. Children 2022, 9, 1217. https://doi.org/10.3390/children9081217
Liu Y-C, Chen Y-W, Chen I-C, Wu Y-H, Lo S-H, Hsu J-S, Hsu J-H, Wu B-N, Cheng Y-F, Dai Z-K. Long-Term Study on Therapeutic Strategy for Treatment of Eisenmenger Syndrome Patients: A Case Series Study. Children. 2022; 9(8):1217. https://doi.org/10.3390/children9081217
Chicago/Turabian StyleLiu, Yi-Ching, Yu-Wen Chen, I-Chen Chen, Yen-Hsien Wu, Shih-Hsing Lo, Jui-Sheng Hsu, Jong-Hau Hsu, Bin-Nan Wu, Yi-Fang Cheng, and Zen-Kong Dai. 2022. "Long-Term Study on Therapeutic Strategy for Treatment of Eisenmenger Syndrome Patients: A Case Series Study" Children 9, no. 8: 1217. https://doi.org/10.3390/children9081217
APA StyleLiu, Y.-C., Chen, Y.-W., Chen, I.-C., Wu, Y.-H., Lo, S.-H., Hsu, J.-S., Hsu, J.-H., Wu, B.-N., Cheng, Y.-F., & Dai, Z.-K. (2022). Long-Term Study on Therapeutic Strategy for Treatment of Eisenmenger Syndrome Patients: A Case Series Study. Children, 9(8), 1217. https://doi.org/10.3390/children9081217