
Cardiac Rhabdoid Tumor—A Rare Foe—Case Report and Literature Review

 , , ,
, , ,  , , ,
, , ,
Abstract
:1. Introduction
1.1. Benign Cardiac Tumors in Infants and Children
1.2. Malignant Tumors of the Heart in Infants and Children—Extrarenal Rhabdoid Tumor
1.3. Diagnosis and Classification
2. Material and Methods
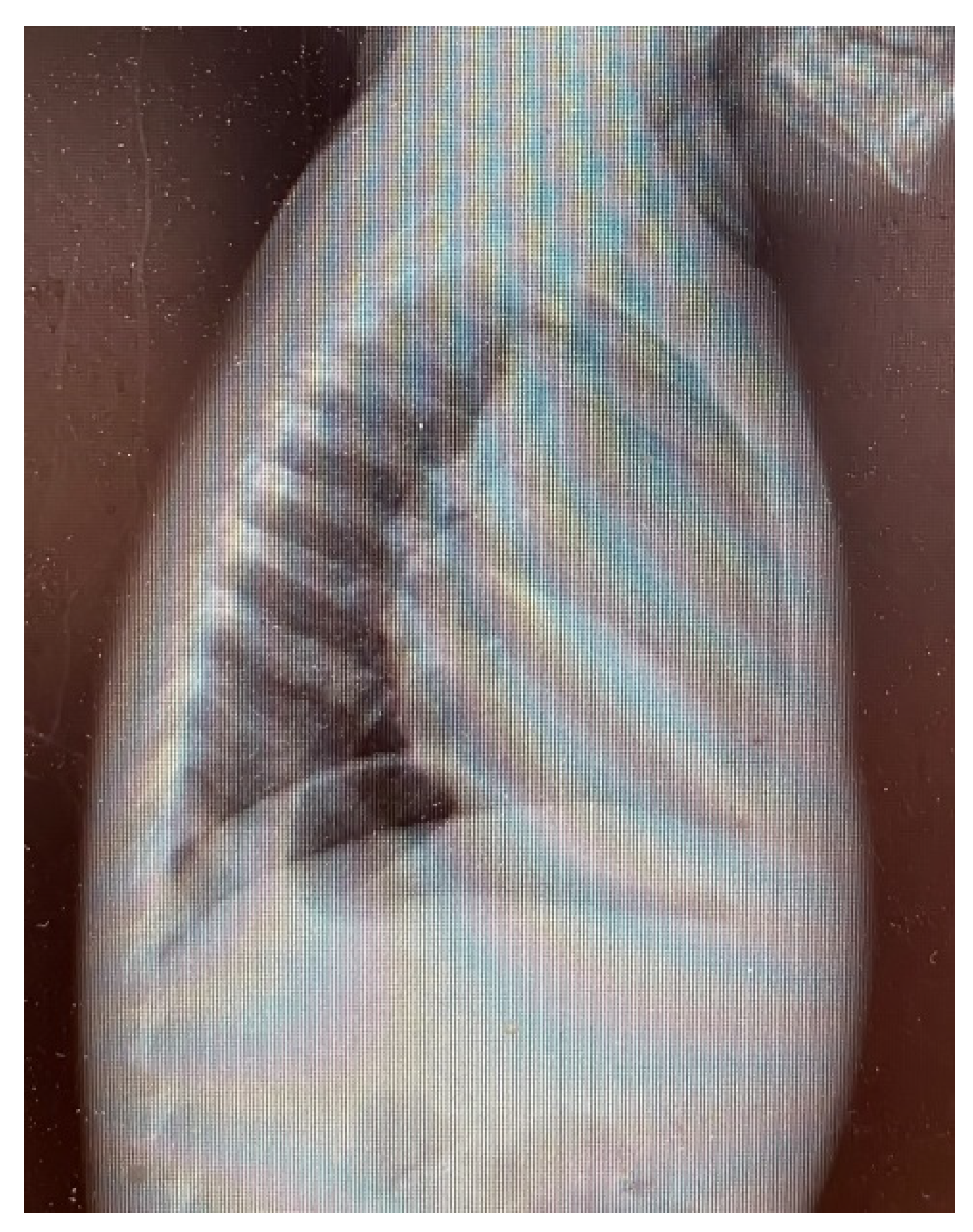
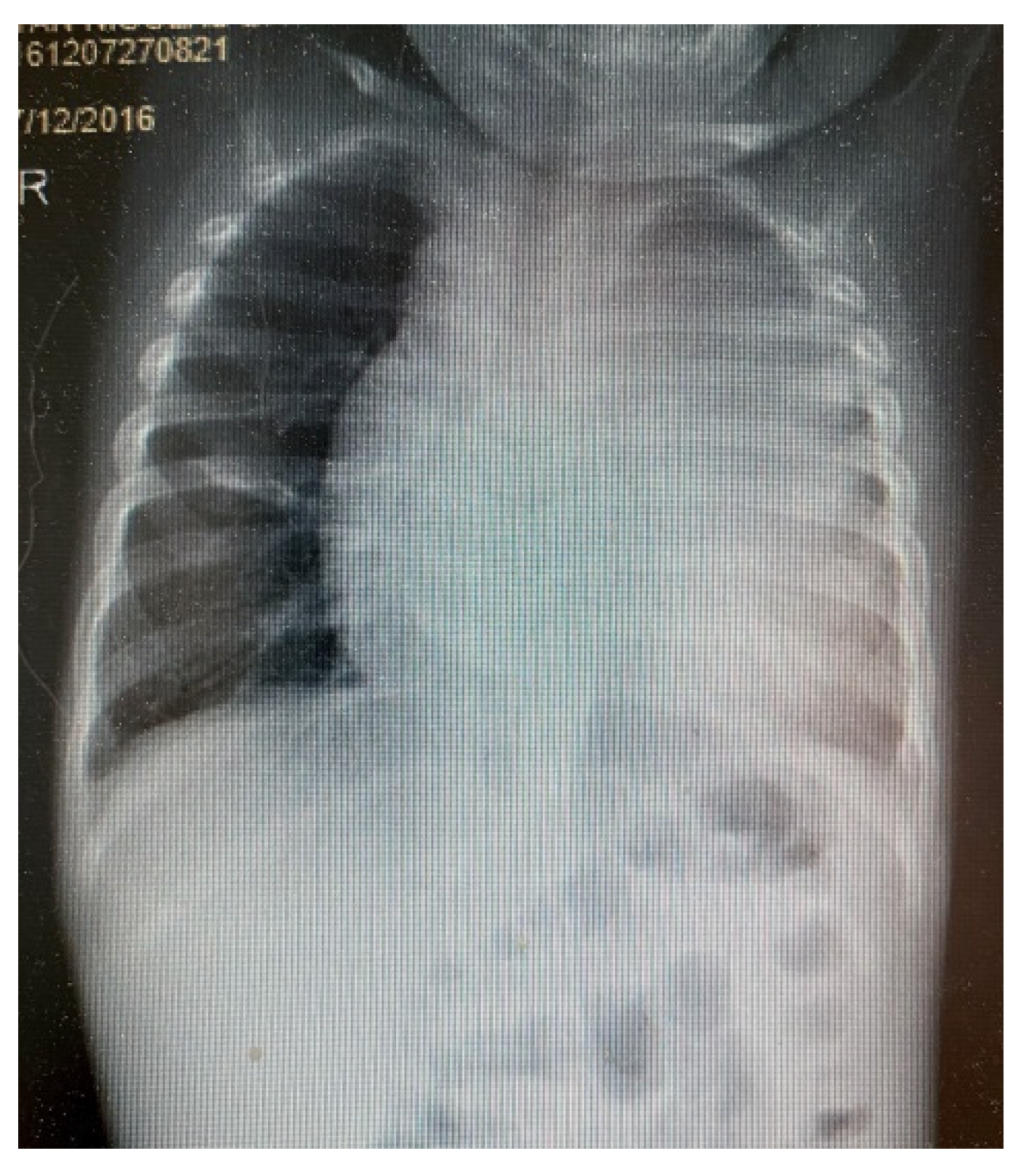
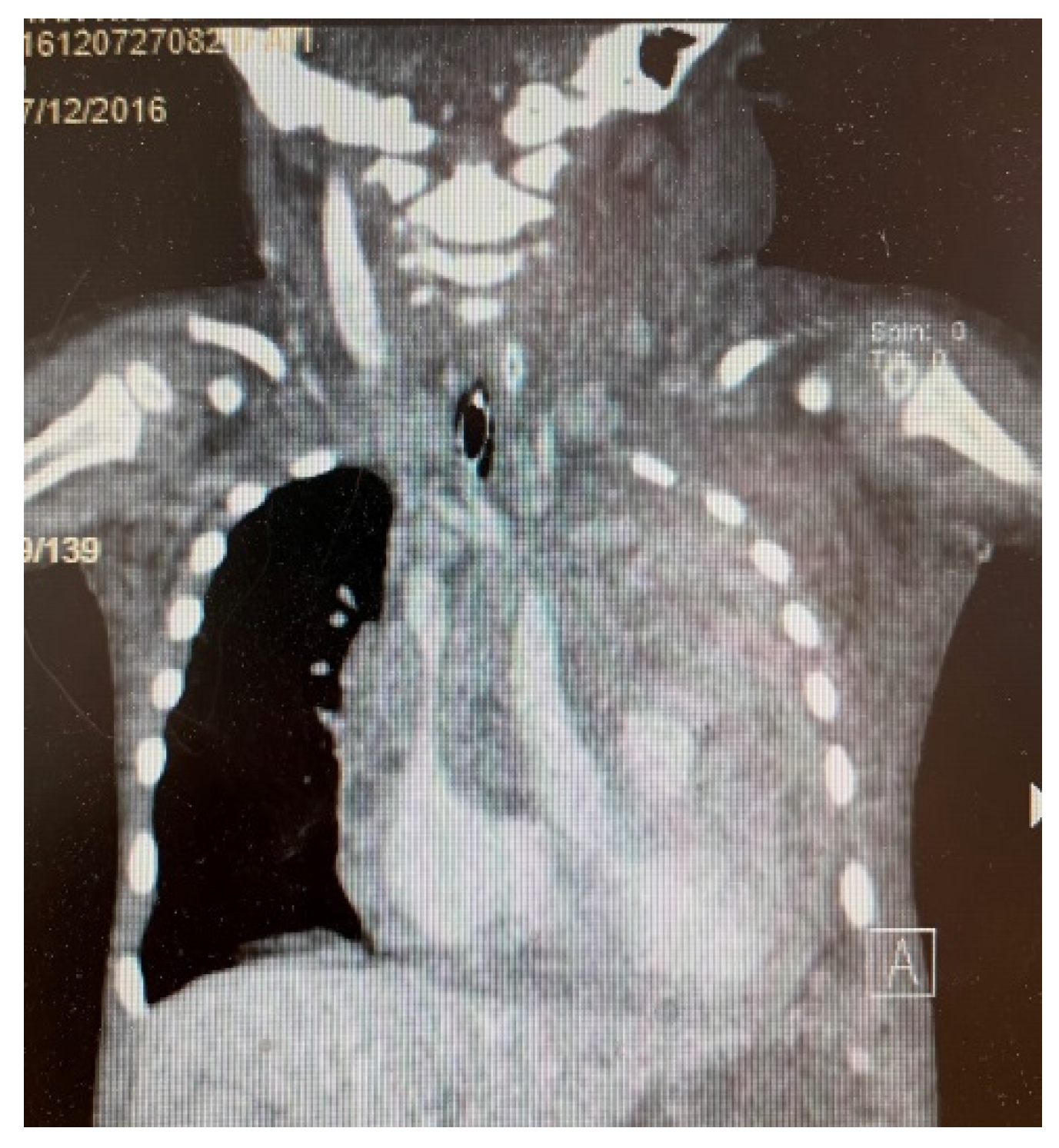
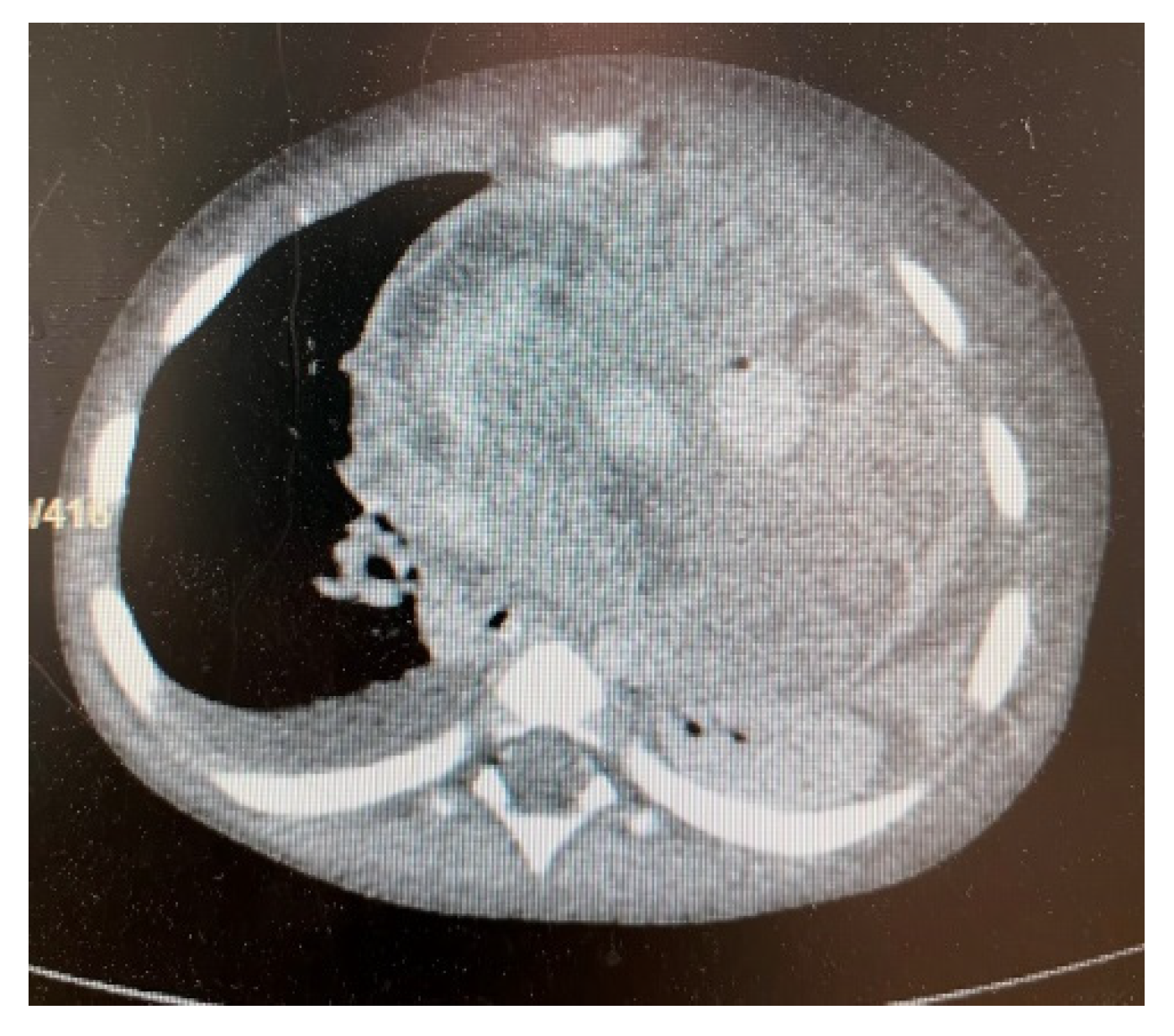
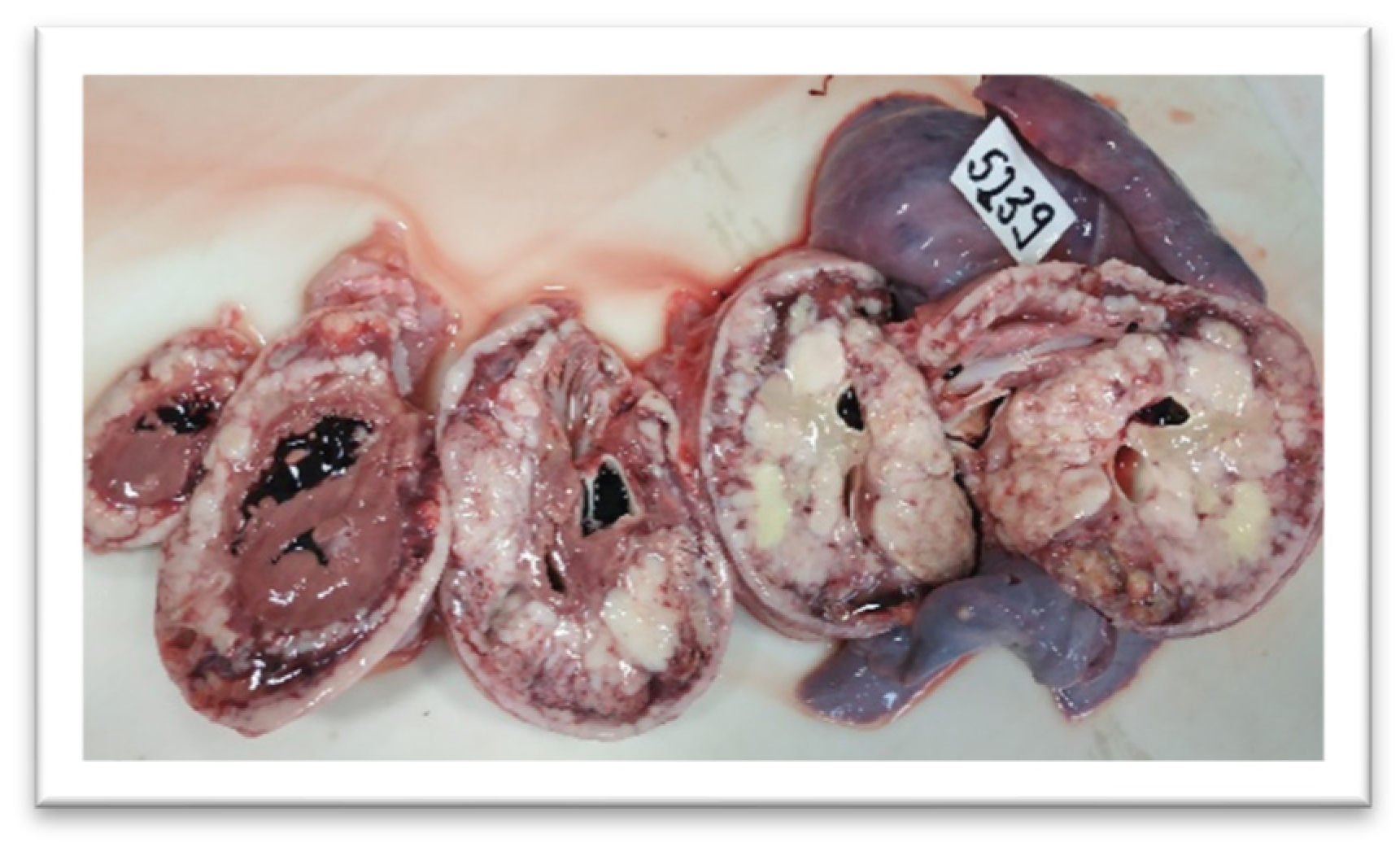
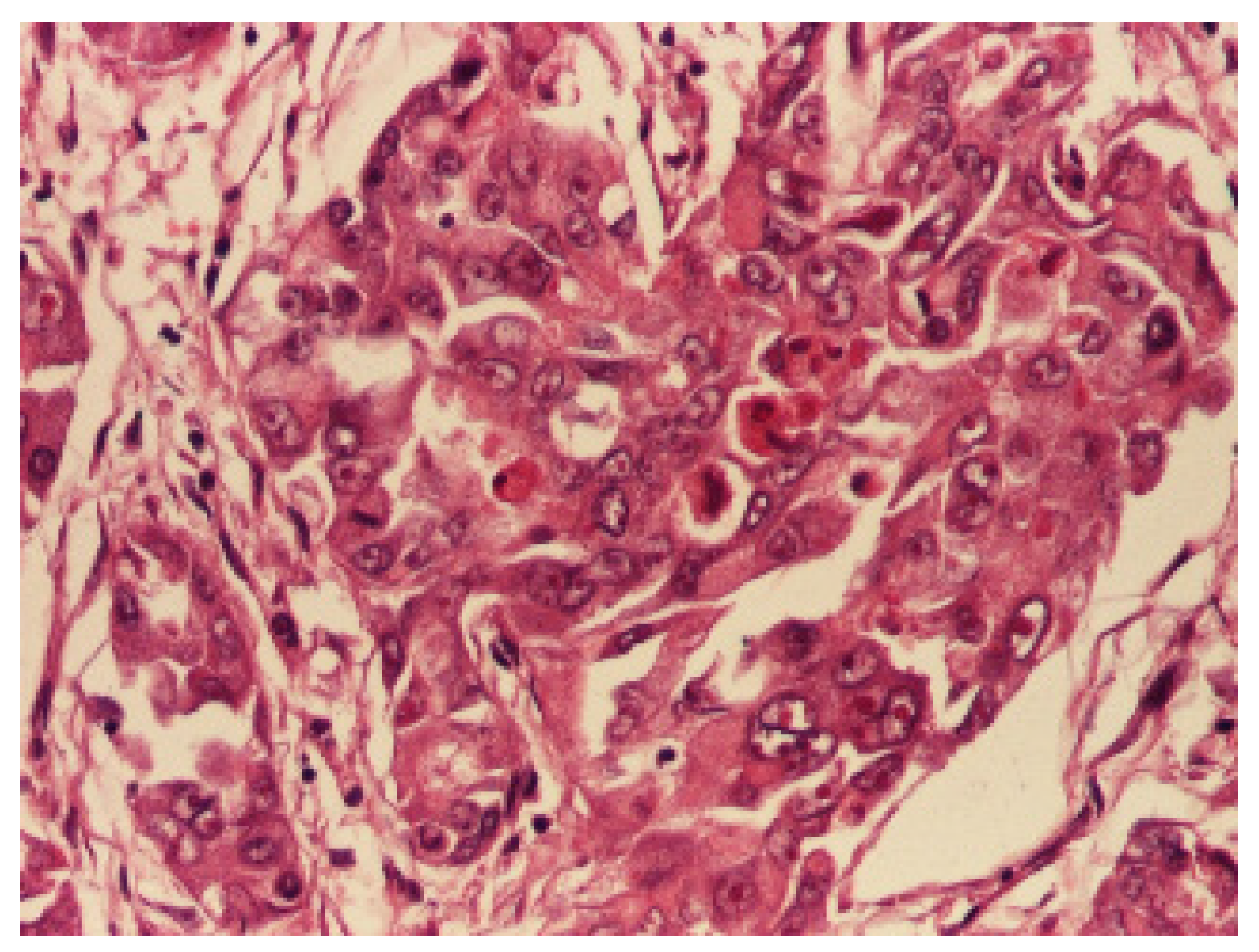
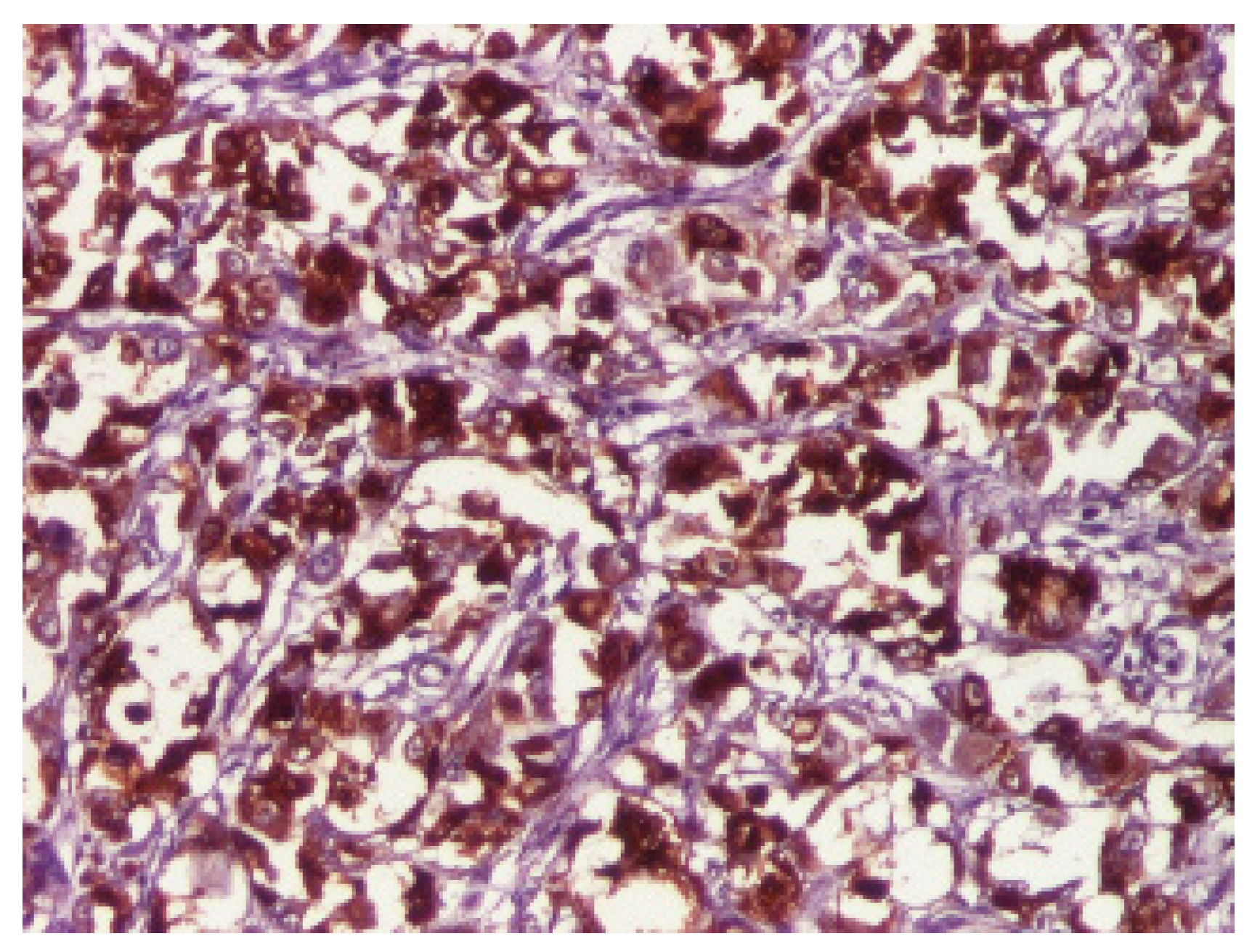
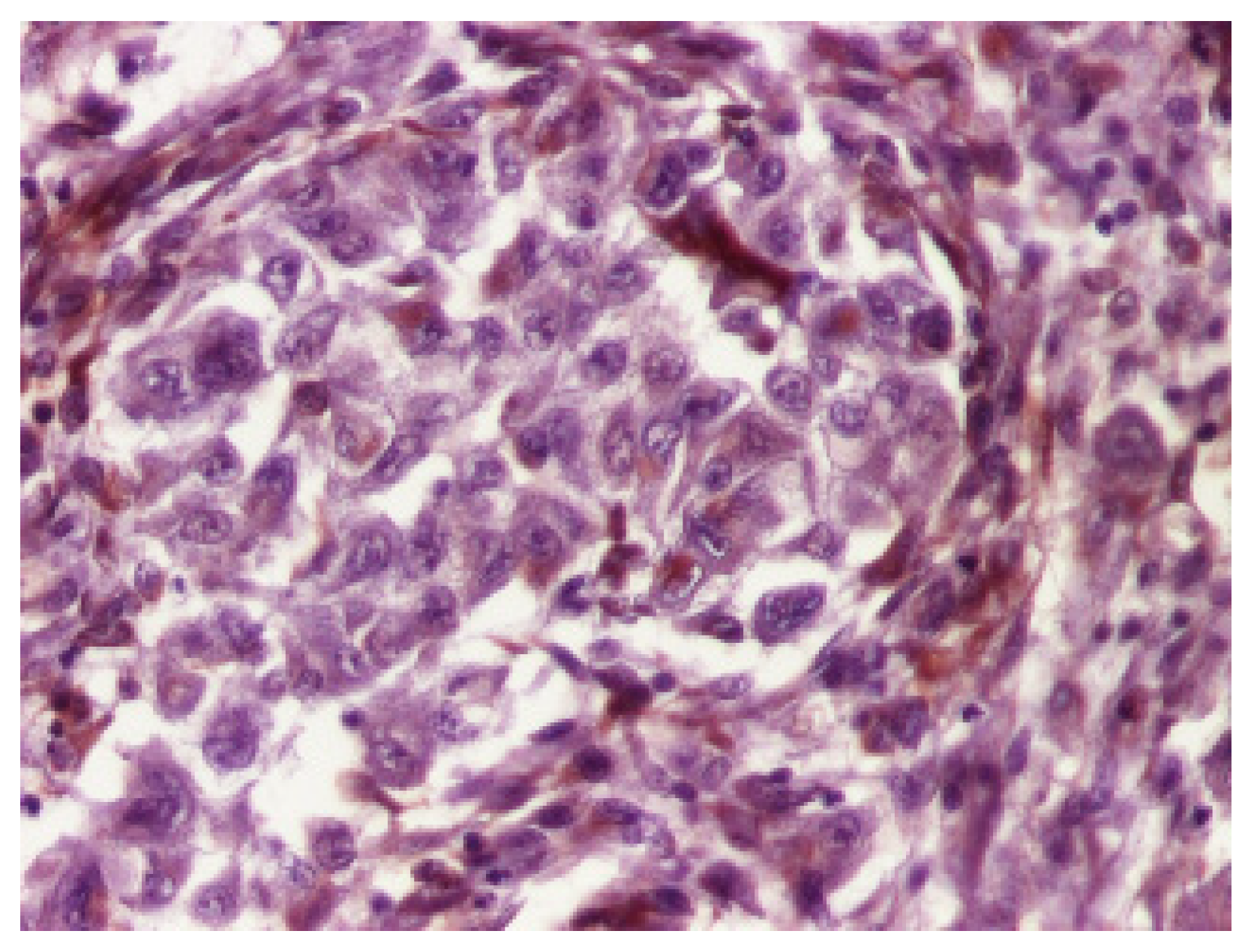
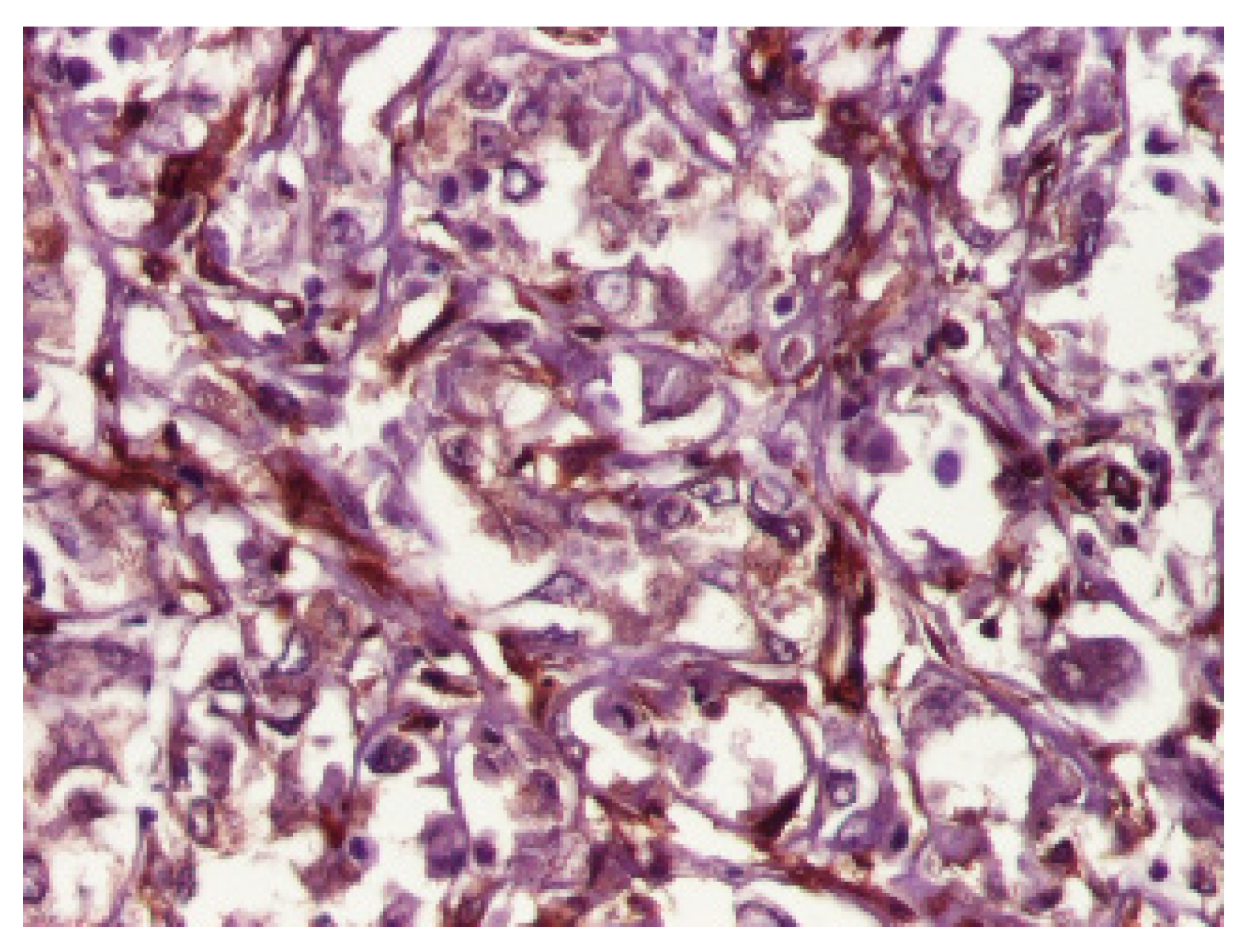
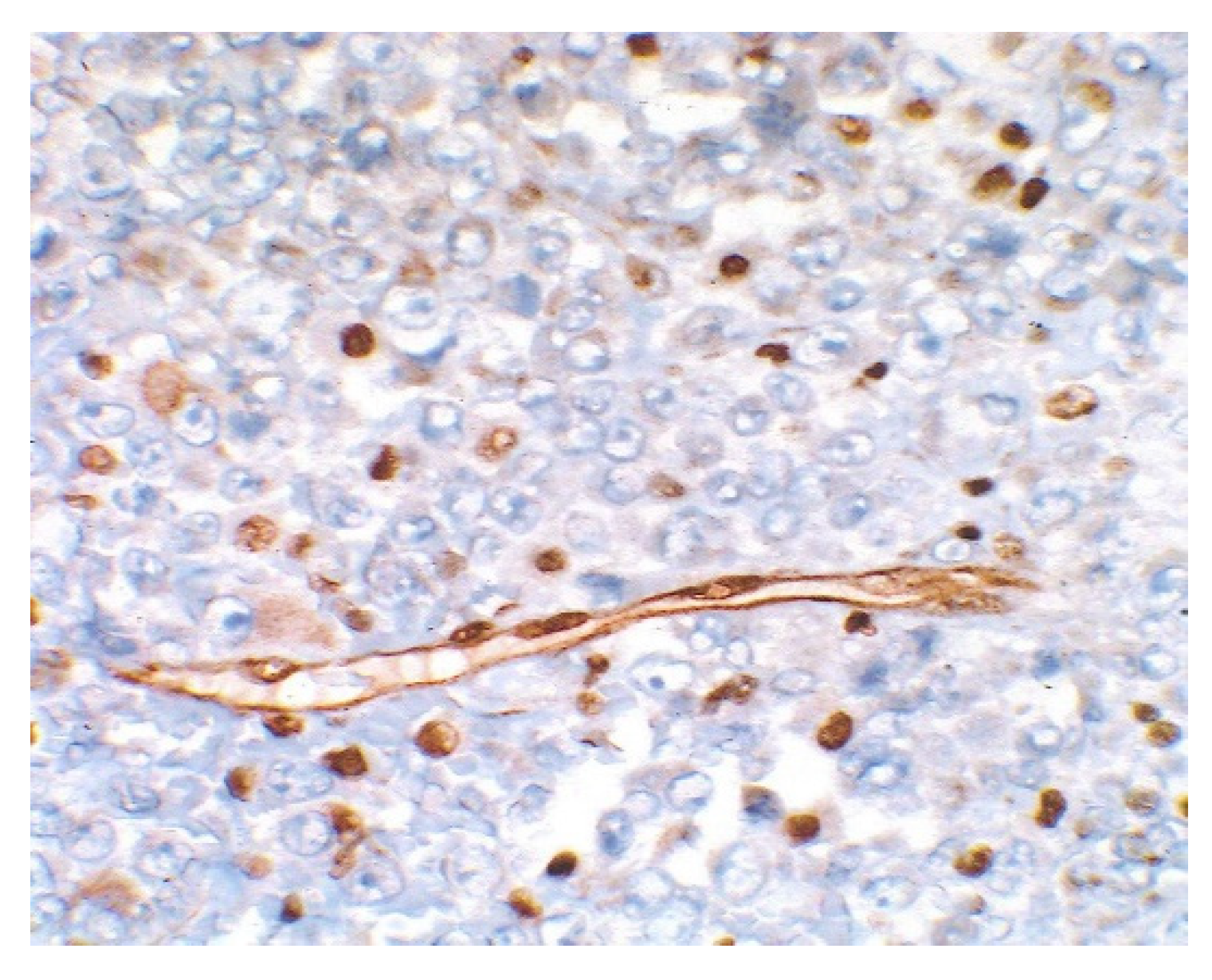
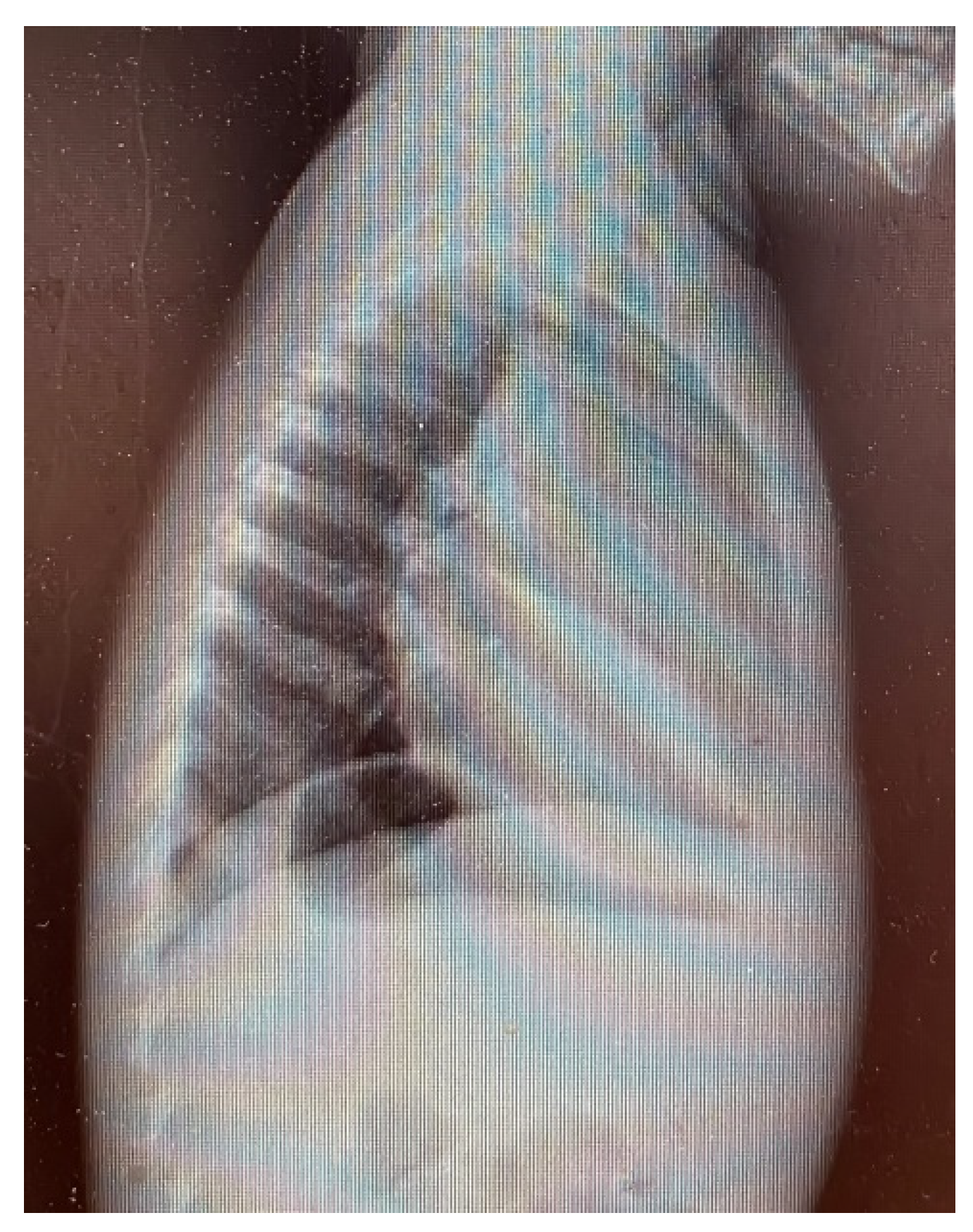
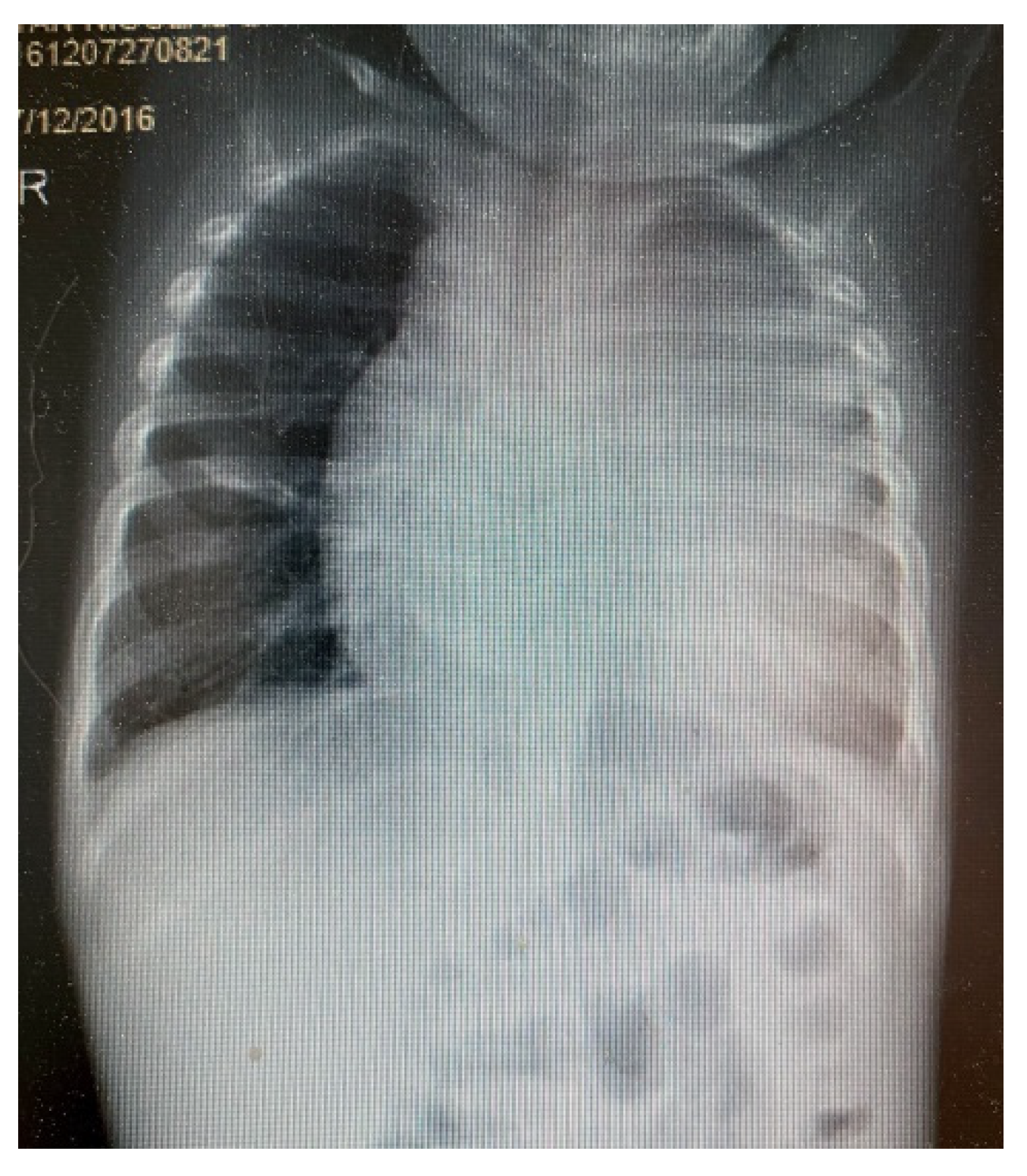
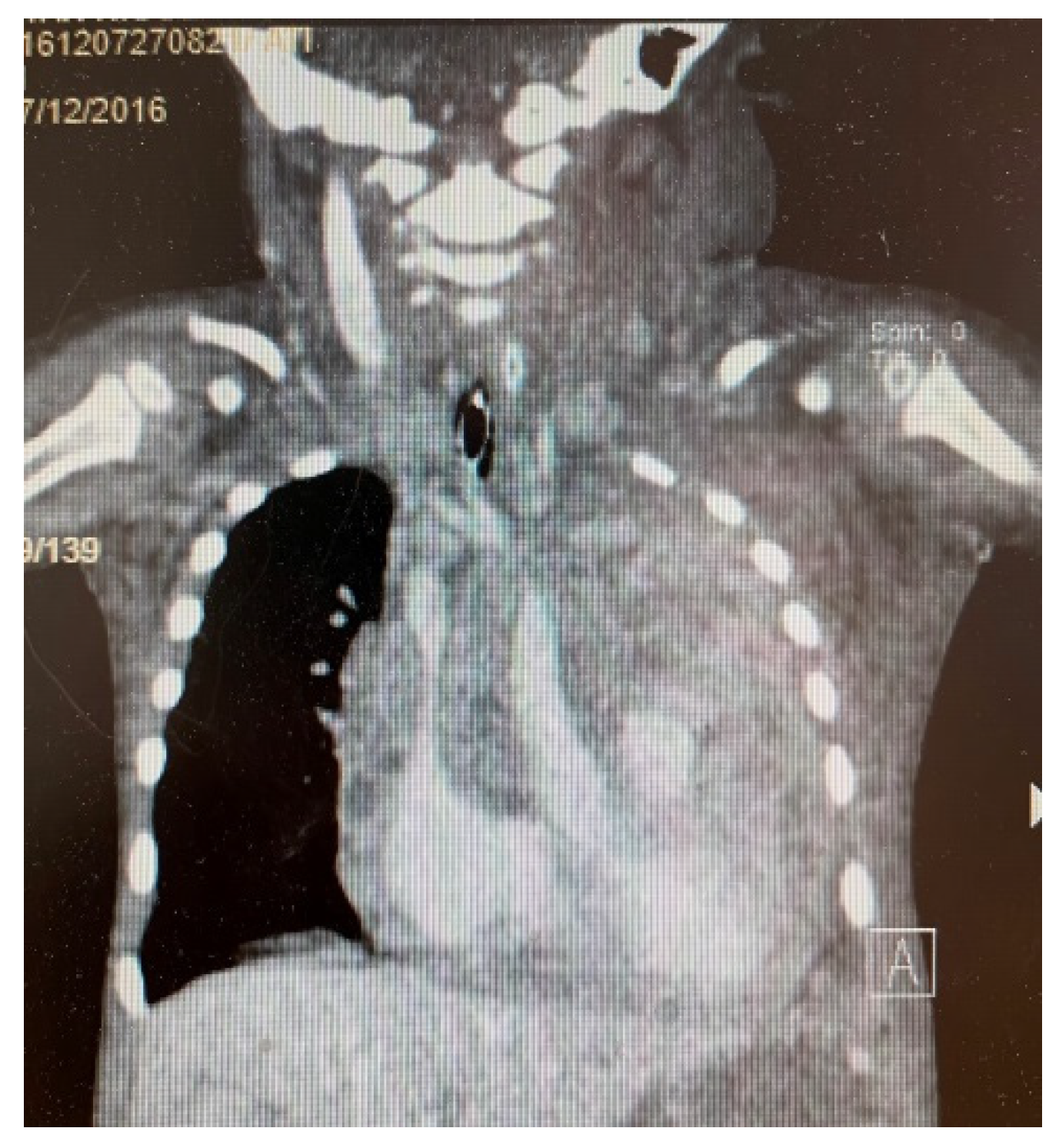
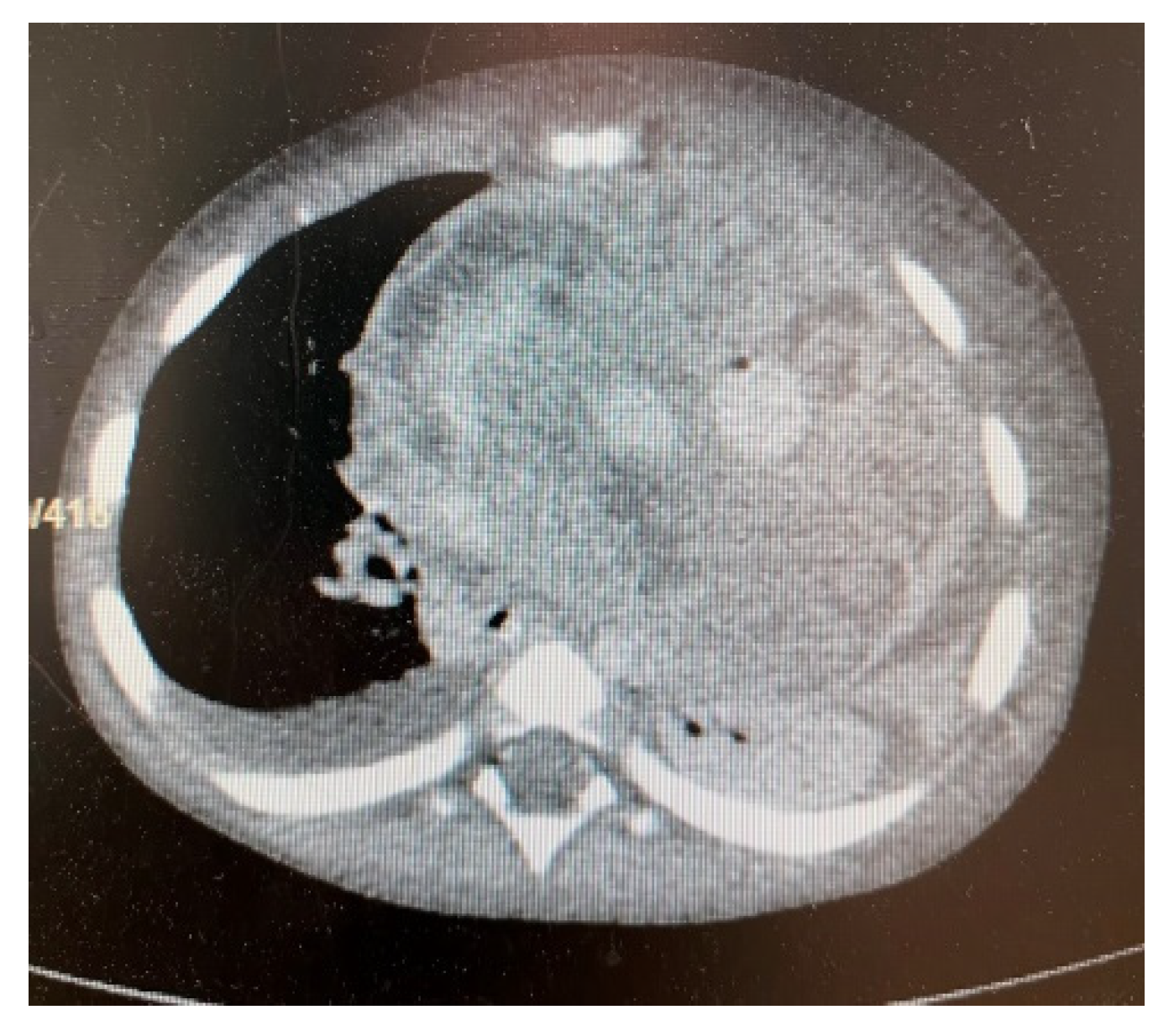
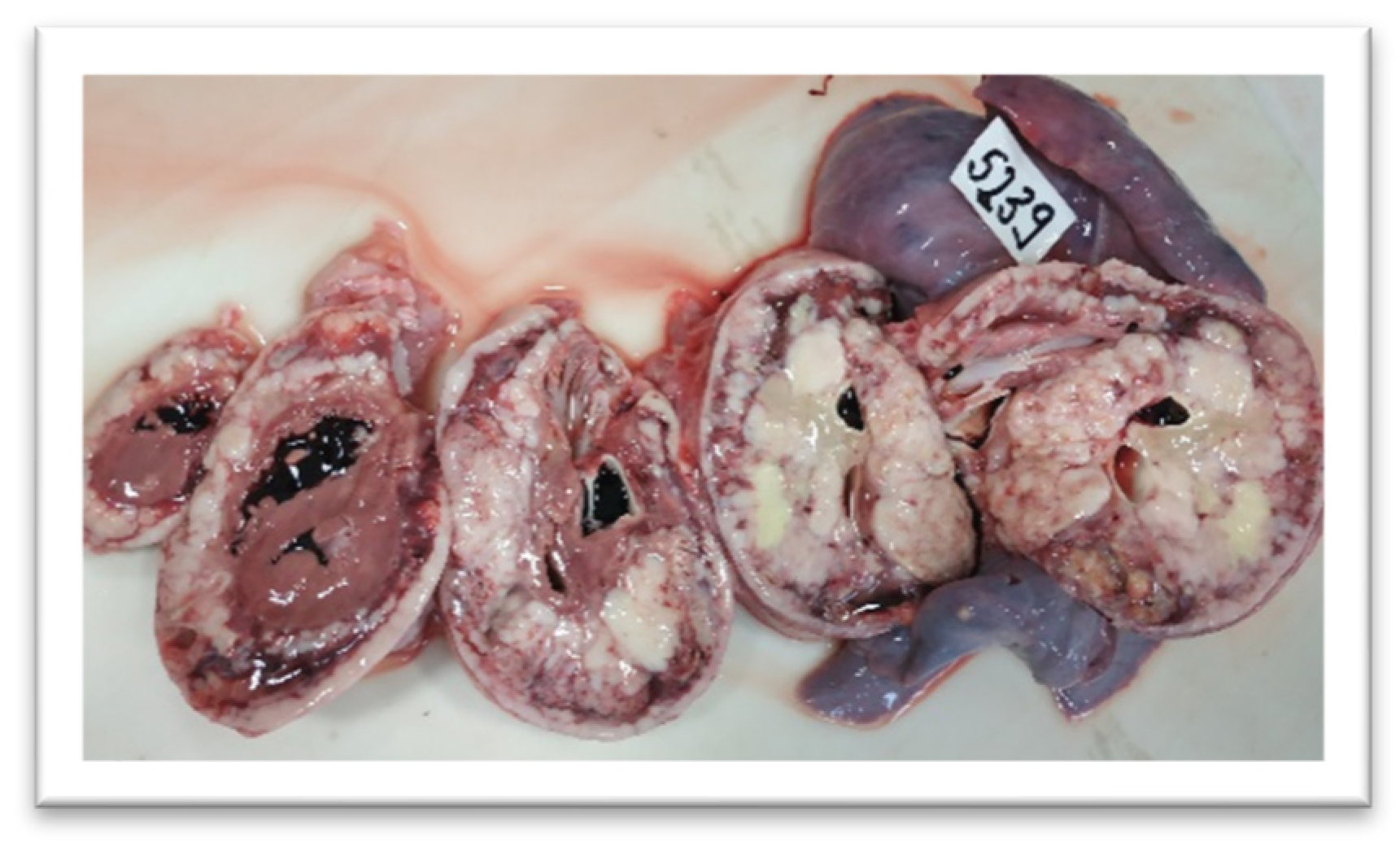
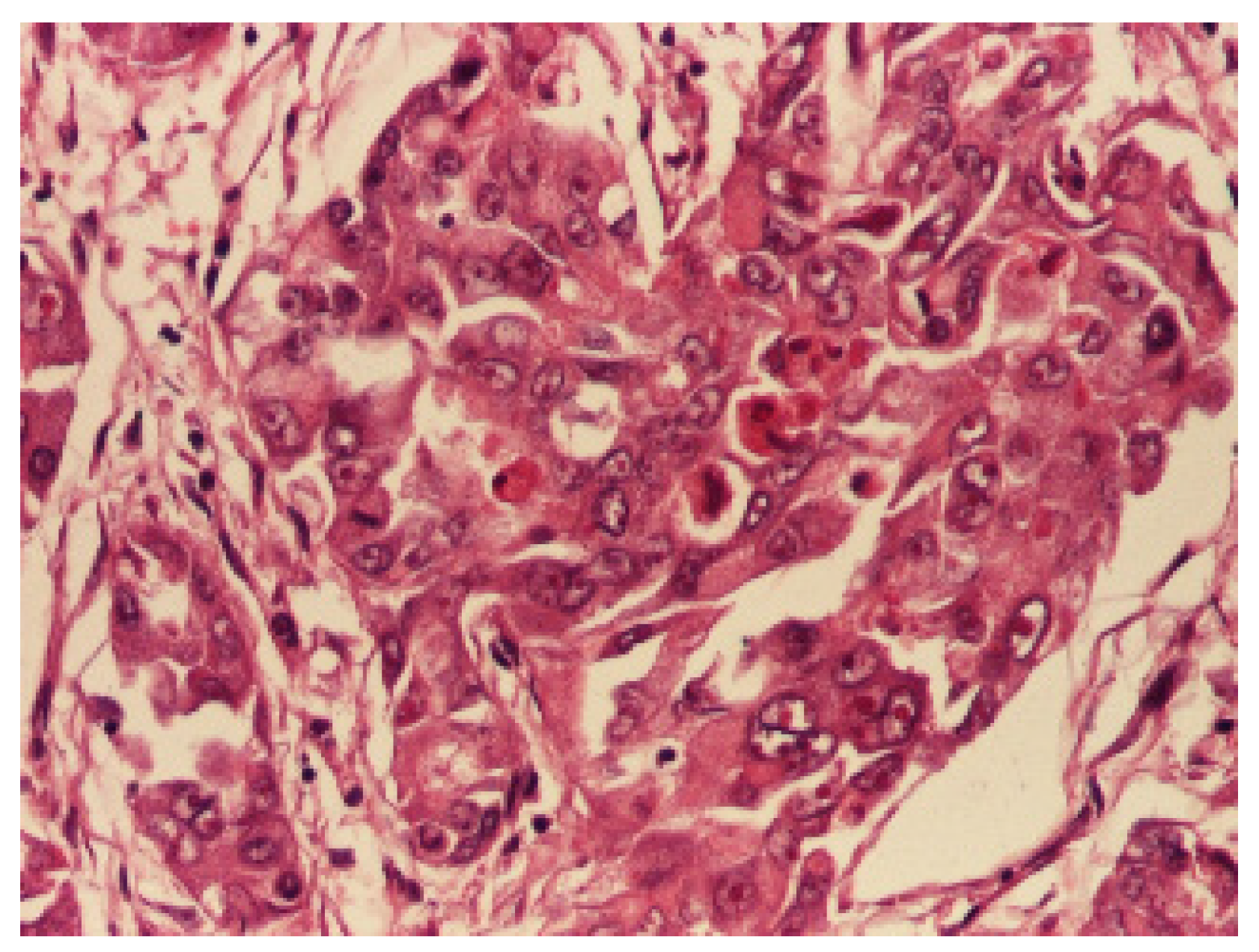
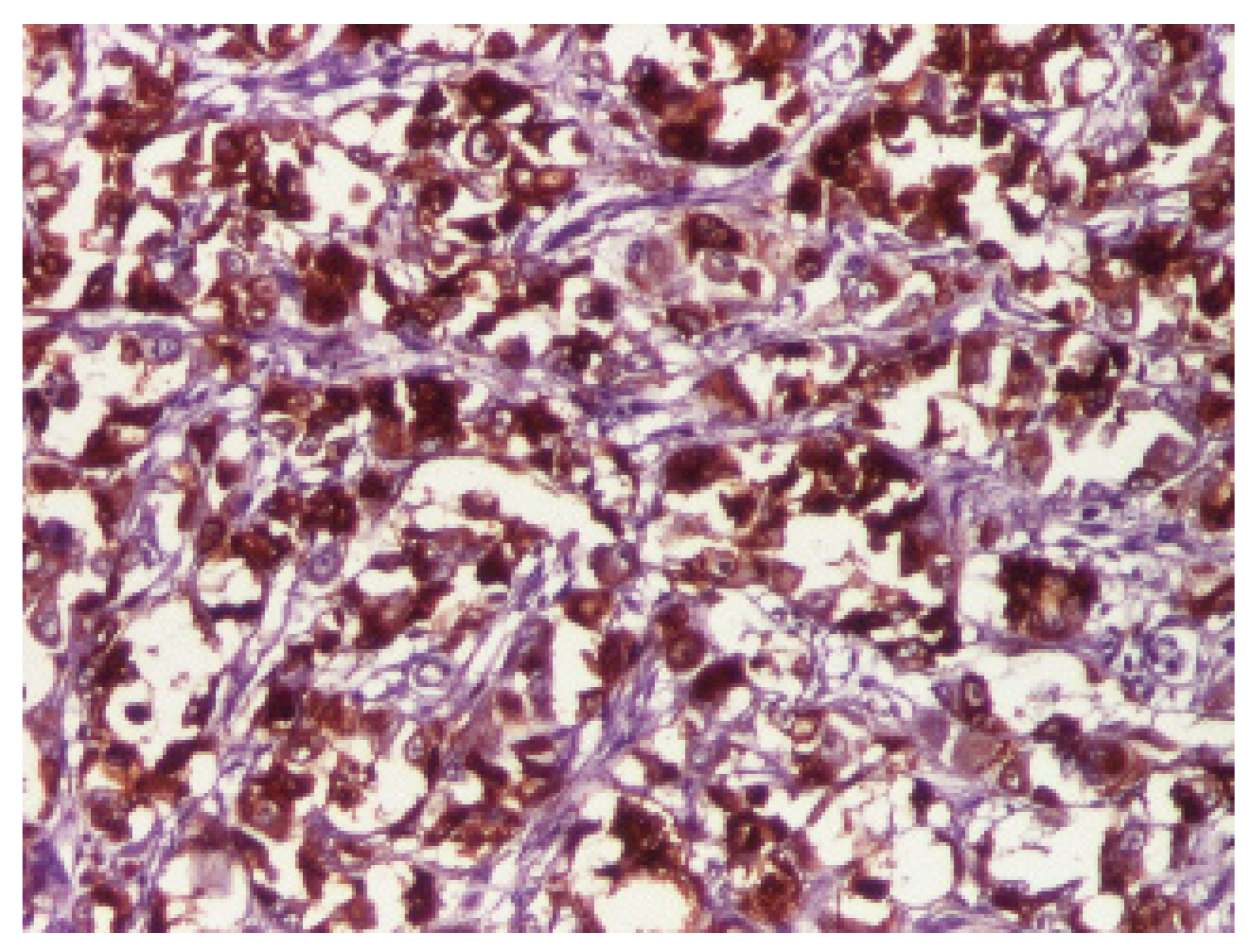
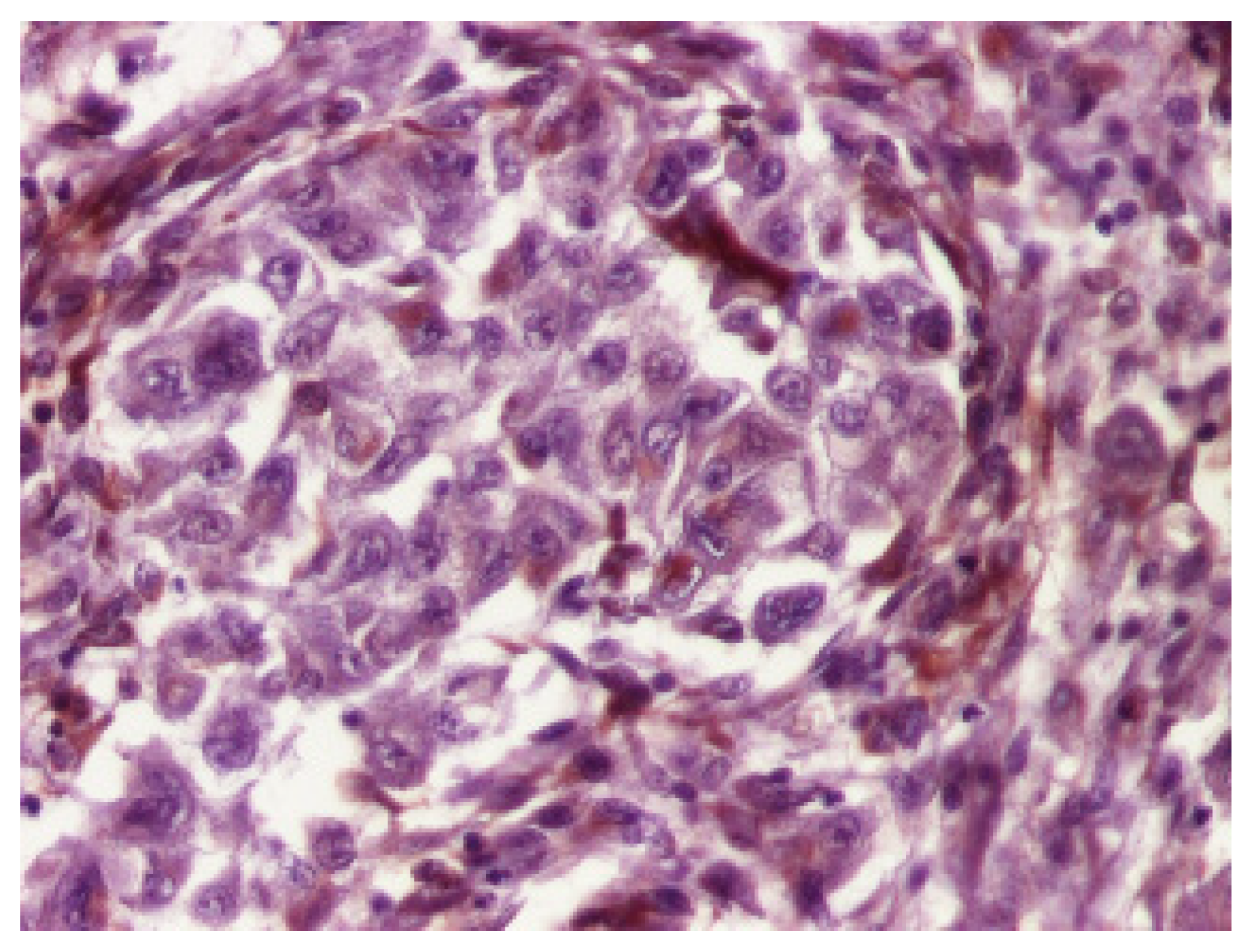
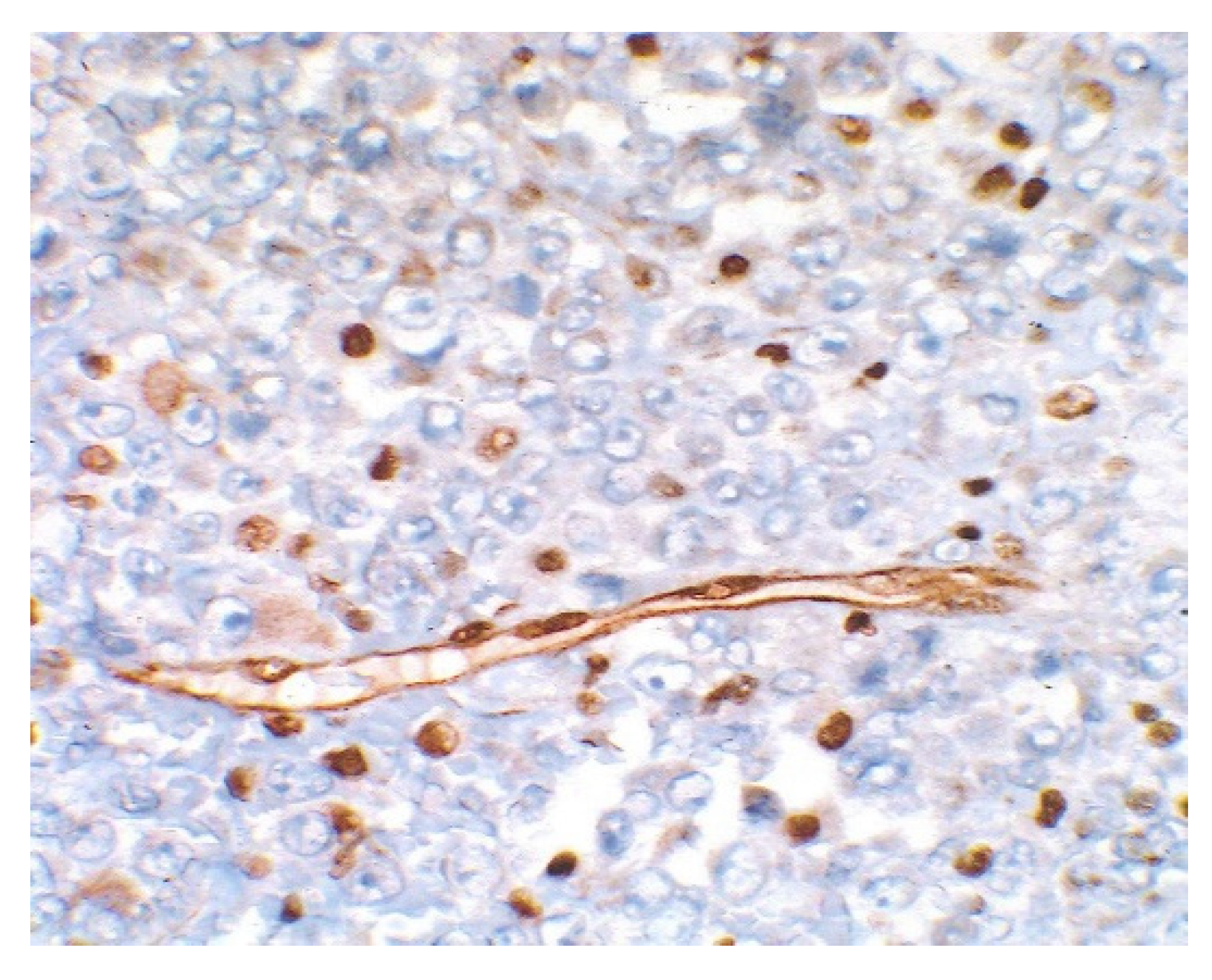
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rosário, M.; Fonseca, A.C.; Sotero, F.D.; Ferro, J.M. Neurological Complications of Cardiac Tumors. Curr. Neurol. Neurosci. Rep. 2019, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Rahouma, M.; Arisha, M.J.; Elmously, A.; El-Sayed Ahmed, M.M.; Spadaccio, C.; Mehta, K.; Baudo, M.; Kamel, M.; Mansor, E.; Ruan, Y.; et al. Cardiac tumors prevalence and mortality: A systematic review and meta-analysis. Int. J. Surg. 2020, 76, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.H.; Shaddy, R.E.; Penny, D.J.; Feltes, T.F.; Cetta, F. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents, Including the Fetus and Young Adult, Wolters Kluwer, 9th ed.; Lippincott Williams & Wlkins: Philadelphia, PA, USA, 2016; pp. 1670–1681. [Google Scholar]
- Butany, J.; Nair, V.; Naseemuddin, A.; Nair, G.M.; Catton, C.; Yau, T. Cardiac tumours: Diagnosis and management. Lancet Oncol. 2005, 6, 219–228. [Google Scholar] [CrossRef]
- Kocabaş, A.; Ekici, F.; Cetin, I.İ.; Emir, S.; Demir, H.A.; Arı, M.E.; Değerliyurt, A.; Güven, A. Cardiac rhabdomyomas associated with tuberous sclerosis complex in 11 children: Presentation to outcome. Pediatr. Hematol. Oncol. 2013, 30, 71–79. [Google Scholar] [CrossRef]
- Ţarcă, E.; Cojocaru, E.; Roşu, S.T.; Butnariu, L.I.; Plămădeală, P.; Moisă, Ş.M. Differential diagnosis difficulties related to infantile hemangioma—Case report and literature review. Rom. J. Morphol. Embryol. 2019, 60, 1375–1379. [Google Scholar]
- Palaskas, N.; Thompson, K.; Gladish, G.; Agha, A.M.; Hassan, S.; Iliescu, C.; Kim, P.; Durand, J.B.; Lopez-Mattei, J.C. Evaluation and Management of Cardiac Tumors. Curr. Treat Options. Cardiovasc Med. 2018, 20, 29. [Google Scholar] [CrossRef]
- Pitsava, G.; Zhu, C.; Sundaram, R.; Mills, J.L.; Stratakis, C.A. Predicting the risk of cardiac myxoma in Carney complex. Genet Med. 2021, 23, 80–85. [Google Scholar] [CrossRef]
- Carreon, C.K.; Sanders, S.P.; Perez-Atayde, A.R.; Del Nido, P.J.; Walsh, E.P.; Geva, T.; Alexander, M.E. Interdigitating Myocardial Tongues in Pediatric Cardiac Fibromas: Plausible Substrate for Ventricular Tachycardia and Cardiac Arrest. JACC Clin. Electrophysiol. 2019, 5, 563–575. [Google Scholar] [CrossRef]
- Bartelheim, K.; Sumerauer, D.; Behrends, U.; Kodetova, D.; Kucera, F.; Leuschner, I.; Neumayer, P.; Oyen, F.; Rübe, C.; Siebert, R.; et al. Clinical and genetic features of rhabdoid tumors of the heart registered with the European Rhabdoid Registry (EU-RHAB). Cancer Genet. 2014, 207, 379–383. [Google Scholar] [CrossRef]
- Mohamed, D.A.; Essaber, H.; Waiss, A.A.; Diekouadio, F.; El Haddad, S.; Fekkar, A.; Lamalmi, N. Pleural effusion revealing a malignant rhabdoid tumor of the chest wall in an infant: A case report and literature review. Int. J. Case Rep. Images 2020, 11, 101125Z01DM2020. [Google Scholar] [CrossRef]
- Nemes, K.; Bens, S.; Bourdeaut, F.; Johann, P.; Kordes, U.; Siebert, R.; Frühwald, M.C. Rhabdoid Tumor Predisposition Syndrome; GeneReviews®: Seattle, WA, USA, 2017; pp. 1993–2020. [Google Scholar]
- Ng, W.K.; Toe, B.P.; Lau, H.Y. Malignant Rhabdoid Tumor of the Mediastinum: A Case Report and Literature Review. J. Clin. Imaging Sci. 2019, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Liu, D. Handbook of Tumor Syndromes; Taylor and Francis Group: Abingdon, UK, 2020; pp. 77–83. [Google Scholar] [CrossRef]
- Aggeli, C.; Dimitroglou, Y.; Raftopoulos, L.; Sarri, G.; Mavrogeni, S.; Wong, J.; Tsiamis, E.; Tsioufis, C. Cardiac Masses: The Role of Cardiovascular Imaging in the Differential Diagnosis. Diagnostics 2020, 10, 1088. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Kamihara, J.; Evans, D.G.R.; Brugières, L.; Bourdeaut, F.; Molenaar, J.J.; Walsh, M.F.; Brodeur, G.M.; Diller, L. Cancer Surveillance in Gorlin Syndrome and Rhabdoid Tumor Predisposition Syndrome. Clin. Cancer Res. 2017, 23, e62–e67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frühwald, M.C.; Nemes, K.; Boztug, H.; Cornips, M.C.A.; Evans, D.G.; Farah, R.; Glentis, S.; Jorgensen, M.; Katsibardi, K.; Hirsch, S.; et al. Current recommendations for clinical surveillance and genetic testing in rhabdoid tumor predisposition: A report from the SIOPE Host Genome Working Group. Fam. Cancer 2021, 20, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Delmo Walter, E.M.; Javier, M.F.; Sander, F.; Hartmann, B.; Ekkernkamp, A.; Hetzer, R. Primary Cardiac Tumors in Infants and Children: Surgical Strategy and Long-Term Outcome. Ann. Thorac. Surg. 2016, 102, 2062–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenthar, J. Clinical presentations, diagnosis, and management of arrhythmias associated with cardiac tumors. J. Arrhythmia 2018, 34, 384–393. [Google Scholar] [CrossRef]
- Richardson, R.R. Atlas of Acquired Cardiovascular Disease Imaging in Children; Springer: Berlin/Heidelberg, Germany, 2017; pp. 95–119. [Google Scholar]
- Miyake, C.Y.; Del Nido, P.J.; Alexander, M.E.; Cecchin, F.; Berul, C.I.; Triedman, J.K.; Geva, T.; Walsh, E.P. Cardiac tumors and associated arrhythmias in pediatric patients, with observations on surgical therapy for ventricular tachycardia. J. Am. Coll. Cardiol. 2011, 58, 1903–1909. [Google Scholar] [CrossRef] [Green Version]
- Wren, C. Concise Guide to Pediatric Arrhythmias; Wiley-Blackwell: West Sussex, UK, 2012; ISBN 978-0-470-65855-0. [Google Scholar]
- Grebenc, M.L.; Rosado de Christenson, M.L.; Burke, A.P.; Green, C.E.; Galvin, J.R. Primary cardiac and pericardial neoplasms: Radiologic-pathologic correlation. Radiographics 2000, 20, 1073–1103. [Google Scholar] [CrossRef] [Green Version]
- Mocellin, S. Soft Tissue Tumors—A practical and Comprehensive Guide to Sarcomas and Benign Neoplasms; Springer: Berlin/Heidelberg, Germany, 2021; pp. 155–163. [Google Scholar]
- Careddu, L.; Oppido, G.; Petridis, F.D.; Liberi, R.; Ragni, L.; Pacini, D.; Pace Napoleone, C.; Angeli, E.; Gargiulo, G. Primary cardiac tumours in the paediatric population. Multimed. Man. Cardiothorac. Surg. 2013, 2013, mmt013. [Google Scholar] [CrossRef]
- Wu, C.M.; Bergquist, P.J.; Srichai, M.B. Multimodality Imaging in the Evaluation of Intracardiac Masses. Curr. Treat. Options Cardiovasc. Med. 2019, 21, 55. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.; Liu, J.; Xu, L.; Li, Y.; Liu, D.; Sun, Z.; Wen, Z. Cardiac magnetic resonance imaging of primary cardiac tumors. Quant Imaging Med. Surg. 2020, 10, 294–313. [Google Scholar] [CrossRef] [PubMed]
- Tzani, A.; Doulamis, I.P.; Mylonas, K.S.; Avgerinos, D.V.; Nasioudis, D. Cardiac Tumors in Pediatric Patients: A Systematic Review. World J. Pediatr. Congenit. Heart Surg. 2017, 8, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, N.; Cheezum, M.K.; Aghayev, A.; Padera, R.; Vita, T.; Steigner, M.; Hulten, E.; Bittencourt, M.S.; Dorbala, S.; Di Carli, M.F.; et al. Assessment of Cardiac Masses by Cardiac Magnetic Resonance Imaging: Histological Correlation and Clinical Outcomes. J. Am. Heart Assoc. 2019, 8, e007829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Zhao, H.; Liu, Y.; Chen, D.; Hacker, M.; Wei, Y.; Li, X.; Zhang, X.; Kreissl, M.C. Assessment of cardiac tumors by 18F-FDG PET/CT imaging: Histological correlation and clinical outcomes. J. Nucl. Cardiol. 2020, 28, 2233–2243. [Google Scholar] [CrossRef] [PubMed]
- Fathala, A.; Abouzied, M.; AlSugair, A.-A. Cardiac and pericardial tumors: A potential application of positron emission tomography-magnetic resonance imaging. World J. Cardiol. 2017, 9, 600. [Google Scholar] [CrossRef] [PubMed]
- Seeringer, A.; Bartelheim, K.; Kerl, K.; Hasselblatt, M.; Leuschner, I.; Rutkowski, S.; Timmermann, B.; Kortmann, R.D.; Koscielniak, E.; Schneppenheim, R.; et al. Feasibility of intensive multimodal therapy in infants affected by rhabdoid tumors—Experience of the EU-RHAB registry. Klin. Padiatr. 2014, 226, 143–148. [Google Scholar] [CrossRef]
- Beck, J.D.; Bokemeyer, C.; Langer, T. Late Treatment Effects and Cancer Survivor Care in the Young; Springer: Berlin/Heidelberg, Germany, 2021; pp. 336–338. [Google Scholar]
- Tomlinson, G.E.; Breslow, N.E.; Dome, J.; Guthrie, K.A.; Norkool, P.; Li, S.; Thomas, P.R.; Perlman, E.; Beckwith, J.B.; D’Angio, G.J.; et al. Rhabdoid tumor of the kidney in the National Wilms’ Tumor Study: Age at diagnosis as a prognostic factor. J. Clin. Oncol. 2005, 23, 7641–7645. [Google Scholar] [CrossRef]
- Hollmann, T.J.; Hornick, J.L. INI1-deficient tumors: Diagnostic features and molecular genetics. Am. J. Surg. Pathol. 2011, 35, e47–e63. [Google Scholar] [CrossRef]
- Herpel, E.; Rieker, R.J.; Dienemann, H.; Muley, T.; Meister, M.; Hartmann, A.; Warth, A.; Agaimy, A. SMARCA4 and SMARCA2 deficiency in non-small cell lung cancer: Immunohistochemical survey of 316 consecutive specimens. Ann. Diagn. Pathol. 2017, 26, 47–51. [Google Scholar] [CrossRef]
- Reddy, A.; Strother, D.; Judkins, A.; Krailo, M.; Gao, Y.; Douglas, J.; Mahajan, A.; Lewis, V.; Mazewski, C.; Laningham, F.; et al. Treatment of atypical teratoid rhabdoid tumors (atrt) of the central nervous system with surgery, intensive chemotherapy, and 3-d conformal radiation. A report from the children’s oncology group. Neuro Oncol. 2016, 18 (Suppl. S3), iii2. [Google Scholar] [CrossRef] [Green Version]
- Bartelheim, K.; Nemes, K.; Seeringer, A.; Kerl, K.; Buechner, J.; Boos, J.; Graf, N.; Dürken, M.; Gerss, J.; Hasselblatt, M.; et al. Improved 6-year overall survival in AT/RT—Results of the registry study Rhabdoid 2007. Cancer Med. 2016, 5, 1765–1775. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | Imagistic Studies | Frequency |
|---|---|---|
| All | Whole-body MRI | After SMARCAB1 mutation discovered |
| 0–6 months | Whole-body MRI or CNS MRI Abdomen and soft tissue ultrasound | Every 4 weeks, not less than every 2–3 months |
| 7–18 months | Whole-body MRI or CNS MRI Abdomen and soft tissue ultrasound | Every 2–3 months |
| 19 months–5 years | Whole-body MRI or CNS MRI Abdomen and soft tissue ultrasound | Every 3 months |
| >5 years | Whole-body MRI | Yearly |
| Tumor Type | Clinical Manifestations [19] | *ECG [20,21,22] | *TTE/ TEE [23] | *CT [20,23] | *CMR [20,23] | *Biomarkers [24] | Differential Diagnosis [24] | Therapy [20,24,25] |
|---|---|---|---|---|---|---|---|---|
| Myxoma | Flow-Obstruction; Emboli; Systemic symptoms; | Left atrial enlargement; Ventricular tachycardia; | Narrow stalk; Hyperechoic mass in characteristic location; Calcifications; Dynamic tumor; | Low-attenuation heterogeneous mass compared with myocardium; Pulmonary infarction; Intratumoral calcification; | T1 hypointense, T2 hyperintense Heterogeneously enhancing isointense or hyperintense on delayed imaging; | CD31 + CD34+ Calretinin + CD68-Cytokeratins- | Left atrial thrombus; Metastatic carcinoma; Myxoid sarcoma; Papillary fibroelastoma; Fibroma; | Surgical excision |
| Fibroma | Heart murmurs; Congestive heart failure; Arrhythmias; Sudden death; | T-wave abnormalities; Ventricular tachycardia; Atrioventricular block; | Large, solid, heterogeneous mass that is noncontractile | Central calcification within a discrete mass; Non-specific low attenuation mass; | Encapsulated mass; Delayed enhancement; Hypointense to isointense in T1; Hypointense in T2; | Vimentin + Ki-67- CD34- S100- HMB45- | Cardiac rhabdomyoma; Myxoma; Teratoma; Lipoma; Hemangioma; Hypertrophic cardiomyopathy; Metastatic disease | Amiodarone and/or beta-blockers; Surgical excision; Single ventricle palliation; Cardiac transplant; |
| Rhabdomyoma | Flow obstruction; Heart failure; Arrhythmias; Decreased peripheral pulses and/or cyanosis | Extrasystoles; Ventricular Tachycardia; Supraventricular tachycardia; Wolff–Parkinson–White syndrome | Solid, hyperechoic, avascular mass; Focal abnormality of cardiac wall motion | Hypodense compared with adjacent myocardium | T1 isointense/slightly hyperintense; T2 hyperintense; No fat suppression | Myoglobin + Actin + Desmin + Vimentin + S100- | Glycogen storage disease; Granular cell tumor; Lipoma; | mTOR inhibitors; Surgical excision if located in the left ventricle; |
| Rhabdomyosarcoma | Systemic illness; Syncope; Arrhythmias; Sudden death; Pericardial disease or tamponade; Embolic phenomena | Ventricular arrhythmias | Solid, hyperechoic mass with irregular borders | Hypoattenuating mass involving any cardiac chamber; Smooth or irregular borders | Heterogeneous mass with high signal intensity in T2 | Myogenin+ MSA + MYOD1 + Desmin+ | Angiosarcoma; Fibrosarcoma; Osteosarcoma; Leiomyosarcoma; Liposarcoma; Lymphoma; Intrapericardial pheochromocytoma; Metastatic disease | Surgical resection; Heart transplantation; USA chemotherapy: Vincristine + Actinomycin-D + Cyclophosphamide; EU chemotherapy: Ifosfamide + Vincristine + Actinomycin-D |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luca, A.C.; Miron, I.C.; Cojocaru, E.; Țarcă, E.; Curpan, A.-S.; Mihăila, D.; Mihaela Trandafir, L.; Iordache, A.-C.; Lupu, V.-V.; Tazelaar, H.D.; et al. Cardiac Rhabdoid Tumor—A Rare Foe—Case Report and Literature Review. Children 2022, 9, 942. https://doi.org/10.3390/children9070942
Luca AC, Miron IC, Cojocaru E, Țarcă E, Curpan A-S, Mihăila D, Mihaela Trandafir L, Iordache A-C, Lupu V-V, Tazelaar HD, et al. Cardiac Rhabdoid Tumor—A Rare Foe—Case Report and Literature Review. Children. 2022; 9(7):942. https://doi.org/10.3390/children9070942
Chicago/Turabian StyleLuca, Alina Costina, Ingrith Crenguța Miron, Elena Cojocaru, Elena Țarcă, Alexandrina-Stefania Curpan, Doina Mihăila, Laura Mihaela Trandafir, Alin-Constantin Iordache, Vasile-Valeriu Lupu, Henry D. Tazelaar, and et al. 2022. "Cardiac Rhabdoid Tumor—A Rare Foe—Case Report and Literature Review" Children 9, no. 7: 942. https://doi.org/10.3390/children9070942
APA StyleLuca, A. C., Miron, I. C., Cojocaru, E., Țarcă, E., Curpan, A.-S., Mihăila, D., Mihaela Trandafir, L., Iordache, A.-C., Lupu, V.-V., Tazelaar, H. D., & Pădureț, I. A. (2022). Cardiac Rhabdoid Tumor—A Rare Foe—Case Report and Literature Review. Children, 9(7), 942. https://doi.org/10.3390/children9070942