
22q11.2 Deletion Syndrome: Impact of Genetics in the Treatment of Conotruncal Heart Defects

 , , ,
, , ,  , and
, and
Abstract
1. Introduction
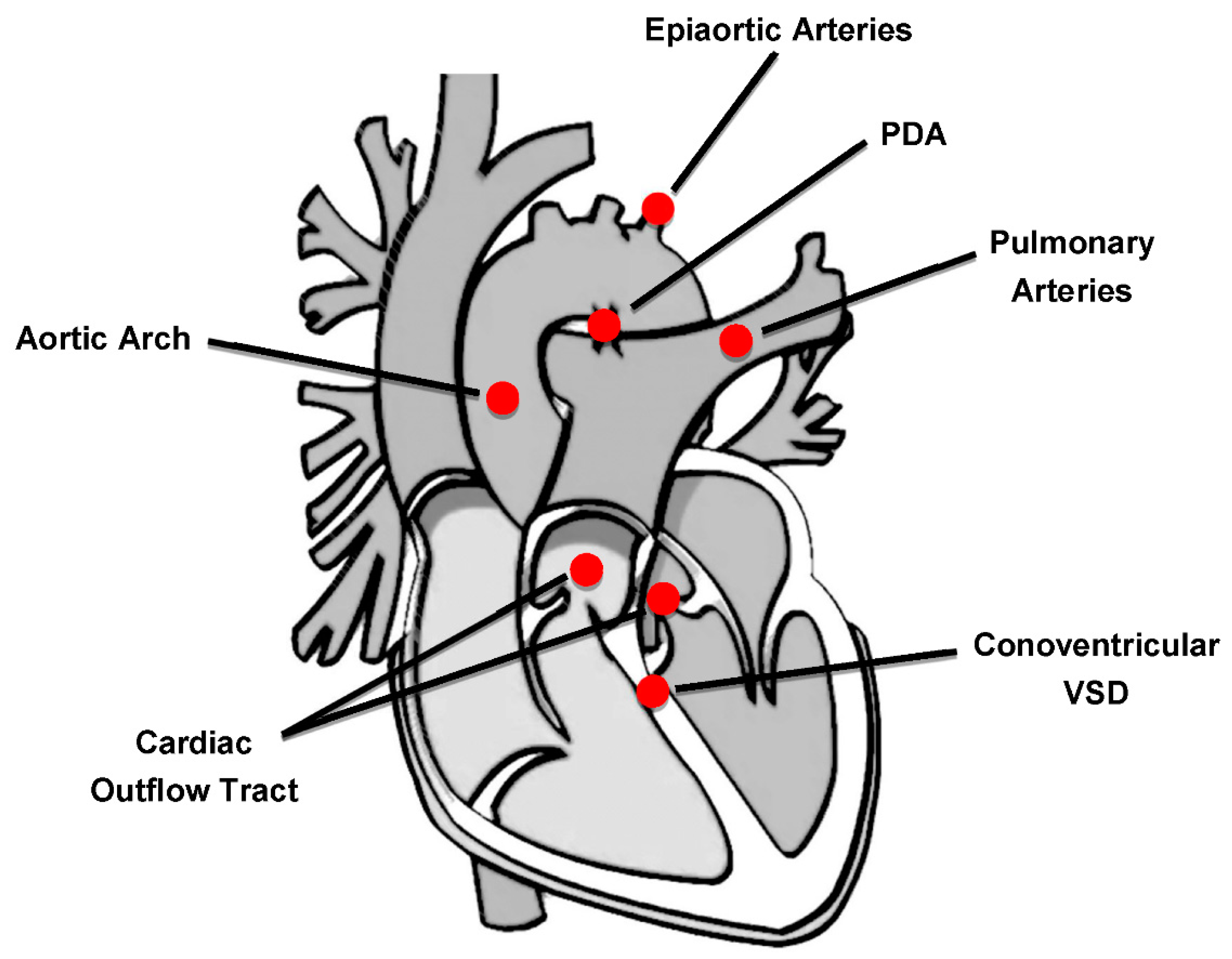
2. Congenital Heart Defects in 22q11.2 Deletion Syndrome
3. Outcome of Cardiac Surgery in 22q11.2 Deletion Syndrome
3.1. Tetralogy of Fallot (ToF)
3.2. Pulmonary Atresia with Ventricular Septal Defect +/− Major Aortopulmonary Collateral Arteries (PA-VSD+/−MAPCAs)
3.3. Truncus Arteriosus (TA)
3.4. Interrupted Aortic Arch (IAA)
4. Perioperative Management
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Donofrio, M.T.; Moon-Grady, A.J.; Hornberger, L.K.; Copel, J.A.; Sklansky, M.S.; Abuhamad, A.; Cuneo, B.F.; Huhta, J.C.; Jonas, R.A.; Krishnan, A.; et al. Diagnosis and treatment of fetal cardiac disease: A scientific statement from the American Heart Association. Circulation 2014, 129, 2183–2242. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.P.; He, X.; Mayer, J.E., Jr.; Austin, E.H., 3rd; Quintessenza, J.A.; Karl, T.R.; Vricella, L.; Mavroudis, C.; O’Brien, S.M.; Pasquali, S.K.; et al. Mortality Trends in Pediatric and Congenital Heart Surgery: An Analysis of The Society of Thoracic Surgeons Congenital Heart Surgery Database. Ann. Thorac. Surg. 2016, 102, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Calcagni, G.; Pugnaloni, F.; Digilio, M.C.; Unolt, M.; Putotto, C.; Niceta, M.; Baban, A.; Piceci Sparascio, F.; Drago, F.; De Luca, A.; et al. Cardiac Defects and Genetic Syndromes: Old Uncertainties and New Insights. Genes 2021, 12, 1047. [Google Scholar] [CrossRef] [PubMed]
- Marino, B.; Digilio, M.C. Congenital heart disease and genetic syndromes: Specific correlation between cardiac phenotype and genotype. Cardiovasc. Pathol. 2000, 9, 303–315. [Google Scholar] [CrossRef]
- Formigari, R.; Michielon, G.; Digilio, M.C.; Piacentini, G.; Carotti, A.; Giardini, A.; Di Donato, R.M.; Marino, B. Genetic syndromes and congenital heart defects: How is surgical management affected? Eur. J. Cardiothorac. Surg. 2009, 35, 606–614. [Google Scholar] [CrossRef]
- Botto, L.D.; May, K.; Fernhoff, P.M.; Correa, A.; Coleman, K.; Rasmussen, S.A.; Merritt, R.K.; O’Leary, L.A.; Wong, L.Y.; Elixson, E.M.; et al. A population-based study of the 22q11.2 deletion: Phenotype, incidence, and contribution to major birth defects in the population. Pediatrics 2003, 112, 101–107. [Google Scholar] [CrossRef]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 Deletion Syndrome. Nat. Rev. Dis. Primers 2015, 1, 15071. [Google Scholar] [CrossRef]
- Marino, B.; Digilio, M.C.; Toscano, A.; Anaclerio, S.; Giannotti, A.; Feltri, C.; de Ioris, M.A.; Angioni, A.; Dallapiccola, B. Anatomic patterns of conotruncal defects associated with deletion 22q11. Genet. Med. 2001, 3, 45–48. [Google Scholar] [CrossRef]
- Momma, K. Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome. Am. J. Cardiol. 2010, 105, 1617–1624. [Google Scholar] [CrossRef]
- McElhinney, D.B.; Clark, B.J., 3rd; Weinberg, P.M.; Kenton, M.L.; McDonald-McGinn, D.; Driscoll, D.A.; Zackai, E.H.; Goldmuntz, E. Association of chromosome 22q11 deletion with isolated anomalies of aortic arch laterality and branching. J. Am. Coll. Cardiol. 2001, 37, 2114–2119. [Google Scholar] [CrossRef]
- Lin, A.E.; Basson, C.T.; Goldmuntz, E.; Magoulas, P.L.; McDermott, D.A.; McDonald-McGinn, D.M.; McPherson, E.; Morris, C.A.; Noonan, J.; Nowak, C.; et al. Adults with genetic syndromes and cardiovascular abnormalities: Clinical history and management. Genet. Med. 2008, 10, 469–494. [Google Scholar] [CrossRef] [PubMed]
- Repetto, G.M.; Guzman, M.L.; Delgado, I.; Loyola, H.; Palomares, M.; Lay-Son, G.; Vial, C.; Benavides, F.; Espinoza, K.; Alvarez, P. Case fatality rate and associated factors in patients with 22q11 microdeletion syndrome: A retrospective cohort study. BMJ Open 2014, 4, e005041. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.S.; Chow, E.W.; Husted, J.; Hodgkinson, K.A.; Oechslin, E.; Harris, L.; Silversides, C. Premature death in adults with 22q11.2 deletion syndrome. J. Med. Genet. 2009, 46, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Van, L.; Heung, T.; Graffi, J.; Ng, E.; Malecki, S.; Van Mil, S.; Boot, E.; Corral, M.; Chow, E.W.C.; Hodgkinson, K.A.; et al. All-cause mortality and survival in adults with 22q11.2 deletion syndrome. Genet. Med. 2019, 21, 2328–2335. [Google Scholar] [CrossRef]
- Marino, B.; Vairo, U.; Corno, A.; Nava, S.; Guccione, P.; Calabrò, R.; Marcelletti, C. Atrioventricular canal in Down syndrome. Prevalence of associated cardiac malformations compared with patients without Down syndrome. Am. J. Dis. Child. 1990, 144, 1120–1122. [Google Scholar] [CrossRef]
- Levin, S.E.; Dansky, R.; Milner, S.; Benatar, A.; Govendrageloo, K.; du Plessis, J. Atrioventricular septal defect and type A postaxial polydactyly without other major associated anomalies: A specific association. Pediatr. Cardiol. 1995, 16, 242–246. [Google Scholar] [CrossRef]
- Pierpont, M.E.; Digilio, M.C. Cardiovascular disease in Noonan syndrome. Curr. Opin. Pediatr. 2018, 30, 601–608. [Google Scholar] [CrossRef]
- Anaclerio, S.; Marino, B.; Carotti, A.; Digilio, M.C.; Toscano, A.; Gitto, P.; Giannotti, A.; Di Donato, R.; Dallapiccola, B. Pulmonary atresia with ventricular septal defect: Prevalence of deletion 22q11 in the different anatomic patterns. Ital. Heart J. 2001, 2, 384–387. [Google Scholar]
- Toscano, A.; Anaclerio, S.; Digilio, M.C.; Giannotti, A.; Fariello, G.; Dallapiccola, B.; Marino, B. Ventricular septal defect and deletion of chromosome 22q11: Anatomical types and aortic arch anomalies. Eur. J. Pediatr. 2002, 161, 116–117. [Google Scholar] [CrossRef]
- Marmon, L.M.; Balsara, R.K.; Chen, R.; Dunn, J.M. Congenital cardiac anomalies associated with the DiGeorge syndrome: A neonatal experience. Ann. Thorac. Surg. 1984, 38, 146–150. [Google Scholar] [CrossRef]
- Michielon, G.; Marino, B.; Formigari, R.; Gargiulo, G.; Picchio, F.; Digilio, M.C.; Anaclerio, S.; Oricchio, G.; Sanders, S.P.; Di Donato, R.M. Genetic syndromes and outcome after surgical correction of tetralogy of Fallot. Ann. Thorac. Surg. 2006, 81, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Michielon, G.; Marino, B.; Oricchio, G.; Digilio, M.C.; Iorio, F.; Filippelli, S.; Placidi, S.; Di Donato, R.M. Impact of DEL22q11, trisomy 21, and other genetic syndromes on surgical outcome of conotruncal heart defects. J. Thorac. Cardiovasc. Surg. 2009, 138, 565–570.e2. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Simsic, J.M.; Coleman, K.; Maher, K.O.; Cuadrado, A.; Kirshbom, P.M. Do neonates with genetic abnormalities have an increased morbidity and mortality following cardiac surgery? Congenit. Heart Dis. 2009, 4, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Anaclerio, S.; Di Ciommo, V.; Michielon, G.; Digilio, M.C.; Formigari, R.; Picchio, F.M.; Gargiulo, G.; Di Donato, R.; De Ioris, M.A.; Marino, B. Conotruncal heart defects: Impact of genetic syndromes on immediate operative mortality. Ital. Heart J. 2004, 5, 624–628. [Google Scholar] [PubMed]
- Carotti, A.; Digilio, M.C.; Piacentini, G.; Saffirio, C.; Di Donato, R.M.; Marino, B. Cardiac defects and results of cardiac surgery in 22q11.2 deletion syndrome. Dev. Disabil. Res. Rev. 2008, 14, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Kyburz, A.; Bauersfeld, U.; Schinzel, A.; Riegel, M.; Hug, M.; Tomaske, M.; Valsangiacomo Buchel, E.R. The fate of children with microdeletion 22q11.2 syndrome and congenital heart defect: Clinical course and cardiac outcome. Pediatr. Cardiol. 2008, 29, 76–83. [Google Scholar] [CrossRef]
- Ziolkowska, L.; Kawalec, W.; Turska-Kmiec, A.; Krajewska-Walasek, M.; Brzezinska-Rajszys, G.; Daszkowska, J.; Maruszewski, B.; Burczynski, P. Chromosome 22q11.2 microdeletion in children with conotruncal heart defects: Frequency, associated cardiovascular anomalies, and outcome following cardiac surgery. Eur. J. Pediatr. 2008, 167, 1135–1140. [Google Scholar] [CrossRef]
- McDonald, R.; Dodgen, A.; Goyal, S.; Gossett, J.M.; Shinkawa, T.; Uppu, S.C.; Blanco, C.; Garcia, X.; Bhutta, A.T.; Imamura, M.; et al. Impact of 22q11.2 deletion on the postoperative course of children after cardiac surgery. Pediatr. Cardiol. 2013, 34, 341–347. [Google Scholar] [CrossRef]
- Koth, A.; Sidell, D.; Bauser-Heaton, H.; Wise-Faberowski, L.; Hanley, F.L.; McElhinney, D.B.; Asija, R. Deletion of 22q11 chromosome is associated with postoperative morbidity after unifocalisation surgery. Cardiol. Young 2019, 29, 19–22. [Google Scholar] [CrossRef]
- Mercer-Rosa, L.; Pinto, N.; Yang, W.; Tanel, R.; Goldmuntz, E. 22q11.2 Deletion syndrome is associated with perioperative outcome in tetralogy of Fallot. J. Thorac. Cardiovasc. Surg. 2013, 146, 868–873. [Google Scholar] [CrossRef]
- O’Byrne, M.L.; Yang, W.; Mercer-Rosa, L.; Parnell, A.S.; Oster, M.E.; Levenbrown, Y.; Tanel, R.E.; Goldmuntz, E. 22q11.2 Deletion syndrome is associated with increased perioperative events and more complicated postoperative course in infants undergoing infant operative correction of truncus arteriosus communis or interrupted aortic arch. J. Thorac. Cardiovasc. Surg. 2014, 148, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, T.Y.; Scavonetto, F.; Hamlin, R.J.; Burkhart, H.M.; Sprung, J.; Weingarten, T.N. Perioperative management of patients with DiGeorge syndrome undergoing cardiac surgery. J. Cardiothorac. Vasc. Anesth. 2014, 28, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Carotti, A.; Albanese, S.B.; Filippelli, S.; Rava, L.; Guccione, P.; Pongiglione, G.; Di Donato, R.M. Determinants of outcome after surgical treatment of pulmonary atresia with ventricular septal defect and major aortopulmonary collateral arteries. J. Thorac. Cardiovasc. Surg. 2010, 140, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Besseau-Ayasse, J.; Violle-Poirsier, C.; Bazin, A.; Gruchy, N.; Moncla, A.; Girard, F.; Till, M.; Mugneret, F.; Coussement, A.; Pelluard, F.; et al. A French collaborative survey of 272 fetuses with 22q11.2 deletion: Ultrasound findings, fetal autopsies and pregnancy outcomes. Prenat. Diagn. 2014, 34, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Swillen, A.; Feys, H.; Adriaens, T.; Nelissen, L.; Mertens, L.; Gewillig, M.; Devriendt, K.; Fryns, J.P. Early motor development in young children with 22q.11 deletion syndrome and a conotruncal heart defect. Dev. Med. Child. Neurol. 2005, 47, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Atallah, J.; Joffe, A.R.; Robertson, C.M.; Leonard, N.; Blakley, P.M.; Nettel-Aguirre, A.; Sauve, R.S.; Ross, D.B.; Rebeyka, I.M. Western Canadian Complex Pediatric Therapies Project Follow-up Group Two-year general and neurodevelopmental outcome after neonatal complex cardiac surgery in patients with deletion 22q11.2: A comparative study. J. Thorac. Cardiovasc. Surg. 2007, 134, 772–779. [Google Scholar] [CrossRef]
- Maharasingam, M.; Ostman-Smith, I.; Pike, M.G. A cohort study of neurodevelopmental outcome in children with DiGeorge syndrome following cardiac surgery. Arch. Dis. Child. 2003, 88, 61–64. [Google Scholar] [CrossRef][Green Version]
- Matsuoka, R.; Kimura, M.; Scambler, P.J.; Morrow, B.E.; Imamura, S.; Minoshima, S.; Shimizu, N.; Yamagishi, H.; Joh-o, K.; Watanabe, S.; et al. Molecular and clinical study of 183 patients with conotruncal anomaly face syndrome. Hum. Genet. 1998, 103, 70–80. [Google Scholar] [CrossRef]
- McElhinney, D.B.; Driscoll, D.A.; Levin, E.R.; Jawad, A.F.; Emanuel, B.S.; Goldmuntz, E. Chromosome 22q11 deletion in patients with ventricular septal defect: Frequency and associated cardiovascular anomalies. Pediatrics 2003, 112, e472. [Google Scholar] [CrossRef]
- Momma, K.; Matsuoka, R.; Takao, A. Aortic arch anomalies associated with chromosome 22q11 deletion (CATCH 22). Pediatr. Cardiol. 1999, 20, 97–102. [Google Scholar] [CrossRef]
- Digilio, M.C.; Marino, B.; Giannotti, A.; Novelli, G.; Dallapiccola, B. Conotruncal heart defects and chromosome 22q11 microdeletion. J. Pediatr. 1997, 130, 675–677. [Google Scholar] [CrossRef]
- Goldmuntz, E.; Clark, B.J.; Mitchell, L.E.; Jawad, A.F.; Cuneo, B.F.; Reed, L.; McDonald-McGinn, D.; Chien, P.; Feuer, J.; Zackai, E.H.; et al. Frequency of 22q11 deletions in patients with conotruncal defects. J. Am. Coll. Cardiol. 1998, 32, 492–498. [Google Scholar] [CrossRef]
- Marino, B.; Digilio, M.C.; Persiani, M.; Di Donato, R.; Toscano, A.; Giannotti, A.; Dallapiccola, B. Deletion 22q11 in patients with interrupted aortic arch. Am. J. Cardiol. 1999, 84, 360–361. [Google Scholar] [CrossRef]
- Chessa, M.; Butera, G.; Bonhoeffer, P.; Iserin, L.; Kachaner, J.; Lyonnet, S.; Munnich, A.; Sidi, D.; Bonnet, D. Relation of genotype 22q11 deletion to phenotype of pulmonary vessels in tetralogy of Fallot and pulmonary atresia-ventricular septal defect. Heart 1998, 79, 186–190. [Google Scholar] [CrossRef]
- Galindo, A.; Gutierrez-Larraya, F.; Martinez, J.M.; Del Rio, M.; Graneras, A.; Velasco, J.M.; Puerto, B.; Gratacos, E. Prenatal diagnosis and outcome for fetuses with congenital absence of the pulmonary valve. Ultrasound Obs. Gynecol. 2006, 28, 32–39. [Google Scholar] [CrossRef]
- Babaoglu, K.; Altun, G.; Binnetoglu, K.; Donmez, M.; Kayabey, O.; Anik, Y. Crossed pulmonary arteries: A report on 20 cases with an emphasis on the clinical features and the genetic and cardiac abnormalities. Pediatr. Cardiol. 2013, 34, 1785–1790. [Google Scholar] [CrossRef]
- Momma, K.; Kondo, C.; Matsuoka, R. Tetralogy of Fallot with pulmonary atresia associated with chromosome 22q11 deletion. J. Am. Coll. Cardiol. 1996, 27, 198–202. [Google Scholar] [CrossRef][Green Version]
- Marino, B.; Digilio, M.C.; Dallapiccola, B. Severe truncal valve dysplasia: Association with DiGeorge syndrome? Ann. Thorac. Surg. 1998, 66, 980. [Google Scholar]
- Marino, B.; Digilio, M.C.; Toscano, A.; Dallapiccola, B. Deficiency of the infundibular septum in patients with interrupted aortic arch and del 22q11. Cardiol. Young 2000, 10, 428–429. [Google Scholar] [CrossRef][Green Version]
- Momma, K.; Ando, M.; Matsuoka, R.; Joo, K. Interruption of the aortic arch associated with deletion of chromosome 22q11 is associated with a subarterial and doubly committed ventricular septal defect in Japanese patients. Cardiol. Young 1999, 9, 463–467. [Google Scholar] [CrossRef]
- John, A.S.; McDonald-McGinn, D.M.; Zackai, E.H.; Goldmuntz, E. Aortic root dilation in patients with 22q11.2 deletion syndrome. Am. J. Med. Genet. A 2009, 149A, 939–942. [Google Scholar] [CrossRef] [PubMed]
- De Rinaldis, C.P.; Butensky, A.; Patel, S.; Edman, S.; Wasserman, M.; McGinn, D.E.; Bailey, A.; Zackai, E.H.; Crowley, T.B.; McDonald-McGinn, D.M.; et al. Aortic Root Dilation in Patients with 22q11.2 Deletion Syndrome without Intracardiac Anomalies. Pediatr. Cardiol. 2021, 42, 1594–1600. [Google Scholar] [CrossRef] [PubMed]
- Niwa, K. Aortic dilatation in complex congenital heart disease. Cardiovasc. Diagn. Ther. 2018, 8, 725–738. [Google Scholar] [CrossRef] [PubMed]
- John, A.S.; Rychik, J.; Khan, M.; Yang, W.; Goldmuntz, E. 22q11.2 deletion syndrome as a risk factor for aortic root dilation in tetralogy of Fallot. Cardiol. Young 2014, 24, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Merscher, S.; Funke, B.; Epstein, J.A.; Heyer, J.; Puech, A.; Lu, M.M.; Xavier, R.J.; Demay, M.B.; Russell, R.G.; Factor, S.; et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 2001, 104, 619–629. [Google Scholar] [CrossRef]
- Lindsay, E.A.; Vitelli, F.; Su, H.; Morishima, M.; Huynh, T.; Pramparo, T.; Jurecic, V.; Ogunrinu, G.; Sutherland, H.F.; Scambler, P.J.; et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 2001, 410, 97–101. [Google Scholar] [CrossRef]
- Xu, H.; Morishima, M.; Wylie, J.N.; Schwartz, R.J.; Bruneau, B.G.; Lindsay, E.A.; Baldini, A. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development 2004, 131, 3217–3227. [Google Scholar] [CrossRef]
- Yagi, H.; Furutani, Y.; Hamada, H.; Sasaki, T.; Asakawa, S.; Minoshima, S.; Ichida, F.; Joo, K.; Kimura, M.; Imamura, S.; et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 2003, 362, 1366–1373. [Google Scholar] [CrossRef]
- Mastromoro, G.; Calcagni, G.; Versacci, P.; Putotto, C.; Chinali, M.; Lambiase, C.; Unolt, M.; Pelliccione, E.; Anaclerio, S.; Caprio, C.; et al. Left pulmonary artery in 22q11.2 deletion syndrome. Echocardiographic evaluation in patients without cardiac defects and role of Tbx1 in mice. PLoS ONE 2019, 14, e0211170. [Google Scholar] [CrossRef]
- Goldmuntz, E. 22q11.2 Deletion Syndrome and Congenital Heart Disease. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 64–72. [Google Scholar] [CrossRef]
- Zhao, Y.; Diacou, A.; Johnston, H.R.; Musfee, F.I.; McDonald-McGinn, D.M.; McGinn, D.; Crowley, T.B.; Repetto, G.M.; Swillen, A.; Breckpot, J.; et al. Complete Sequence of the 22q11.2 Allele in 1,053 Subjects with 22q11.2 Deletion Syndrome Reveals Modifiers of Conotruncal Heart Defects. Am. J. Hum. Genet. 2020, 106, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Chapnik, E.; Sasson, V.; Blelloch, R.; Hornstein, E. Dgcr8 controls neural crest cells survival in cardiovascular development. Dev. Biol. 2012, 362, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.S.; McDonald-McGinn, D.M.; Devriendt, K.; Digilio, M.C.; Goldenberg, P.; Habel, A.; Marino, B.; Oskarsdottir, S.; Philip, N.; Sullivan, K.; et al. International 22q11.2 Deletion Syndrome Consortium Practical guidelines for managing patients with 22q11.2 deletion syndrome. J. Pediatr. 2011, 159, 332–339.e1. [Google Scholar] [CrossRef] [PubMed]
- Fung, W.L.; Butcher, N.J.; Costain, G.; Andrade, D.M.; Boot, E.; Chow, E.W.; Chung, B.; Cytrynbaum, C.; Faghfoury, H.; Fishman, L.; et al. Practical guidelines for managing adults with 22q11.2 deletion syndrome. Genet. Med. 2015, 17, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Unolt, M.; Versacci, P.; Anaclerio, S.; Lambiase, C.; Calcagni, G.; Trezzi, M.; Carotti, A.; Crowley, T.B.; Zackai, E.H.; Goldmuntz, E.; et al. Congenital heart diseases and cardiovascular abnormalities in 22q11.2 deletion syndrome: From well-established knowledge to new frontiers. Am. J. Med. Genet. A 2018, 176, 2087–2098. [Google Scholar] [CrossRef]
- Digilio, M.C.; Marino, B.; Grazioli, S.; Agostino, D.; Giannotti, A.; Dallapiccola, B. Comparison of occurrence of genetic syndromes in ventricular septal defect with pulmonic stenosis (classic tetralogy of Fallot) versus ventricular septal defect with pulmonic atresia. Am. J. Cardiol. 1996, 77, 1375–1376. [Google Scholar] [CrossRef]
- Peyvandi, S.; Ingall, E.; Woyciechowski, S.; Garbarini, J.; Mitchell, L.E.; Goldmuntz, E. Risk of congenital heart disease in relatives of probands with conotruncal cardiac defects: An evaluation of 1620 families. Am. J. Med. Genet. A 2014, 164A, 1490–1495. [Google Scholar] [CrossRef]
- Mercer-Rosa, L.; Elci, O.U.; Pinto, N.M.; Tanel, R.E.; Goldmuntz, E. 22q11.2 Deletion Status and Perioperative Outcomes for Tetralogy of Fallot with Pulmonary Atresia and Multiple Aortopulmonary Collateral Vessels. Pediatr. Cardiol. 2018, 39, 906–910. [Google Scholar] [CrossRef]
- Carotti, A.; Di Donato, R.M.; Squitieri, C.; Guccione, P.; Catena, G. Total repair of pulmonary atresia with ventricular septal defect and major aortopulmonary collaterals: An integrated approach. J. Thorac. Cardiovasc. Surg. 1998, 116, 914–923. [Google Scholar] [CrossRef][Green Version]
- Mahle, W.T.; Crisalli, J.; Coleman, K.; Campbell, R.M.; Tam, V.K.; Vincent, R.N.; Kanter, K.R. Deletion of chromosome 22q11.2 and outcome in patients with pulmonary atresia and ventricular septal defect. Ann. Thorac. Surg. 2003, 76, 567–571. [Google Scholar] [CrossRef]
- Momma, K.; Ando, M.; Matsuoka, R. Truncus arteriosus communis associated with chromosome 22q11 deletion. J. Am. Coll. Cardiol. 1997, 30, 1067–1071. [Google Scholar] [CrossRef]
- Marino, B.; Digilio, M.C.; Toscano, A. Common arterial trunk, DiGeorge syndrome and microdeletion 22q11. Prog. Ped Card 2002, 15, 9–17. [Google Scholar] [CrossRef]
- Ghimire, L.V.; Devoe, C.; Moon-Grady, A.J. 22q11.2 Deletion Status Influences Resource Utilization in Infants Requiring Repair of Tetralogy of Fallot and Common Arterial Trunk. Pediatr. Cardiol. 2020, 41, 918–924. [Google Scholar] [CrossRef]
- Alsoufi, B.; Gillespie, S.; Mahle, W.T.; Deshpande, S.; Kogon, B.; Maher, K.; Kanter, K. The Effect of Noncardiac and Genetic Abnormalities on Outcomes Following Neonatal Congenital Heart Surgery. Semin. Thorac. Cardiovasc. Surg. 2016, 28, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Aggarwal, A.; Shaw, M.; Gulati, G.S.; Kothari, S.S.; Ramakrishnan, S.; Saxena, A.; Devagourou, V.; Talwar, S.; Sharma, S.; et al. Clarifying the anatomy of common arterial trunk: A clinical study of 70 patients. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 914–922. [Google Scholar] [CrossRef]
- Hamzah, M.; Othman, H.F.; Daphtary, K.; Komarlu, R.; Aly, H. Outcomes of truncus arteriosus repair and predictors of mortality. J. Card. Surg. 2020, 35, 1856–1864. [Google Scholar] [CrossRef] [PubMed]
- Mastropietro, C.W.; Amula, V.; Sassalos, P.; Buckley, J.R.; Smerling, A.J.; Iliopoulos, I.; Riley, C.M.; Jennings, A.; Cashen, K.; Narasimhulu, S.S.; et al. Collaborative Research in Pediatric Cardiac Intensive Care Investigators Characteristics and operative outcomes for children undergoing repair of truncus arteriosus: A contemporary multicenter analysis. J. Thorac. Cardiovasc. Surg. 2019, 157, 2386–2398.e4. [Google Scholar] [CrossRef]
- Russell, H.M.; Pasquali, S.K.; Jacobs, J.P.; Jacobs, M.L.; O’Brien, S.M.; Mavroudis, C.; Backer, C.L. Outcomes of repair of common arterial trunk with truncal valve surgery: A review of the society of thoracic surgeons congenital heart surgery database. Ann. Thorac. Surg. 2012, 93, 164–169, discussion 169. [Google Scholar] [CrossRef]
- McCrindle, B.W.; Tchervenkov, C.I.; Konstantinov, I.E.; Williams, W.G.; Neirotti, R.A.; Jacobs, M.L.; Blackstone, E.H.; Congenital Heart Surgeons Society. Risk factors associated with mortality and interventions in 472 neonates with interrupted aortic arch: A Congenital Heart Surgeons Society study. J. Thorac. Cardiovasc. Surg. 2005, 129, 343–350. [Google Scholar] [CrossRef]
- Sanchez Mejia, A.A.; Cambronero, N.; Dongarwar, D.; Salihu, H.M.; Vigil-Mallette, M.A.; Garcia, B.Y.; Morris, S.A. Hospital Outcomes among Infants with Interrupted Aortic Arch with Simple and Complex Associated Heart Defects. Am. J. Cardiol. 2022, 166, 97–106. [Google Scholar] [CrossRef]
- Shen, L.; Gu, H.; Wang, D.; Yang, C.; Xu, Z.; Jing, H.; Jiang, Y.; Ding, Y.; Hou, H.; Ge, Z.; et al. Influence of chromosome 22q11.2 microdeletion on postoperative calcium level after cardiac-correction surgery. Pediatr. Cardiol. 2011, 32, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kosaka, K.; Kimura, M.; Imamura, S.; Yamada, O.; Iwai, K.; Matsuoka, R. Thrombocytopenia in patients with 22q11.2 deletion syndrome and its association with glycoprotein Ib-beta. Genet. Med. 2003, 5, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Stransky, C.; Basta, M.; McDonald-McGinn, D.M.; Solot, C.B.; Drummond, D.; Zackai, E.; LaRossa, D.; Kirschner, R.; Jackson, O. Perioperative risk factors in patients with 22q11.2 deletion syndrome requiring surgery for velopharyngel dysfunction. Cleft Palate Craniofac. J. 2015, 52, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Sacca, R.; Zur, K.B.; Crowley, T.B.; Zackai, E.H.; Valverde, K.D.; McDonald-McGinn, D.M. Association of airway abnormalities with 22q11.2 deletion syndrome. Int. J. Pediatr. Otorhinolaryngol. 2017, 96, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Shashi, V.; Berry, M.N.; Hines, M.H. Vasomotor instability in neonates with chromosome 22q11 deletion syndrome. Am. J. Med. Genet. A 2003, 121A, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Wylam, M.E.; Feldt, R.H.; Porter, C.J.; Dewald, G.; Scanlon, P.D.; Driscoll, D.J. Pulmonary atresia with ventricular septal defect and persistent airway hyperresponsiveness. J. Thorac. Cardiovasc. Surg. 2001, 122, 169–177. [Google Scholar] [CrossRef][Green Version]


{kind=link}
| Congenital Heart Diseases | % 22q11.2DS Patients (*) | % Cases Associated with 22q11.2DS (*) |
|---|---|---|
| Tetralogy of Fallot | 20–45% | 10–15% |
| Pulmonary atresia + VSD | 10–25% | 30–45% |
| Aortic arch interruption | 5–20% | 50–80% |
| Truncus arteriosus | 5–10% | 30–50% |
| Conoventricular VSD | 10–50% | 5% |
| Isolated aortic arch anomalies | 10% | 25% |
| Problem | Perioperative Management (*) |
|---|---|
| Immunological disorders |
|
| Hypocalcemia |
|
| Thrombocytopenia |
|
| Velopharyngeal, upper cervical spine, and neck vessels abnormalities |
|
| Risk of pulmonary hyper-reactivity and vasomotor instability |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Putotto, C.; Pugnaloni, F.; Unolt, M.; Maiolo, S.; Trezzi, M.; Digilio, M.C.; Cirillo, A.; Limongelli, G.; Marino, B.; Calcagni, G.; et al. 22q11.2 Deletion Syndrome: Impact of Genetics in the Treatment of Conotruncal Heart Defects. Children 2022, 9, 772. https://doi.org/10.3390/children9060772
Putotto C, Pugnaloni F, Unolt M, Maiolo S, Trezzi M, Digilio MC, Cirillo A, Limongelli G, Marino B, Calcagni G, et al. 22q11.2 Deletion Syndrome: Impact of Genetics in the Treatment of Conotruncal Heart Defects. Children. 2022; 9(6):772. https://doi.org/10.3390/children9060772
Chicago/Turabian StylePutotto, Carolina, Flaminia Pugnaloni, Marta Unolt, Stella Maiolo, Matteo Trezzi, Maria Cristina Digilio, Annapaola Cirillo, Giuseppe Limongelli, Bruno Marino, Giulio Calcagni, and et al. 2022. "22q11.2 Deletion Syndrome: Impact of Genetics in the Treatment of Conotruncal Heart Defects" Children 9, no. 6: 772. https://doi.org/10.3390/children9060772
APA StylePutotto, C., Pugnaloni, F., Unolt, M., Maiolo, S., Trezzi, M., Digilio, M. C., Cirillo, A., Limongelli, G., Marino, B., Calcagni, G., & Versacci, P. (2022). 22q11.2 Deletion Syndrome: Impact of Genetics in the Treatment of Conotruncal Heart Defects. Children, 9(6), 772. https://doi.org/10.3390/children9060772