
The Evaluation of FGFR1, FGFR2 and FOXO1 in Orofacial Cleft Tissue



Abstract
1. Introduction
2. Materials and Methods
2.1. Information about the Patients
2.2. Chromogenic In Situ Hybridization
2.3. Statistical Analysis
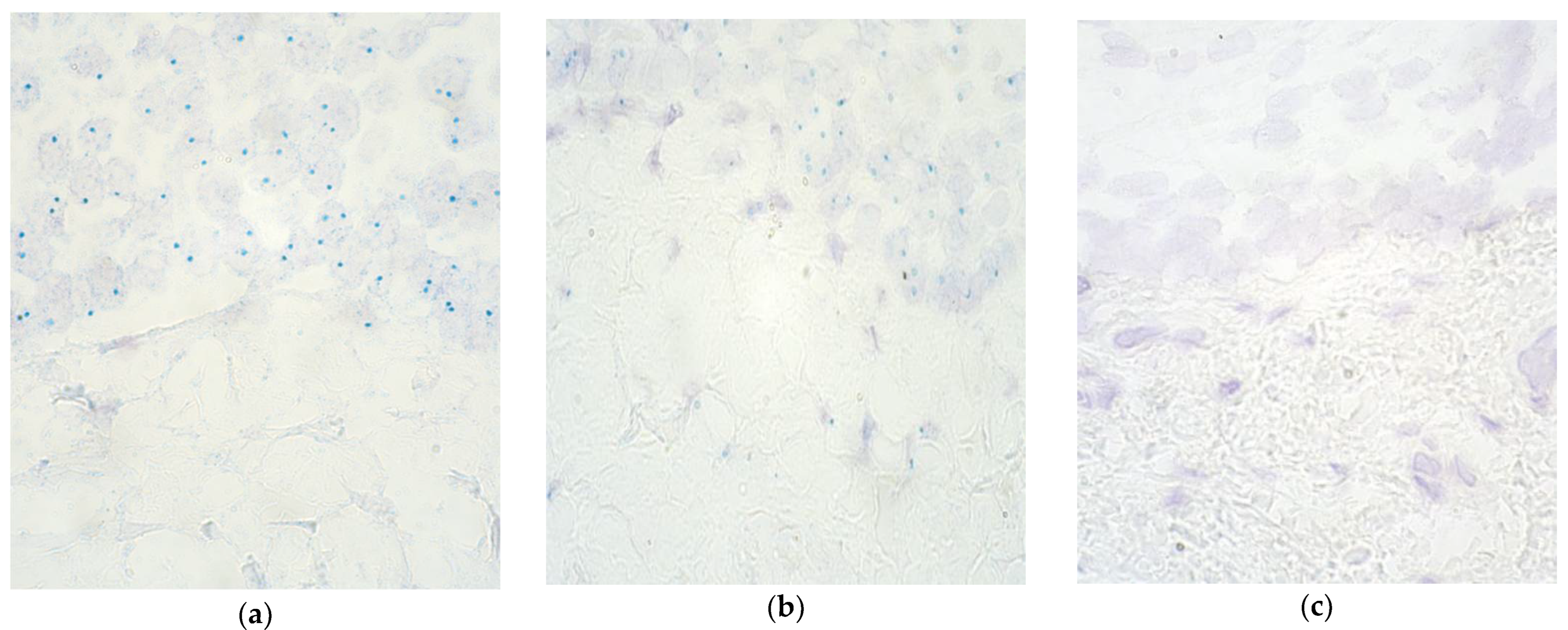
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahimov, F.; Jugessur, A.; Murray, J.C. Genetics of Nonsyndromic Orofacial Clefts. Cleft Palate-Craniofacial J. 2012, 49, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Sadler, T.W. Langman’s Medical Embryology, 12th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; pp. 275–278. [Google Scholar]
- Meng, L.; Bian, Z.; Torensma, R.; Von den Hoff, J.W. Biological Mechanisms in Palatogenesis and Cleft Palate. J. Dent. Res. 2009, 88, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Stanier, P.; Pauws, E. Development of the Lip and Palate: FGF Signalling. Front. Oral Biol. 2012, 16, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Mossey, P.A.; Modell, B. Epidemiology of Oral Clefts 2012: An International Perspective. Front. Oral Biol. 2012, 16, 1–18. [Google Scholar] [CrossRef]
- Mulliken, J.B. The changing faces of children with cleft lip and palate. N. Engl. J. Med. 2004, 351, 745–747. [Google Scholar] [CrossRef]
- Durning, P.; Chestnutt, I.G.; Morgan, M.Z.; Lester, N.J. The relationship between orofacial clefts and material deprivation in Wales. Cleft Palate-Craniofacial J. 2007, 44, 203–207. [Google Scholar] [CrossRef]
- Clark, J.D.; Mossey, P.A.; Sharp, L.; Little, J. Socioeconomic status and orofacial clefts in Scotland, 1989 to 1998. Cleft Palate-Craniofacial J. 2003, 40, 481–485. [Google Scholar] [CrossRef]
- Mossey, P.A.; Little, J.; Munger, R.G.; Dixon, M.J.; Shaw, W.C. Cleft lip and palate. Lancet 2009, 374, 1773–1785. [Google Scholar] [CrossRef]
- Yin, X.; Li, J.; Li, Y.; Zou, S. Maternal alcohol consumption and oral clefts: A meta-analysis. Br. J. Oral Maxillofac. Surg. 2019, 57, 839–846. [Google Scholar] [CrossRef]
- Little, J.; Cardy, A.; Munger, R.G. Tobacco smoking and oral clefts: A meta-analysis. Bull. World Health Organ. 2004, 82, 213–218. [Google Scholar]
- Millacura, N.; Pardo, R.; Cifuentes, L.; Suazo, J. Effects of folic acid fortification on orofacial clefts prevalence: A meta-analysis. Public Health Nutr. 2017, 20, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.J.; Marazita, M.L.; Beaty, T.H.; Murray, J.C. Cleft lip and palate: Understanding genetic and environmental influences. Nat. Rev. Genet. 2011, 12, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.Q.; Zuo, X.B.; He, M.; Gao, J.P.; Fu, Y.C.; Qin, C.Q.; Meng, L.Y.; Wang, W.J.; Song, Y.L.; Cheng, Y.; et al. Genome-wide analyses of non-syndromic cleft lip with palate identify 14 novel loci and genetic heterogeneity. Nat. Commun. 2017, 8, 14364. [Google Scholar] [CrossRef] [PubMed]
- Conte, F.; Oti, M.; Dixon, J.; Carels, C.E.L.; Rubini, M.; Zhou, H.Q. Systematic analysis of copy number variants of a large cohort of orofacial cleft patients identifies candidate genes for orofacial clefts. Hum. Genet. 2016, 135, 41–59. [Google Scholar] [CrossRef]
- Scapoli, L.; Palmieri, A.; Martinelli, M.; Pezzetti, F.; Carinci, P.; Tognon, M.; Carinci, F. Strong evidence of linkage disequilibrium between polymorphisms at the IRF6 locus and nonsyndromic cleft lip with or without cleft palate, in an Italian population. Am. J. Hum. Genet. 2005, 76, 180–183. [Google Scholar] [CrossRef]
- Park, J.W.; McIntosh, I.; Hetmanski, J.B.; Jabs, E.W.; Vander Kolk, C.A.; Wu-Chou, Y.H.; Chen, P.K.; Chong, S.S.; Yeow, V.; Jee, S.H.; et al. Association between IRF6 and nonsyndromic cleft lip with or without cleft palate in four populations. Genet. Med. 2007, 9, 219–227. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef]
- Kelleher, F.C.; O’Sullivan, H.; Smyth, E.; McDermott, R.; Viterbo, A. Fibroblast growth factor receptors, developmental corruption and malignant disease. Carcinogenesis 2013, 34, 2198–2205. [Google Scholar] [CrossRef]
- Trueb, B. Biology of FGFRL1, the fifth fibroblast growth factor receptor. Cell Mol. Life Sci. 2011, 68, 951–964. [Google Scholar] [CrossRef]
- Yu, K.; Karuppaiah, K.; Ornitz, D.M. Mesenchymal fibroblast growth factor receptor signaling regulates palatal shelf elevation during secondary palate formation. Dev. Dyn. 2015, 244, 1427–1438. [Google Scholar] [CrossRef]
- Riley, B.M.; Mansilla, M.A.; Ma, J.; Daack-Hirsch, S.; Maher, B.S.; Raffensperger, L.M.; Russo, E.T.; Vieira, A.R.; Dode, C.; Mohammadi, M.; et al. Impaired FGF signaling contributes to cleft lip and palate. Proc. Natl. Acad. Sci. USA 2007, 104, 4512–4517. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, T.X.; Wu, T.; Hetmanski, J.B.; Ruczinski, I.G.; Schwender, H.; Liang, K.Y.; Murray, T.; Fallin, D.; Redett, R.J.; et al. The FGF and FGFR Gene Family and Risk of Cleft Lip With or Without Cleft Palate. Cleft Palate-Craniofacial J. 2013, 50, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Pilmane, M.; Jain, V.N.; Vitenberga-Verza, Z. Expression Analysis of FGF/FGFR and FOX Family Proteins in Mucosal Tissue Obtained from Orofacial Cleft-Affected Children. Biology 2021, 10, 423. [Google Scholar] [CrossRef] [PubMed]
- Rafiqdoost, Z.; Rafiqdoost, A.; Rafiqdoost, H.; Hashemi, M.; Khayatzadeh, J.; Eskandari-Nasab, E. Investigation of FGF1 and FGFR gene polymorphisms in a group of Iranian patients with nonsyndromic cleft lip with or without cleft palate. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 731–736. [Google Scholar] [CrossRef]
- Mostowska, A.; Hozyasz, K.K.; Wojcicki, P.; Biedziak, B.; Paradowska, P.; Jagodzinski, P.P. Association between Genetic Variants of Reported Candidate Genes or Regions and Risk of Cleft Lip with or without Cleft Palate in the Polish Population. Birth Defects Res. Part A Clin. Mol. Teratol. 2010, 88, 538–545. [Google Scholar] [CrossRef]
- Leslie, E.J.; Taub, M.A.; Liu, H.; Steinberg, K.M.; Koboldt, D.C.; Zhang, Q.Y.; Carlson, J.C.; Hetmanski, J.B.; Wang, H.; Larson, D.E.; et al. Identification of Functional Variants for Cleft Lip with or without Cleft Palate in or near PAX7, FGFR2, and NOG by Targeted Sequencing of GWAS Loci. Am. J. Hum. Genet. 2015, 96, 397–411. [Google Scholar] [CrossRef]
- Jackson, B.C.; Carpenter, C.; Nebert, D.W.; Vasiliou, V. Update of human and mouse forkhead box (FOX) gene families. Hum. Genom. 2010, 4, 345–352. [Google Scholar] [CrossRef]
- Schuff, M.; Siegel, D.; Bardine, N.; Oswald, F.; Donow, C.; Knoechel, W. FoxO genes are dispensable during gastrulation but required for late embryogenesis in Xenopus laevis. Dev. Biol. 2010, 337, 259–273. [Google Scholar] [CrossRef]
- Hosaka, T.; Biggs, W.H.; Tieu, D.; Boyer, A.D.; Varki, N.M.; Cavenee, W.K.; Arden, K.C. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc. Natl. Acad. Sci. USA 2004, 101, 2975–2980. [Google Scholar] [CrossRef]
- Furuyama, T.; Kitayama, K.; Shimoda, Y.; Ogawa, M.; Sone, K.; Yoshida-Araki, K.; Hisatsune, H.; Nishikawa, S.; Nakayama, K.; Nakayama, K.; et al. Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J. Biol. Chem. 2004, 279, 34741–34749. [Google Scholar] [CrossRef]
- Poche, R.A.; Sharma, R.; Garcia, M.D.; Wada, A.M.; Nolte, M.J.; Udan, R.S.; Paik, J.H.; DePinho, R.A.; Bartlett, J.D.; Dickinson, M.E. Transcription Factor FoxO1 Is Essential for Enamel Biomineralization. PLoS ONE 2012, 7, e30357. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, F.; Xiong, Z.Y.; Huo, J.W.; Li, W.; Jiang, B.H.; Mao, W.; He, B.; Wang, X.J.; Li, G.Z. The cleft palate candidate gene BAG6 supports FoxO1 acetylation to promote FasL-mediated apoptosis during palate fusion. Exp. Cell Res. 2020, 396, 112310. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.L.; Li, R.; Ren, W.; Wang, Z.C.; Wang, Z.H.; Yang, M.S.; Zhang, S.H. The FOXO1 Gene-Obesity Interaction Increases the Risk of Type 2 Diabetes Mellitus in a Chinese Han Population. J. Korean Med. Sci. 2017, 32, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Reliable and Simple Detection of Genomic Alterations Using Light Microscopy. ZytoDotR 2CTM-2-Color CISH for the Detection of Genomic Alterations. A User Manual Provided by ZytoVision GmbH-Fischkai 1, 27572 Bremerhaven- Germany. ZytoVision Molecular Diagnostics Simplified. Available online: www.Zytovision.com (accessed on 13 December 2021).
- Rosa, F.E.; Silveira, S.M.; Silveira, C.G.T.; Bergamo, N.A.; Neto, F.A.M.; Domingues, M.A.C.; Soares, F.A.; Caldeira, J.R.F.; Rogatto, S.R. Quantitative real-time RT-PCR and chromogenic in situ hybridization: Precise methods to detect HER-2 status in breast carcinoma. BMC CANCER 2009, 9, 90. [Google Scholar] [CrossRef]
- Rosa, F.E.; Santos, R.M.; Rogatto, S.R.; Domingues, M.A.C. Chromogenic in situ hybridization compared with other approaches to evaluate HER2/neu status in breast carcinomas. Braz. J. Med. Biol. Res. 2013, 46, 207–216. [Google Scholar] [CrossRef]
- Ayatollahi, H.; Fani, A.; Ghayoor Karimiani, E.; Homaee, F.; Shajiei, A.; Sheikh, M.; Shakeri, S.; Shams, S.F. Chromogenic in situ Hybridization Compared with Real Time Quantitative Polymerase Chain Reaction to Evaluate HER2/neu Status in Breast Cancer. Iran. J. Pathol. 2017, 12, 128–134. [Google Scholar] [CrossRef]
- Pilmane, M.; Rumba, I.; Sundler, F.; Luts, A. Patterns of distribution and occurrence of neuroendocrine elements in lungs of humans with chronic lung disease. Proc. Latv. Acad. Sci. 1998, 52, 144–152. [Google Scholar]
- Vitenberga, Z.; Pilmane, M.; Babjoniseva, A. The evaluation of inflammatory, anti-inflammatory and regulatory factors contrib- uting to the pathogenesis of COPD in airways. Pathol. Res. Pract. 2019, 215, 97–105. [Google Scholar] [CrossRef]
- Bush, J.O.; Jiang, R.L. Palatogenesis: Morphogenetic and molecular mechanisms of secondary palate development. Development 2012, 139, 231–243. [Google Scholar] [CrossRef]
- Stoler, J.M.; Rosen, H.; Desai, U.; Mulliken, J.B.; Meara, J.G.; Rogers, G.F. Cleft Palate in Pfeiffer Syndrome. J. Craniofacial Surg. 2009, 20, 1375–1377. [Google Scholar] [CrossRef]
- Snyder-Warwick, A.K.; Perlyn, C.A.; Pan, J.; Yu, K.; Zhang, L.J.; Ornitz, D.M. Analysis of a gain-of-function FGFR2 Crouzon mutation provides evidence of loss of function activity in the etiology of cleft palate. Proc. Natl. Acad. Sci. USA 2010, 107, 2515–2520. [Google Scholar] [CrossRef] [PubMed]
- Pitteloud, N.; Acierno, J.S.; Meysing, A.; Eliseenkova, A.V.; Ma, J.H.; Ibrahimi, O.A.; Metzger, D.L.; Hayes, F.J.; Dwyer, A.A.; Hughes, V.A.; et al. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA 2006, 103, 6281–6286. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Herrick, S.R.; Lemyre, E.; Kishikawa, S.; Salisz, J.A.; Seminara, S.; MacDonald, M.E.; Bruns, G.A.P.; Morton, C.C.; Quade, B.J.; et al. Hypogonadotropic hypogonadism and cleft lip and palate caused by a balanced translocation producing haploinsufficiency for FGFR1. J. Med. Genet. 2005, 42, 666–672. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dode, C.; Levilliers, J.; Dupont, J.M.; De Paepe, A.; Le Du, N.; Soussi-Yanicostas, N.; Coimbra, R.S.; Delmaghani, S.; Compain-Nouaille, S.; Baverel, F.; et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 2003, 33, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Cammarata-Scalisi, F.; Yilmaz, E.; Callea, M.; Avendano, A.; Mihci, E.; Alper, O.M. Clinical and genetic findings of two cases with Apert syndrome. Bol. Med. Hosp. Infant. Mex. 2019, 76, 44–48. [Google Scholar] [CrossRef]
- Wang, C.; Chang, J.Y.F.; Yang, C.F.; Huang, Y.Q.; Liu, J.C.; You, P.; McKeehan, W.L.; Wang, F.; Li, X.K. Type 1 Fibroblast Growth Factor Receptor in Cranial Neural Crest Cell-derived Mesenchyme Is Required for Palatogenesis. J. Biol. Chem. 2013, 288, 22174–22183. [Google Scholar] [CrossRef]
- Welsh, I.C.; Hagge-Greenberg, A.; O’Brien, T.P. A dosage-dependent role for Spry2 in growth and patterning during palate development. Mech. Dev. 2007, 124, 746–761. [Google Scholar] [CrossRef]
- Pilmane, M.; Sidhoma, E.; Akota, I.; Kazoka, D. Characterization of Cytokines and Proliferation Marker Ki67 in Cleft Affected Lip Tissue. Med. Lith. 2019, 55, 518. [Google Scholar] [CrossRef]
- Li, J.P.; Shi, S.; Srivastava, S.P.; Kitada, M.; Nagai, T.; Nitta, K.; Kohno, M.; Kanasaki, K.; Koya, D. FGFR1 is critical for the anti-endothelial mesenchymal transition effect of N-acetyl-seryl-aspartyl-lysyl-proline via induction of the MAP4K4 pathway. Cell Death Dis. 2017, 8, e2965. [Google Scholar] [CrossRef]
- Li, J.P.; Liu, H.J.; Srivastava, S.P.; Hu, Q.Y.; Gao, R.F.; Li, S.L.; Kitada, M.; Wu, G.S.; Koya, D.; Kanasaki, K. Endothelial FGFR1 (Fibroblast Growth Factor Receptor 1) Deficiency Contributes Differential Fibrogenic Effects in Kidney and Heart of Diabetic Mice. Hypertension 2020, 76, 1935–1944. [Google Scholar] [CrossRef]
- Lou, D.Y.; Han, J.B.; Zhou, L.Q.; Ma, H.J.; Xv, J.J.; Shou, J.W.; Xu, Z.X.; Jiang, L.Q.; Qian, Y.Y. Fibroblast growth factor receptor 1 antagonism attenuates lipopolysaccharide-induced activation of hepatic stellate cells via suppressing inflammation. Exp. Ther. Med. 2018, 16, 2909–2916. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, R.; Rajendran, V.; Giraldo-Velasquez, M.; Megalofonou, F.F.; Gurski, F.; Stadelmann, C.; Karnati, S.; Berghoff, M. Oligodendrocyte-Specific Deletion of FGFR1 Reduces Cerebellar Inflammation and Neurodegeneration in MOG(35-55)-Induced EAE. Int. J. Mol. Sci. 2021, 22, 9495. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Wang, F.; Li, H.; Xu, S.Y.; Xu, W.W.; Pan, X.J.; Hu, Y.F.; Mao, L.J.; Qian, S.Z.; Pan, J.Y. Inhibition of Fibroblast Growth Factor Receptor by AZD4547 Protects Against Inflammation in Septic Mice. Inflammation 2019, 42, 1957–1967. [Google Scholar] [CrossRef]
- Goida, J.; Pilmane, M. Characterization of Macrophages and TNF-alpha in Cleft Affected Lip Tissue. Cosmetics 2021, 8, 42. [Google Scholar] [CrossRef]
- Rice, R.; Spencer-Dene, B.; Connor, E.C.; Gritli-Linde, A.; McMahon, A.P.; Dickson, C.; Thesleff, I.; Rice, D.P.C. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J. Clin. Investig. 2004, 113, 1692–1700. [Google Scholar] [CrossRef]
- Edlund, R.K.; Ohyama, T.; Kantarci, H.; Riley, B.B.; Groves, A.K. Foxi transcription factors promote pharyngeal arch development by regulating formation of FGF signaling centers. Dev. Biol. 2014, 390, 1–13. [Google Scholar] [CrossRef]
- Shirokova, V.; Jussila, M.; Hytonen, M.K.; Perala, N.; Drogemuller, C.; Leeb, T.; Lohi, H.; Sainio, K.; Thesleff, I.; Mikkola, M.L. Expression of Foxi3 is regulated by ectodysplasin in skin appendage placodes. Dev. Dyn 2013, 242, 593–603. [Google Scholar] [CrossRef]
- Shirokova, V.; Biggs, L.C.; Jussila, M.; Ohyama, T.; Groves, A.K.; Mikkola, M.L. Foxi3 Deficiency Compromises Hair Follicle Stem Cell Specification and Activation. Stem Cells 2016, 34, 1896–1908. [Google Scholar] [CrossRef]
- Hosokawa, R.; Deng, X.M.; Takamori, K.; Xu, X.; Urata, M.; Bringas, P.; Chai, Y. Epithelial-Specific Requirement of FGFR2 Signaling During Tooth and Palate Development. J. Exp. Zool. Part B Mol. Dev. Evol. 2009, 312, 343–350. [Google Scholar] [CrossRef]
- Martinez-Abadias, N.; Holmes, G.; Pankratz, T.; Wang, Y.L.; Zhou, X.Y.; Jabs, E.W.; Richtsmeier, J.T. From shape to cells: Mouse models reveal mechanisms altering palate development in Apert syndrome. Dis. Model. Mech. 2013, 6, 768–779. [Google Scholar] [CrossRef]
- Villarejo-Balcells, B.; Guichard, S.; Rigby, P.M.; Carvajal, J.J. Expression pattern of the FoxO1 gene during mouse embryonic development. Gene Expr. Patterns 2011, 11, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Albahrani, Y.; Zeichner, J.A. Acneiform Eruptions in Dermatology: A Differential Diagnosis; Springer: New York, NY, USA, 2014; pp. 179–181. [Google Scholar]
- Cammarata-Scalisi, F.; Avendano, A.; Callea, M. Main genetic entities associated with supernumerary teeth. Arch. Argent. Pediatr. 2018, 116, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.C.; Liu, Y.X.; Thant, L.M.; Pang, J.; Palmer, G.; Alikhani, M. Foxo1, a Novel Regulator of Osteoblast Differentiation and Skeletogenesis. J. Biol. Chem. 2010, 285, 31055–31065. [Google Scholar] [CrossRef] [PubMed]
- Shirai, Y.; Kawabe, K.; Tosa, I.; Tsukamoto, S.; Yamada, D.; Takarada, T. Runx2 function in cells of neural crest origin during intramembranous ossification. Biochem. Biophys. Res. Commun. 2019, 509, 1028–1033. [Google Scholar] [CrossRef]
- Cammarata-Scalisi, F.; Girardi, K.; Strocchio, L.; Merli, P.; Bernardin, A.G.; Galeotti, A.; Magliarditi, F.; Inserra, A.; Callea, M. Oral Manifestations and Complications in Childhood Acute Myeloid Leukemia. Cancers 2020, 12, 1634. [Google Scholar] [CrossRef]
- Callea, M.; Fattori, F.; Bertini, E.S.; Cammarata-Scalisi, F.; Callea, F.; Bellacchio, E. Blood malignancies presenting with mutations at equivalent residues in RUNX1–2 suggest a common leukemogenic pathway. Leuk. Lymphoma 2017, 58, 2002–2004. [Google Scholar] [CrossRef]
- Kawane, T.; Qin, X.; Jiang, Q.; Miyazaki, T.; Komori, H.; Yoshida, C.A.; Matsuura-Kawata, V.K.D.; Sakane, C.; Matsuo, Y.; Nagai, K.; et al. Runx2 is required for the proliferation of osteoblast progenitors and induces proliferation by regulating Fgfr2 and Fgfr3. Sci. Rep. 2018, 8, 13551. [Google Scholar] [CrossRef]
- Komori, T. Molecular Mechanism of Runx2-Dependent Bone Development. Mol. Cells 2020, 43, 168–175. [Google Scholar] [CrossRef]
- Kim, W.J.; Shin, H.L.; Kim, B.S.; Kim, H.J.; Ryoo, H.M. RUNX2-modifying enzymes: Therapeutic targets for bone diseases. Exp. Mol. Med. 2020, 52, 1178–1184. [Google Scholar] [CrossRef]
- Wilhelm, K.; Happel, K.; Eelen, G.; Schoors, S.; Oellerich, M.F.; Lim, R.; Zimmermann, B.; Aspalter, I.M.; Franco, C.A.; Boettger, T.; et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 2016, 529, 216–220. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Lim, J.S.; Liu, J.; Ponugoti, B.; Alsadun, S.; Tian, C.; Vafa, R.; Graves, D.T. FOXO1 expression in keratinocytes promotes connective tissue healing. Sci. Rep. 2017, 7, 42834. [Google Scholar] [CrossRef] [PubMed]
- Ponugoti, B.; Xu, F.X.; Zhang, C.Y.; Tian, C.; Pacios, S.; Graves, D.T. FOXO1 promotes wound healing through the up-regulation of TGF-beta 1 and prevention of oxidative stress. J. Cell Biol. 2013, 203, 327–343. [Google Scholar] [CrossRef] [PubMed]
- Krivicka-Uzkurele, B.; Pilmane, M.; Akota, I. Barx1, growth factors and apoptosis in facial tissue of children with clefts. Stomatologija 2008, 10, 62–66. [Google Scholar] [PubMed]
- Vinuela, A.; Brown, A.A.; Buil, A.; Tsai, P.C.; Davies, M.N.; Bell, J.T.; Dermitzakis, E.T.; Spector, T.D.; Small, K.S. Age-dependent changes in mean and variance of gene expression across tissues in a twin cohort. Hum. Mol. Genet. 2018, 27, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.L.; Huang, T.; Petralia, F.; Long, Q.; Zhang, B.; Argmann, C.; Zhao, Y.; Mobbs, C.V.; Schadt, E.E.; Zhu, J.; et al. Synchronized age-related gene expression changes across multiple tissues in human and the link to complex diseases. Sci. Rep. 2015, 5, 15145. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| No. | Gender | Diagnosis | Procedure | Age (Months) | Remarks |
|---|---|---|---|---|---|
| 1. | M | Cheilognathouranoschisis dextra | Right cheiloplasty | 3 | - |
| Veloplasty | 10 | ||||
| 2. | M | Cheilognathouranoschisis dextra | Right cheiloplasty | 3 | - |
| Veloplasty | 9 | ||||
| 3. | M | Cheilognathouranoschisis sinistra | Left cheiloplasty | 3 | Mother with a cleft lip and palate. |
| Veloplasty | 8 | ||||
| 4. | M | Cheilognathouranoschisis sinistra | Left cheiloplasty | 3.5 | Paracetamol had been used during pregnancy. Epilepsy in family tree. Father a regular smoker. |
| Veloplasty | 15 | ||||
| 5. | F | Cheilognathouranoschisis sinistra | Left cheiloplasty | 4 | - |
| Veloplasty | 9 | ||||
| 6. | F | Cheilognathouranoschisis dextra | Right cheiloplasty | 4 | - |
| Veloplasty | 8 | ||||
| 7. | M | Cheilognathouranoschisis dextra | Right cheiloplasty | 4 | Hepatitis B during the pregnancy. Cleft lip and palate in the family tree. |
| Veloplasty | 10 | ||||
| 8. | M | Cheilognathouranoschisis sinistra | Left cheiloplasty | 4.5 | Down syndrome present in family history. |
| Veloplasty | 10 | ||||
| 9. | M | Cheilognathouranoschisis sinistra | Left cheiloplasty | 5 | - |
| Veloplasty | 10 | ||||
| 10. | M | Cheilognathouranoschisis bilateralis | Bilateral cheiloplasty | 13 | Multiple anomalies including congenital heart failure. |
| Veloplasty | 36 |
| Lip Mucosa | |||||||||
| Patient’s No. | Epithelium | Connective Tissue | Endothelium | ||||||
| FGFR1 | FGFR2 | FOXO1 | FGFR1 | FGFR2 | FOXO1 | FGFR1 | FGFR2 | FOXO1 | |
| 1. | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2. | 0 | +++ | 0 | 0 | +/++ | 0 | 0 | + | 0 |
| 3. | 0 | +++ | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 4. | +++ | 0/+ | +++ | ++ | 0 | + | + | 0 | ++ |
| 5. | 0 | ++ | +++ | 0 | +/++ | ++/+++ | 0 | 0/+ | ++ |
| 6. | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 7. | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 8. | +++ | 0/+ | ++/+++ | + | 0 | 0/+ | 0/+ | 0 | + |
| 9. | 0 | +++ | 0/+ | 0 | + | 0 | 0 | 0 | 0 |
| 10. | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mode | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Control | 0 | 0 | 0 | 0 | 0/+ | 0 | 0 | 0 | 0 |
| Vomer Mucosa | |||||||||
| Patient’s No. | Epithelium | Connective Tissue | Endothelium | ||||||
| FGFR1 | FGFR2 | FOXO1 | FGFR1 | FGFR2 | FOXO1 | FGFR1 | FGFR2 | FOXO1 | |
| 1. | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2. | +/++ | +++ | 0 | 0 | +/++ | 0 | 0 | + | 0 |
| 3. | 0 | +++ | 0 | 0 | 0/+ | 0 | 0 | 0/+ | 0 |
| 4. | ++/+++ | 0 | ++/+++ | +/++ | 0 | 0 | + | 0 | 0/+ |
| 5. | 0 | 0/+ | ++/+++ | 0 | 0/+ | 0/+ | 0 | 0 | 0/+ |
| 6. | ++ | + | 0 | 0/+ | 0 | 0 | 0 | 0 | 0 |
| 7. | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 8. | +++ | +++ | 0/+ | +/++ | + | 0 | +/++ | 0/+ | 0 |
| 9. | 0/+ | +++ | 0 | 0 | 0/+ | 0 | 0 | 0 | 0 |
| 10. | 0/+ | 0/+ | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mode | 0 | +++ | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Control | 0 | 0 | 0 | 0 | 0/+ | 0 | 0 | 0 | 0 |
| Lip Mucosa vs. Vomer Mucosa | |||||||||
| Epithelium | Connective Tissue | Endothelium | |||||||
| FGFR1 | FGFR2 | FOXO1 | FGFR1 | FGFR2 | FOXO1 | FGFR1 | FGFR2 | FOXO1 | |
| Mann–Whitney U | 65 | 56 | 42.5 | 54 | 55 | 39 | 51.5 | 54.5 | 42 |
| p-Value | 0.280 | 0.684 | 0.579 | 0.796 | 0.739 | 0.436 | 0.912 | 0.739 | 0.579 |
| Lip Mucosa vs. Control | |||||||||
| Epithelium | Connective Tissue | Endothelium | |||||||
| FGFR1 | FGFR2 | FOXO1 | FGFR1 | FGFR2 | FOXO1 | FGFR1 | FGFR2 | FOXO1 | |
| Mann–Whitney U | 16 | 31 | 9 | 12 | 35 | 10.5 | 12 | 39.5 | 10.5 |
| p-Value | 1.000 | 1.000 | 0.371 | 0.692 | 0.635 | 0.469 | 0.692 | 0.313 | 0.469 |
| Vomer Mucosa vs. Control | |||||||||
| Epithelium | Connective Tissue | Endothelium | |||||||
| FGFR1 | FGFR2 | FOXO1 | FGFR1 | FGFR2 | FOXO1 | FGFR1 | FGFR2 | FOXO1 | |
| Mann–Whitney U | 10 | 28.5 | 10.5 | 10.5 | 31 | 13.5 | 12 | 37.5 | 12 |
| p-Value | 0.469 | 0.875 | 0.469 | 0.469 | 1.000 | 0.811 | 0.692 | 0.428 | 0.692 |
| Factor 1 | Factor 2 | R1 | p-Value1 | R2 | p-Value2 |
| Very Strong Correlation | |||||
| FGFR1 in epithelium | FGFR1 in connective tissue | 0.994 | 0.000 | 0.837 | 0.002 |
| FGFR1 in connective tissue | FGFR1 in endothelium | 1.000 | 0.000 | 0.855 | 0.002 |
| FGFR2 in connective tissue | FGFR2 in endothelium | 0.855 | 0.002 | 0.802 | 0.005 |
| FOXO1 in epithelium | FOXO1 in endothelium | 0.915 | 0.000 | 0.861 | 0.001 |
| Very Strong-Strong Correlation | |||||
| FGFR1 in epithelium | FGFR1 in endothelium | 0.994 | 0.000 | 0.725 | 0.018 |
| FOXO1 in connective tissue | FOXO1 in endothelium | 0.995 | 0.000 | 0.667 | 0.035 |
| FGFR2 in epithelium | FGFR2 in connective tissue | 0.652 | 0.041 | 0.811 | 0.004 |
| Strong Correlation | |||||
| FOXO1 in epithelium | FGFR1 in endothelium | 0.651 | 0.042 | 0.679 | 0.031 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goida, J.; Pilmane, M. The Evaluation of FGFR1, FGFR2 and FOXO1 in Orofacial Cleft Tissue. Children 2022, 9, 516. https://doi.org/10.3390/children9040516
Goida J, Pilmane M. The Evaluation of FGFR1, FGFR2 and FOXO1 in Orofacial Cleft Tissue. Children. 2022; 9(4):516. https://doi.org/10.3390/children9040516
Chicago/Turabian StyleGoida, Jana, and Mara Pilmane. 2022. "The Evaluation of FGFR1, FGFR2 and FOXO1 in Orofacial Cleft Tissue" Children 9, no. 4: 516. https://doi.org/10.3390/children9040516
APA StyleGoida, J., & Pilmane, M. (2022). The Evaluation of FGFR1, FGFR2 and FOXO1 in Orofacial Cleft Tissue. Children, 9(4), 516. https://doi.org/10.3390/children9040516