
Segmental Absence of Intestinal Musculature in a Child with Type IV Ehlers–Danlos Syndrome


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
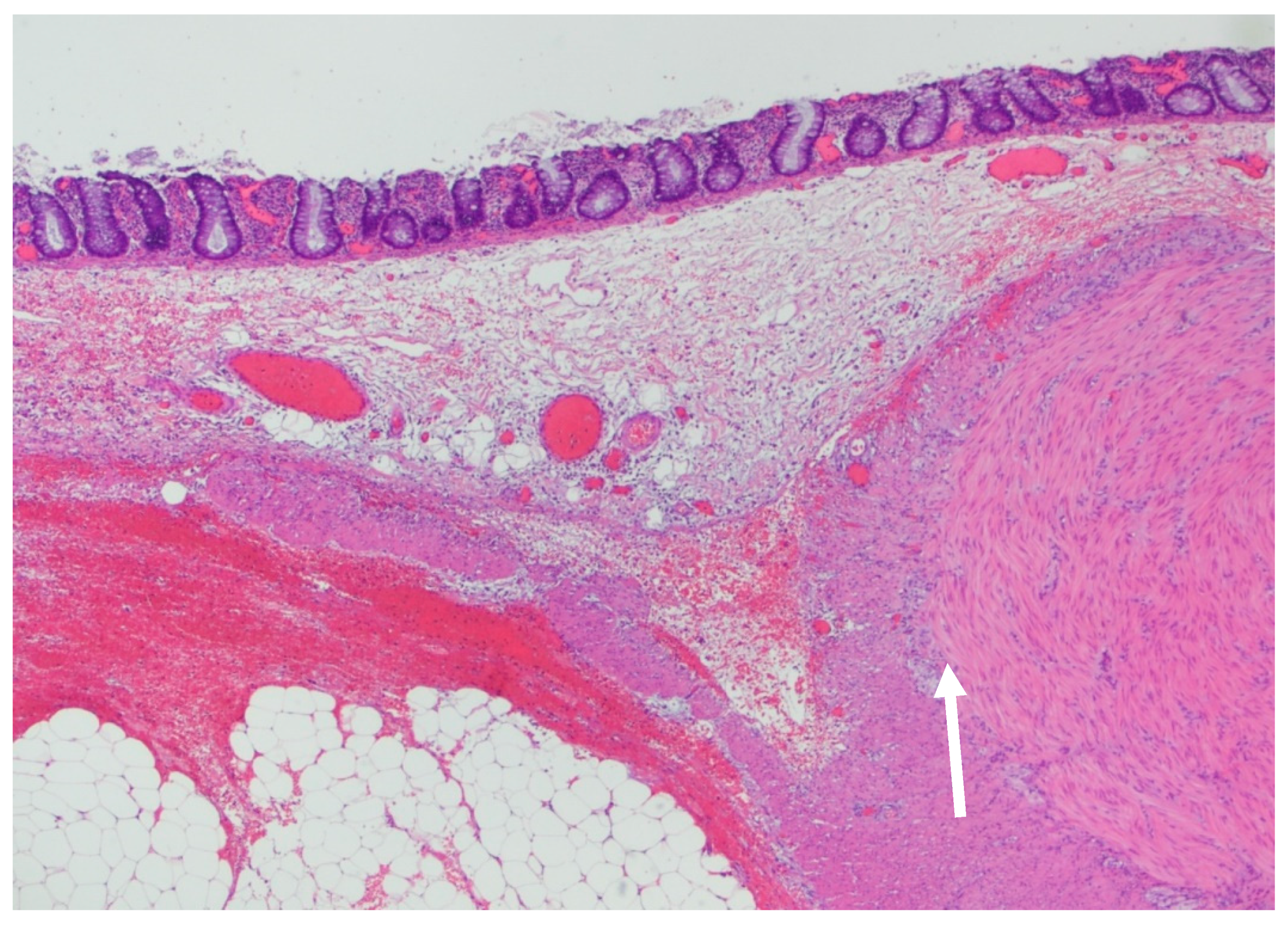
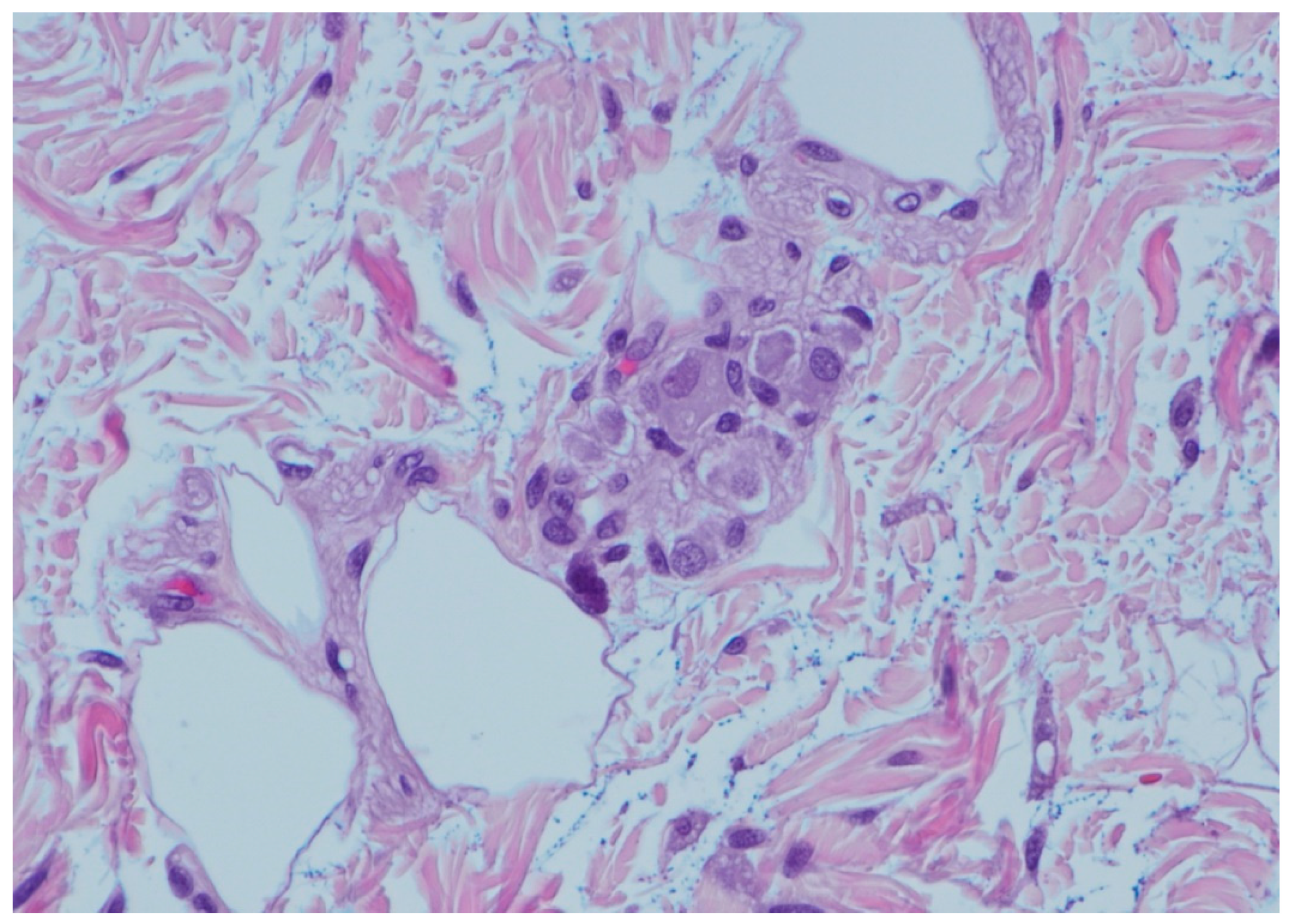
2. Case Reports
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kashiwagi, K.; Jimbo, K.; Hosoi, K.; Miyano, G.; Kudo, T.; Yamataka, A.; Shimizu, T. A novel segmental absence of intestinal musculature with small intestinal stenosis: A case report. BMC Gastroenterol. 2020, 20, 272. [Google Scholar] [CrossRef]
- Aldalati, O.; Phelan, C.; Ibrahim, H.N. Segmental absence of intestinal musculature (SAIM): A case report in an adult. BMJ Case Rep. 2009, 2009. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.S.; Ryan, M.L.; Shields, J.M.; Sola, J.; Perez, E.A.; Neville, H.L.; Rodriguez, M.M. Segmental absence of intestinal musculature: An increasingly reported pathology. J. Pediatr. Surg. 2012, 47, 1566–1571. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, B.; Gault, J.; Sanson, J. Neonatal intestinal obstruction due to absence of intestinal musculature: A new entity. J. Pediatr. Surg. 1967, 2, 332–335. [Google Scholar] [CrossRef]
- Oretti, C.; Bussani, R.; Janes, A.; DeMarini, S. Multiple segmental absence of intestinal musculature presenting as spontaneous isolated perforation in an extremely low-birth-weight infant. J. Pediatr. Surg. 2010, 45, e25–e27. [Google Scholar] [CrossRef]
- Tsuyuki, T.; Satou, A.; Takahara, T.; Nakajima, K.; Tsuzuki, T. Prevalence and Clinicopathologic Features of Intestinal Perforation Caused by Segmental Absence of the Intestinal Musculature in Adults. Am. J. Surg. Pathol. 2021, 45, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Athanazio, D.A.; Rocha, M.T.A.; Mattos, A.P.; Ribeiro, T.C.M.; Sorte, N.C.B.; Freitas, L.A.R.; Silva, L.R. Segmental absence of intestinal musculature-presentation in a 10-year-old boy with an extensive involved segment. Fetal Pediatr. Pathol. 2014, 34, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Malukani, K.; Nandedkar, S.S.; Patidar, E.; Nayak, R. Segmental absence of intestinal musculature: A rare case report. Int. J. Appl. Basic Med. Res. 2015, 5, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, O.; Newell, B.; Lee, K.R. Case Report: Segmental Absence of Intestinal Musculature in an Adult. Dig. Dis. Sci. 1998, 43, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Darcha, C.; Orliaguet, T.; Levrel, O.; Pezet, D.; Lointier, P.; Chipponi, J.; Dechelotte, P. Segmental absence of colonic muscularis propria. Report of a case in an adult. Ann. Pathol. 1997, 17, 31–33. [Google Scholar] [PubMed]
- Morikawa, N.; Namba, S.; Fujii, Y.; Sato, Y.; Fukuba, K. Intrauterine volvulus without malrotation associated with segmental absence of small intestinal musculature. J. Pediatr. Surg. 1999, 34, 1549–1551. [Google Scholar] [CrossRef]
- Wyllie, R.; Hyams, J.S.; Kay, M. Pediatric Gastrointestinal and Liver Disease; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar] [CrossRef]
- Park, K.Y.; Gill, K.G.; Kohler, J.E. Intestinal Perforation in Children as an Important Differential Diagnosis of Vascular Ehlers-Danlos Syndrome. Am. J. Case Rep. 2019, 20, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N. Engl. J. Med. 2000, 342, 673–680, Erratum in 2001, 334, 392. [Google Scholar] [CrossRef] [PubMed]
- Adham, S.M.; Zinzindohoué, F.; Jeunemaitre, X.; Frank, M. Natural history and surgical management of colonic perforations in vascular Ehlers-Danlos syndrome: A retrospective review. Dis. Colon Rectum. 2019, 62, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.; Adham, S.; Zinzindohoué, F.; Jeunemaitre, X. Natural history of gastrointestinal manifestations in vascular Ehlers–Danlos syndrome: A 17-year retrospective review. J. Gastroenterol. Hepatol. 2018, 34, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Leake, T.F.; Singhal, T.; Chandra, A.; Ashcroft, A.; Doddi, S.; Hussain, A.; Smedley, F. Occult small bowel perforation in a patient with Ehlers Danlos syndrome: A case report and review of the literature. Cases J. 2010, 3, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuivaniemi, H.; Tromp, G. Type III collagen (COL3A1): Gene and protein structure, tissue distribution, and associated diseases. Gene 2019, 707, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Speake, D.; Dvorkin, L.; Vaizey, C.J.; Carlson, G.L. Management of colonic complications of type IV Ehlers–Danlos syndrome: A systematic review and evidence-based management strategy. Colorectal Dis. 2019, 22, 129–135. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeky, N.; Short, C.; Keith, B.; Craver, R.D.; Zagory, J.A. Segmental Absence of Intestinal Musculature in a Child with Type IV Ehlers–Danlos Syndrome. Children 2021, 8, 680. https://doi.org/10.3390/children8080680
Zeky N, Short C, Keith B, Craver RD, Zagory JA. Segmental Absence of Intestinal Musculature in a Child with Type IV Ehlers–Danlos Syndrome. Children. 2021; 8(8):680. https://doi.org/10.3390/children8080680
Chicago/Turabian StyleZeky, Nicole, Celia Short, Brent Keith, Randall D. Craver, and Jessica A. Zagory. 2021. "Segmental Absence of Intestinal Musculature in a Child with Type IV Ehlers–Danlos Syndrome" Children 8, no. 8: 680. https://doi.org/10.3390/children8080680
APA StyleZeky, N., Short, C., Keith, B., Craver, R. D., & Zagory, J. A. (2021). Segmental Absence of Intestinal Musculature in a Child with Type IV Ehlers–Danlos Syndrome. Children, 8(8), 680. https://doi.org/10.3390/children8080680