
Calprotectin: An Ignored Biomarker of Neutrophilia in Pediatric Respiratory Diseases



Abstract
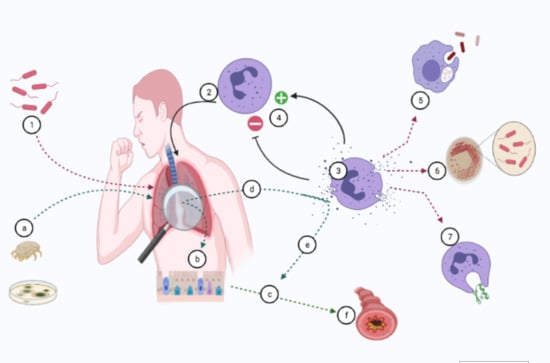
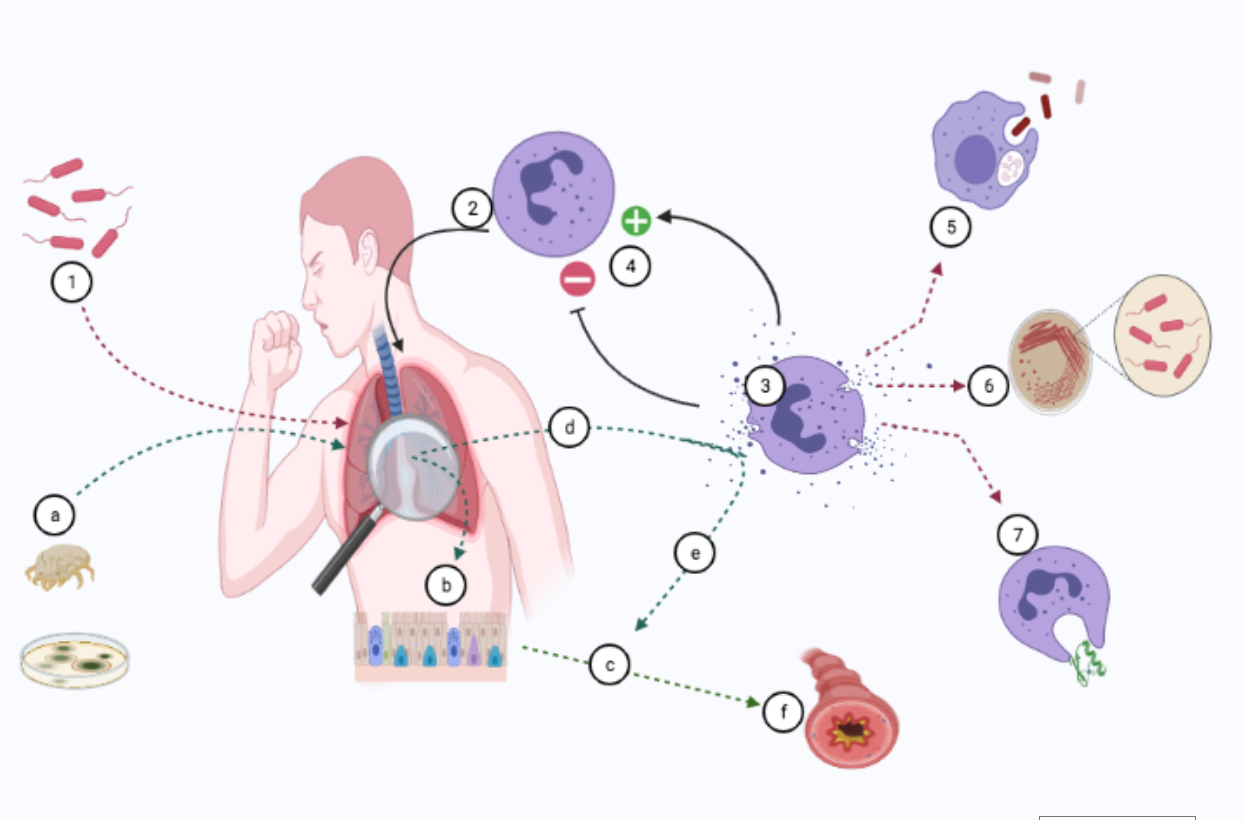
1. Introduction
2. CP Structure and Genes
3. Mechanisms of CP Release
4. Distribution and Reference Range of CP in Humans
5. The Importance of Membrane-CP Interaction
6. The Importance of Soluble CP
7. CP and Respiratory Infections
7.1. CP in Cystic Fibrosis (CF) Patients
7.2. CP in Non-CF Bronchiectasis (Non-CF BE)
7.3. CP in Asthmatics
7.4. CP in Other Lung Diseases
7.5. CP in Children with Obstructive Sleep Apnoea (OSA)
8. Conclusions
Funding
Conflicts of Interest
Abbreviations
References
- Moore, B.W. A soluble protein characteristic of the nervous system. Biochem. Biophys. Res. Commun. 1965, 19, 739–744. [Google Scholar] [CrossRef]
- Vogl, T.; Gharibyan, A.L.; Morozova-Roche, L.A. Pro-inflammatory S100A8 and S100A9 proteins: Self-assembly into multifunctional native and amyloid complexes. Int. J. Mol. Sci. 2012, 13, 2893–2917. [Google Scholar] [CrossRef]
- Rammes, A.; Roth, J.; Goebeler, M.; Klempt, M.; Hartmann, M.; Sorg, C. Myeloid-related protein (MRP) 8 and MRP14, calcium-binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin-dependent pathway. J. Biol. Chem. 1997, 272, 9496–9502. [Google Scholar] [CrossRef] [PubMed]
- Pruenster, M.; Vogl, T.; Roth, J.; Sperandio, M. S100A8/A9: From basic science to clinical application. Pharmacol. Ther. 2016, 167, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.; Fagerhol, M.K.; Bakken, J.S.; Dale, I. Plasma levels of the leucocyte L1 protein in febrile conditions: Relation to aetiology, number of leucocytes in blood, blood sedimentation reaction and C-reactive protein. Scand. J. Clin. Lab. Investig. 1984, 44, 357–362. [Google Scholar] [CrossRef]
- Marenholz, I.; Lovering, R.C.; Heizmann, C.W. An update of the S100 nomenclature. Biochim. Biophys. Acta 2006, 1763, 1282–1283. [Google Scholar] [CrossRef]
- Loomans, H.J.; Hahn, B.L.; Li, Q.Q.; Phadnis, S.H.; Sohnle, P.G. Histidine-based zinc-binding sequences and the antimicrobial activity of calprotectin. J. Infect. Dis. 1998, 177, 812–814. [Google Scholar] [CrossRef]
- Roth, J.; Burwinkel, F.; van den Bos, C.; Goebeler, M.; Vollmer, E.; Sorg, C. MRP8 and MRP14, S-100-like proteins associated with myeloid differentiation, are translocated to plasma membrane and intermediate filaments in a calcium-dependent manner. Blood 1993, 82, 1875–1883. [Google Scholar] [CrossRef]
- Fritz, G.; Botelho, H.M.; Morozova-Roche, L.A.; Gomes, C.M. Natural and amyloid self-assembly of S100 proteins: Structural basis of functional diversity. FEBS J. 2010, 277, 4578–4590. [Google Scholar] [CrossRef]
- Damo, S.M.; Kehl-Fie, T.E.; Sugitani, N.; Holt, M.E.; Rathi, S.; Murphy, W.J.; Zhang, Y.; Betz, C.; Hench, L.; Fritz, G.; et al. Molecular basis for manganese sequestration by calprotectin and roles in the innate immune response to invading bacterial pathogens. Proc. Natl. Acad. Sci. USA 2013, 110, 3841–3846. [Google Scholar] [CrossRef]
- Corbin, B.D.; Seeley, E.H.; Raab, A.; Feldmann, J.; Miller, M.R.; Torres, V.J.; Anderson, K.L.; Dattilo, B.M.; Dunman, P.M.; Gerads, R.; et al. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 2008, 319, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Leukert, N.; Barczyk, K.; Strupat, K.; Roth, J. Biophysical characterization of S100A8 and S100A9 in the absence and presence of bivalent cations. Biochim. Biophys. Acta 2006, 1763, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Leukert, N.; Vogl, T.; Strupat, K.; Reichelt, R.; Sorg, C.; Roth, J. Calcium-dependent tetramer formation of S100A8 and S100A9 is essential for biological activity. J. Mol. Biol. 2006, 359, 961–972. [Google Scholar] [CrossRef]
- Vogl, T.; Tenbrock, K.; Ludwig, S.; Leukert, N.; Ehrhardt, C.; van Zoelen, M.A.; Nacken, W.; Foell, D.; van der Poll, T.; Sorg, C.; et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 2007, 13, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, B.W.; Wicki, R.; Engelkamp, D.; Mattei, M.G.; Heizmann, C.W. Isolation of a YAC clone covering a cluster of nine S100 genes on human chromosome 1q21: Rationale for a new nomenclature of the S100 calcium-binding protein family. Genomics 1995, 25, 638–643. [Google Scholar] [CrossRef]
- Voganatsi, A.; Panyutich, A.; Miyasaki, K.T.; Murthy, R.K. Mechanism of extracellular release of human neutrophil calprotectin complex. J. Leukoc. Biol. 2001, 70, 130–134. [Google Scholar]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef]
- Lemarchand, P.; Vaglio, M.; Mauël, J.; Markert, M. Translocation of a small cytosolic calcium-binding protein (MRP-8) to plasma membrane correlates with human neutrophil activation. J. Biol. Chem. 1992, 267, 19379–19382. [Google Scholar] [CrossRef]
- Hetland, G.; Talgö, G.J.; Fagerhol, M.K. Chemotaxins C5a and fMLP induce release of calprotectin (leucocyte L1 protein) from polymorphonuclear cells in vitro. Mol. Pathol. 1998, 51, 143–148. [Google Scholar] [CrossRef]
- Frosch, M.; Metze, D.; Foell, D.; Vogl, T.; Sorg, C.; Sunderkötter, C.; Roth, J. Early activation of cutaneous vessels and epithelial cells is characteristic of acute systemic onset juvenile idiopathic arthritis. Exp. Dermatol. 2005, 14, 259–265. [Google Scholar] [CrossRef]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Schmidt, S.; Moser, M.; Sperandio, M. The molecular basis of leukocyte recruitment and its deficiencies. Mol. Immunol. 2013, 55, 49–58. [Google Scholar] [CrossRef]
- Pruenster, M.; Kurz, A.R.; Chung, K.J.; Cao-Ehlker, X.; Bieber, S.; Nussbaum, C.F.; Bierschenk, S.; Eggersmann, T.K.; Rohwedder, I.; Heinig, K.; et al. Extracellular MRP8/14 is a regulator of β2 integrin-dependent neutrophil slow rolling and adhesion. Nat. Commun. 2015, 6, 6915. [Google Scholar] [CrossRef]
- Zwadlo, G.; Schlegel, R.; Sorg, C. A monoclonal antibody to a subset of human monocytes found only in the peripheral blood and inflammatory tissues. J. Immunol. 1986, 137, 512–518. [Google Scholar] [PubMed]
- Hessian, P.A.; Edgeworth, J.; Hogg, N. MRP-8 and MRP-14, two abundant Ca (2+)-binding proteins of neutrophils and monocytes. J. Leukoc. Biol. 1993, 53, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P.; Dale, I.; Fagerhol, M.K. Distribution of a formalin-resistant myelomonocytic antigen (L1) in human tissues. I. Comparison with other leukocyte markers by paired immunofluorescence and immunoenzyme staining. Am. J. Clin. Pathol. 1987, 87, 681–699. [Google Scholar] [CrossRef] [PubMed]
- Shabani, F.; Farasat, A.; Mahdavi, M.; Gheibi, N. Calprotectin (S100A8/S100A9): A key protein between inflammation and cancer. Inflamm. Res. 2018, 67, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.; Sundström, J.; Lind, L.; Larsson, A. Serum calprotectin levels in elderly males and females without bacterial or viral infections. Clin. Biochem. 2014, 47, 1065–1068. [Google Scholar] [CrossRef]
- Calcaterra, V.; De Amici, M.; Leonard, M.M.; De Silvestri, A.; Pelizzo, G.; Buttari, N.; Michev, A.; Leggio, M.; Larizza, D.; Cena, H. Serum Calprotectin Level in Children: Marker of Obesity and its Metabolic Complications. Ann. Nutr. Metab. 2018, 73, 177–183. [Google Scholar] [CrossRef]
- Pillay, S.N.; Asplin, J.R.; Coe, F.L. Evidence that calgranulin is produced by kidney cells and is an inhibitor of calcium oxalate crystallization. Am. J. Physiol. 1998, 275, F255-61. [Google Scholar] [CrossRef]
- Garg, M.; Leach, S.T.; Coffey, M.J.; Katz, T.; Strachan, R.; Pang, T.; Needham, B.; Lui, K.; Ali, F.; Day, A.S.; et al. Age-dependent variation of fecal calprotectin in cystic fibrosis and healthy children. J. Cyst. Fibros. 2017, 16, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Li, F.; Wang, J.; Shen, L.; Sheng, X. Fecal Calprotectin in Healthy Children Aged 1-4 Years. PLoS ONE 2016, 11, e0150725. [Google Scholar] [CrossRef]
- Joshi, S.; Lewis, S.J.; Creanor, S.; Ayling, R.M. Age-related faecal calprotectin, lactoferrin and tumour M2-PK concentrations in healthy volunteers. Ann. Clin. Biochem. 2010, 47, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 24, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, G.F.; Mendes, A.; Castro, R.A.; Baú, E.C.; Nader, H.B.; Dietrich, C.P. Distribution of sulfated glycosaminoglycans in the animal kingdom: Widespread occurrence of heparin-like compounds in invertebrates. Biochim. Biophys. Acta 2000, 1475, 287–294. [Google Scholar] [CrossRef]
- Gallagher, J.T.; Lyon, M. Molecular structure of heparan sulfate and interactions with growth factors and morphogens. In Proteoglycans: Structure, Biology and Molecular Interactions; Iozzo, R.V., Ed.; Marcel Dekker Inc.: New York, NY, USA, 2000; pp. 27–59. [Google Scholar]
- Buzza, M.S.; Zamurs, L.; Sun, J.; Bird, C.H.; Smith, A.I.; Trapani, J.A.; Froelich, C.J.; Nice, E.C.; Bird, P.I. Extracellular matrix remodeling by human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J. Biol. Chem. 2005, 280, 23549–23558. [Google Scholar] [CrossRef] [PubMed]
- Hallak, L.K.; Spillmann, D.; Collins, P.L.; Peeples, M.E. Glycosaminoglycan sulfation requirements for respiratory syncytial virus infection. J. Virol. 2000, 74, 10508–10513. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057. [Google Scholar] [CrossRef]
- Robinson, M.J.; Tessier, P.; Poulsom, R.; Hogg, N. The S100 family heterodimer, MRP-8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J. Biol. Chem. 2002, 277, 3658–3665. [Google Scholar] [CrossRef]
- Eue, I.; Sorg, C. Arachidonic acid specifically regulates binding of S100A8/9, a heterodimer complex of the S100 class of calcium binding proteins, to human microvascular endothelial cells. Atherosclerosis 2001, 154, 505–508. [Google Scholar] [CrossRef]
- Newton, R.A.; Hogg, N. The human S100 protein MRP-14 is a novel activator of the beta 2 integrin Mac-1 on neutrophils. J. Immunol. 1998, 160, 1427–1435. [Google Scholar] [PubMed]
- Saenz, S.A.; Taylor, B.C.; Artis, D. Welcome to the neighborhood: Epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol. Rev. 2008, 226, 172–190. [Google Scholar] [CrossRef] [PubMed]
- Kouzaki, H.; O’Grady, S.M.; Lawrence, C.B.; Kita, H. Proteases induce production of thymic stromal lymphopoietin by airway epithelial cells through protease-activated receptor-2. J. Immunol. 2009, 183, 1427–1434. [Google Scholar] [CrossRef]
- Wang, Y.H.; Angkasekwinai, P.; Lu, N.; Voo, K.S.; Arima, K.; Hanabuchi, S.; Hippe, A.; Corrigan, C.J.; Dong, C.; Homey, B.; et al. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J. Exp. Med. 2007, 204, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kouzaki, H.; Matsumoto, K.; Hosoi, J.; Shimizu, T. The effect of calprotectin on TSLP and IL-25 production from airway epithelial cells. Allergol. Int. 2017, 66, 281–289. [Google Scholar] [CrossRef]
- Chan, J.K.; Roth, J.; Oppenheim, J.J.; Tracey, K.J.; Vogl, T.; Feldmann, M.; Horwood, N.; Nanchahal, J. Alarmins: Awaiting a clinical response. J. Clin. Investig. 2012, 122, 2711–2719. [Google Scholar] [CrossRef]
- Watanabe, T.; Asai, K.; Fujimoto, H.; Tanaka, H.; Kanazawa, H.; Hirata, K. Increased levels of HMGB-1 and endogenous secretory RAGE in induced sputum from asthmatic patients. Respir. Med. 2011, 105, 519–525. [Google Scholar] [CrossRef]
- Ullah, M.A.; Loh, Z.; Gan, W.J.; Zhang, V.; Yang, H.; Li, J.H.; Yamamoto, Y.; Schmidt, A.M.; Armour, C.L.; Hughes, J.M.; et al. Receptor for advanced glycation end products and its ligand high-mobility group box-1 mediate allergic airway sensitization and airway inflammation. J. Allergy Clin. Immunol. 2014, 134, 440–450. [Google Scholar] [CrossRef]
- Idzko, M.; Hammad, H.; van Nimwegen, M.; Kool, M.; Willart, M.A.; Muskens, F.; Hoogsteden, H.C.; Luttmann, W.; Ferrari, D.; Di Virgilio, F.; et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat. Med. 2007, 13, 913–919. [Google Scholar] [CrossRef]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef]
- Kehl-Fie, T.E.; Skaar, E.P. Nutritional immunity beyond iron: A role for manganese and zinc. Curr. Opin. Chem. Biol. 2010, 14, 218–224. [Google Scholar] [CrossRef]
- Clark, H.L.; Jhingran, A.; Sun, Y.; Vareechon, C.; de Jesus Carrion, S.; Skaar, E.P.; Chazin, W.J.; Calera, J.A.; Hohl, T.M.; Pearlman, E. Zinc and Manganese Chelation by Neutrophil S100A8/A9 (Calprotectin) Limits Extracellular Aspergillus Fumigatus Hyphal Growth and Corneal Infection. J. Immunol. 2016, 196, 336–344. [Google Scholar] [CrossRef]
- Simard, J.C.; Simon, M.M.; Tessier, P.A.; Girard, D. Damage-associated molecular pattern S100A9 increases bactericidal activity of human neutrophils by enhancing phagocytosis. J. Immunol. 2011, 186, 3622–3631. [Google Scholar] [CrossRef]
- Achouiti, A.; Vogl, T.; Urban, C.F.; Röhm, M.; Hommes, T.J.; van Zoelen, M.A.; Florquin, S.; Roth, J.; van ‘t Veer, C.; de Vos, A.F.; et al. Myeloid-related protein-14 contributes to protective immunity in gram-negative pneumonia derived sepsis. PLoS Pathog. 2012, 8, e1002987. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Y.; Segovia, J.A.; Chang, T.H.; Morris, I.R.; Berton, M.T.; Tessier, P.A.; Tardif, M.R.; Cesaro, A.; Bose, S. DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza a virus infection: Role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog. 2014, 10, e1003848. [Google Scholar] [CrossRef] [PubMed]
- De Jong, H.K.; Achouiti, A.; Koh, G.C.; Parry, C.M.; Baker, S.; Faiz, M.A.; van Dissel, J.T.; Vollaard, A.M.; van Leeuwen, E.M.M.; Roelofs, J.J.T.H.; et al. Expression and function of S100A8/A9 (calprotectin) in human typhoid fever and the murine Salmonella model. PLoS Negl. Trop Dis. 2015, 9, e0003663. [Google Scholar] [CrossRef] [PubMed]
- Raju, M.S.; Kamaraju, R.S.; Sritharan, V.; Rajkumar, K.; Natarajan, S.; Kumar, A.D.; Burgula, S. Continuous evaluation of changes in the serum proteome from early to late stages of sepsis caused by Klebsiella pneumoniae. Mol. Med. Rep. 2016, 13, 4835–4844. [Google Scholar] [CrossRef]
- Ometto, F.; Friso, L.; Astorri, D.; Botsios, C.; Raffeiner, B.; Punzi, L.; Doria, A. Calprotectin in rheumatic diseases. Exp. Biol. Med. 2017, 242, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Coveney, A.P.; Wang, W.; Kelly, J.; Liu, J.H.; Blankson, S.; Wu, Q.D.; Redmond, H.P.; Wang, J.H. Myeloid-related protein 8 induces self-tolerance and cross-tolerance to bacterial infection via TLR4- and TLR2-mediated signal pathways. Sci. Rep. 2015, 5, 13694. [Google Scholar] [CrossRef] [PubMed]
- Lorey, M.B.; Rossi, K.; Eklund, K.K.; Nyman, T.A.; Matikainen, S. Global Characterization of Protein Secretion from Human Macrophages Following Non-canonical Caspase-4/5 Inflammasome Activation. Mol. Cell Proteom. 2017, 16 (Suppl. 1), S187–S199. [Google Scholar] [CrossRef] [PubMed]
- Pouwels, S.D.; Nawijn, M.C.; Bathoorn, E.; Riezebos-Brilman, A.; van Oosterhout, A.J.; Kerstjens, H.A.; Heijink, I.H. Increased serum levels of LL37, HMGB1 and S100A9 during exacerbation in COPD patients. Eur. Respir. J. 2015, 45, 1482–1485. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.L.F.; Rudan, I.; Liu, L.; Nair, H.; Theodoratou, E.; Bhutta, Z.A.; O’Brien, K.L.; Campbell, H.; Black, R.E. Global burden of childhood pneumonia and diarrhoea. Lancet 2013, 381, 1405–1416. [Google Scholar] [CrossRef]
- Global Burden of Disease Child and Adolescent Health Collaboration; Kassebaum, N.; Kyu, H.H.; Zoeckler, L.; Olsen, H.E.; Thomas, K.; Pinho, C. Child and Adolescent Health From 1990 to 2015: Findings From the Global Burden of Diseases, Injuries, and Risk Factors 2015 Study. JAMA Pediatr. 2017, 171, 573–592. [Google Scholar] [CrossRef]
- United Nations Children’s Fund. UNICEF; New York, “The State of the World’s Children 2016: A Fair Chance for Every Child”, 2016. Available online: https://www.unicef.org/publications/files/UNICEF_SOWC_2016.pdf (accessed on 3 March 2021).
- Nair, H.; Simões, E.A.; Rudan, I.; Gessner, B.D.; Azziz-Baumgartner, E.; Zhang, J.S.F.; Feikin, D.R.; Mackenzie, G.A.; Moiisi, J.C.; Roca, A.; et al. Severe Acute Lower Respiratory Infections Working Group. Global and regional burden of hospital admissions for severe acute lower respiratory infections in young children in 2010: A systematic analysis. Lancet 2013, 381, 1380–1390. [Google Scholar] [CrossRef]
- GBD 2015 LRI Collaborators. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory tract infections in 195 countries: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect Dis. 2017, 17, 1133–1161. [Google Scholar] [CrossRef]
- Histoshi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef]
- Dupuy, A.-M.; Philippart, F.; Péan, Y.; Lasocki, S.; Charles, P.-E.; Chalumeau, M.; Claessens, Y.-E.; Quenot, J.-P.; Guen, C.G.-L.; Ruiz, S.; et al. Role of biomarkers in the management of antibiotic therapy: An expert panel review: I—Currently available biomarkers for clinical use in acute infections. Ann. Intensive Care 2013, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Gilbert, D.N. The future of antibiotics and resistance: A tribute to a career of leadership by John Bartlett. Clin. Infect Dis. 2014, 59 (Suppl. 2), S71–S75. [Google Scholar] [CrossRef]
- Morley, D.; Torres, A.; Cillóniz, C.; Martin-Loeches, I. Predictors of treatment failure and clinical stability in patients with community acquired pneumonia. Ann. Transl. Med. 2017, 5, 443. [Google Scholar] [CrossRef]
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Karakioulaki, M.; Stolz, D. Biomarkers in Pneumonia-Beyond Procalcitonin. Int. J. Mol. Sci. 2019, 20, 2004. [Google Scholar] [CrossRef]
- Shaddock, E.J. How and when to use common biomarkers in community-acquired pneumonia. Pneumonia 2016, 8, 17. [Google Scholar] [CrossRef]
- Heiskanen-Kosma, T.; Korppi, M. Serum C-reactive protein cannot differentiate bacterial and viral aetiology of community-acquired pneumonia in children in primary healthcare settings. Scand. J. Infect Dis. 2000, 32, 399–402. [Google Scholar] [CrossRef]
- Ito, A.; Ishida, T. Diagnostic markers for community-acquired pneumonia. Ann. Transl. Med. 2020, 8, 609. [Google Scholar] [CrossRef]
- Lipcsey, M.; Hanslin, K.; Stålberg, J.; Smekal, D.; Larsson, A. The time course of calprotectin liberation from human neutrophil granulocytes after Escherichia coli and endotoxin challenge. Innate Immun. 2019, 25, 369–373. [Google Scholar] [CrossRef]
- Havelka, A.; Sejersen, K.; Venge, P.; Pauksens, K.; Larsson, A. Calprotectin, a new biomarker for diagnosis of acute respiratory infections. Sci. Rep. 2020, 10, 4208. [Google Scholar] [CrossRef]
- Fang, P.; Zheng, L.; Cao, P.; Zhang, C.; Fei, J.; Xu, Z.; Feng, C.-M.; Zhao, H.; Lu, Y.-J.; Fu, L. Serum S100A8 as an early diagnostic biomarker in patients with community-acquired pneumonia. Arch. Med. Sci. 2021. [Google Scholar] [CrossRef]
- Achouiti, A.; Vogl, T.; Van der Meer, A.J.; Stroo, I.; Florquin, S.; de Boer, O.J.; Roth, J.; Zeerleder, S.; van ‘t Veer, C.; de Vos, A.F.; et al. Myeloid-related protein-14 deficiency promotes inflammation in staphylococcal pneumonia. Eur. Respir. J. 2015, 46, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Siljan, W.W.; Holter, J.C.; Michelsen, A.E.; Nymo, S.H.; Lauritzen, T.; Oppen, K.; Husebye, E.; Ueland, T.; Mollnes, T.E.; Aukrust, P.; et al. Inflammatory biomarkers are associated with aetiology and predict outcomes in community-acquired pneumonia: Results of a 5-year follow-up cohort study. ERJ Open Res. 2019, 5, 00014–02019. [Google Scholar] [CrossRef]
- McCauley, L.; Dean, N. Pneumonia and empyema: Causal, casual or unknown. J. Thorac. Dis. 2015, 7, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, J.P.; Wrightson, J.M.; Belcher, E.; DeCamp, M.M.; Feller-Kopman, D.; Rahman, N.M. Pleural infection: Past, present, and future directions. Lancet Respir. Med. 2015, 3, 563–577. [Google Scholar] [CrossRef]
- Light, R.W. Parapneumonic effusions and empyema. Proc. Am. Thorac. Soc. 2006, 3, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Hampson, C.; Lemos, J.A.; Klein, J.S. Diagnosis and management of parapneumonic effusions. Semin. Respir. Crit. Care Med. 2008, 29, 414–426. [Google Scholar] [CrossRef]
- Wu, K.A.; Wu, C.C.; Liu, Y.C.; Hsueh, P.C.; Chin, C.Y.; Wang, C.L.; Chu, C.M.; Shih, L.J.; Yang, C.Y. Combined serum biomarkers in the noninvasive diagnosis of complicated parapneumonic effusions and empyema. BMC Pulm. Med. 2019, 19, 108. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, O.M.; Hussein, K.M.; Ramadan, A.E.; Mahmoud, G.T.; El-Naggar, M.E.; Gaber, N.E.Z. Diagnostic value of calprotectin in differentiation between benign and malignant pleural effusion. Egypt. J. Bronchol. 2019, 13, 382–387. [Google Scholar] [CrossRef]
- Xu, D.; Li, Y.; Li, X.; Wei, L.L.; Pan, Z.; Jiang, T.T.; Chen, Z.L.; Wang, C.; Cao, W.M.; Zhang, X.; et al. Serum protein S100A9, SOD3, and MMP9 as new diagnostic biomarkers for pulmonary tuberculosis by iTRAQ-coupled two-dimensional LC-MS/MS. Proteomics 2015, 15, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Pan, L.; Han, F.; Luo, B.; Jia, H.; Xing, A.; Li, Q.; Zhang, Z. Proteomic profiling for plasma biomarkers of tuberculosis progression. Mol. Med. Rep. 2018, 18, 1551–1559. [Google Scholar] [CrossRef]
- Jerkic, S.P.; Michel, F.; Donath, H.; Herrmann, E.; Schubert, R.; Rosewich, M.; Zielen, S. Calprotectin as a New Sensitive Marker of Neutrophilic Inflammation in Patients with Bronchiolitis Obliterans. Mediat. Inflamm. 2020, 2020, 4641585. [Google Scholar] [CrossRef]
- Wilson, G.B.; Fudenberg, H.H.; Jahn, T.L. Studies on cystic fibrosis using isoelectric focusing. I. An assay for detection of cystic fibrosis homozygotes and heterozygote carriers from serum. Pediatr. Res. 1975, 9, 635–640. [Google Scholar] [CrossRef][Green Version]
- Dorin, J.R.; Novak, M.; Hill, R.E.; Brock, D.J.; Secher, D.S.; van Heyningen, V. A clue to the basic defect in cystic fibrosis from cloning the CF antigen gene. Nature 1987, 326, 614–617. [Google Scholar] [CrossRef]
- Gray, R.D.; Imrie, M.; Boyd, A.C.; Porteous, D.; Innes, J.A.; Greening, A.P. Sputum and serum calprotectin are useful biomarkers during CF exacerbation. J. Cyst. Fibros. 2010, 9, 193–198. [Google Scholar] [CrossRef]
- Schnapp, Z.; Hartman, C.; Livnat, G.; Shteinberg, M.; Elenberg, Y. Decreased Fecal Calprotectin Levels in Cystic Fibrosis Patients After Antibiotic Treatment for Respiratory Exacerbation. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Reid, P.A.; McAllister, D.A.; Boyd, A.C.; Innes, J.A.; Porteous, D.; Greening, A.P.; Gray, R.D. Measurement of serum calprotectin in stable patients predicts exacerbation and lung function decline in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2015, 191, 233–236. [Google Scholar] [CrossRef]
- Gray, R.D.; MacGregor, G.; Noble, D.; Imrie, M.; Dewar, M.; Boyd, A.C.; Innes, J.A.; Porteous, D.J.; Greening, A.P. Sputum proteomics in inflammatory and suppurative respiratory diseases. Am. J. Respir. Crit. Care Med. 2008, 178, 444–452. [Google Scholar] [CrossRef]
- Lee, T.H.; Jang, A.S.; Park, J.S.; Kim, T.H.; Choi, Y.S.; Shin, H.R.; Park, S.W.; Uh, S.T.; Choi, J.S.; Kim, Y.H.; et al. Elevation of S100 calcium binding protein A9 in sputum of neutrophilic inflammation in severe uncontrolled asthma. Ann. Allergy Asthma. Immunol. 2013, 111, 268–275. [Google Scholar] [CrossRef]
- Palmer, L.D.; Maloney, K.N.; Boyd, K.L.; Goleniewska, A.K.; Toki, S.; Maxwell, C.N.; Chazin, W.J.; Peebles, R.S., Jr.; Newcomb, D.C.; Skaar, E.P. The Innate Immune Protein S100A9 Protects from T-Helper Cell Type 2-mediated Allergic Airway Inflammation. Am. J. Respir. Cell Mol. Biol. 2019, 61, 459–468. [Google Scholar] [CrossRef]
- Lee, Y.G.; Hong, J.; Lee, P.H.; Lee, J.; Park, S.W.; Kim, D.; Jang, A.S. Serum Calprotectin Is a Potential Marker in Patients with Asthma. J. Korean Med. Sci. 2020, 35, e362. [Google Scholar] [CrossRef]
- Orivuori, L.; Mustonen, K.; de Goffau, M.C.; Hakala, S.; Paasela, M.; Roduit, C.; Dalphin, J.C.; Genuneit, J.; Lauener, R.; Riedler, J.; et al. High level of fecal calprotectin at age 2 months as a marker of intestinal inflammation predicts atopic dermatitis and asthma by age 6. Clin. Exp. Allergy 2015, 45, 928–939. [Google Scholar] [CrossRef]
- Andréasson, K.; Alrawi, Z.; Persson, A.; Jönsson, G.; Marsal, J. Intestinal dysbiosis is common in systemic sclerosis and associated with gastrointestinal and extraintestinal features of disease. Arthritis Res. Ther. 2016, 18, 278. [Google Scholar] [CrossRef]
- Volkmann, E.R. Intestinal microbiome in scleroderma: Recent progress. Curr. Opin. Rheumatol. 2017, 29, 553–560. [Google Scholar] [CrossRef]
- Raquil, M.A.; Anceriz, N.; Rouleau, P.; Tessier, P.A. Blockade of antimicrobial proteins S100A8 and S100A9 inhibits phagocyte migration to the alveoli in streptococcal pneumonia. J. Immunol. 2008, 180, 3366–3374. [Google Scholar] [CrossRef] [PubMed]
- Kushida, C.A.; Chediak, A.; Berry, R.B.; Brown, L.K.; Gozal, D.; Iber, C.; Parthasarathy, S.; Quan, S.F.; Rowley, J.A.; Positive Airway Pressure Titration Task Force; American Academy of Sleep Medicine. Clinical guidelines for the manual titration of positive airway pressure in patients with obstructive sleep apnea. J. Clin. Sleep Med. 2008, 4, 157–171. [Google Scholar]
- Bhattacharjee, R.; Kheirandish-Gozal, L.; Pillar, G.; Gozal, D. Cardiovascular complications of obstructive sleep apnea syndrome: Evidence from children. Prog. Cardiovasc. Dis. 2009, 51, 416–433. [Google Scholar] [CrossRef] [PubMed]
- Atkeson, A.; Yeh, S.Y.; Malhotra, A.; Jelic, S. Endothelial function in obstructive sleep apnea. Prog. Cardiovasc. Dis. 2009, 51, 351–362. [Google Scholar] [CrossRef]
- Kim, J.; Bhattacharjee, R.; Snow, A.B.; Capdevila, O.S.; Kheirandish-Gozal, L.; Gozal, D. Myeloid-related protein 8/14 levels in children with obstructive sleep apnoea. Eur. Respir. J. 2010, 35, 843–850. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Cell Type | Mode of Release |
|---|---|
| Monocyte | Actively: Independent of Golgi pathway; Endolysosomes and secretory lysosomes are involved. Passively: Necrosis. |
| Neutrophil | Actively: Independent of Golgi pathway; Passively: Necrosis; NET formation. |
| Authors | Age of Cohort | Study Results |
|---|---|---|
| Fang, P. et al. [79] | adults | S100A8 heterodimer is helpful in community-acquired pneumonia diagnosis and severity assessment |
| Siljan, W.W. et al. [81] | adults | CP identifies bacterial vs. viral community acquired pneumonia and predicts the outcome |
| Wu, K.A. et al. [86] | adults | CP contributes in noninvasive diagnosis of complicated parapneumonic effusions and empyema |
| Mohammed, O.M. et al. [87] | adults | CP helps in differentiation between benign and malignant pleural effusion |
| Xu, D. et al. [88] | adults | S100A9 heterodimer contributes to early TB diagnosis |
| Liu, Q. et al. [89] | adults | CP value correlates with TB development |
| Jerkic, S.P. et al. [90] | children and young adults (6.2 to 27.3 years of age) | CP reflects ongoing neutrophilic inflammation in patients with obliterative bronchiolitis |
| Havelka, A. et al. [78] | adults | CP is a better discriminator between bacterial and viral infections compared to HBP and PCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatziparasidis, G.; Kantar, A. Calprotectin: An Ignored Biomarker of Neutrophilia in Pediatric Respiratory Diseases. Children 2021, 8, 428. https://doi.org/10.3390/children8060428
Chatziparasidis G, Kantar A. Calprotectin: An Ignored Biomarker of Neutrophilia in Pediatric Respiratory Diseases. Children. 2021; 8(6):428. https://doi.org/10.3390/children8060428
Chicago/Turabian StyleChatziparasidis, Grigorios, and Ahmad Kantar. 2021. "Calprotectin: An Ignored Biomarker of Neutrophilia in Pediatric Respiratory Diseases" Children 8, no. 6: 428. https://doi.org/10.3390/children8060428
APA StyleChatziparasidis, G., & Kantar, A. (2021). Calprotectin: An Ignored Biomarker of Neutrophilia in Pediatric Respiratory Diseases. Children, 8(6), 428. https://doi.org/10.3390/children8060428