Genetic Predisposition to Neuroblastoma


Abstract
1. Introduction
2. Familial Neuroblastoma
2.1. PHOX2B
2.2. ALK
2.3. KIF1Bβ
2.4. RAS Pathway Mutations and Other Cancer Predisposition Syndromes
2.5. Other Predisposition Syndromes
3. Sporadic Neuroblastoma
3.1. Susceptibility to Sporadic Neuroblastoma
3.2. Predisposition to Neuroblastoma Genotypes
4. Future Directions
4.1. Germline Genetics Predisposing to Adverse Events
4.2. Beyond Caucasians in Sporadic Neuroblastoma
4.3. Collaboration with Therapeutic and Technological Advances
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Seibel, N.L.; Altekruse, S.F.; Ries, L.A.; Melbert, D.L.; O’Leary, M.; Smith, F.O.; Reaman, G.H. Outcomes for children and adolescents with cancer: Challenges for the twenty-first century. J. Clin. Oncol. 2010, 28, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.L.; Schmidt, M.L.; Cohn, S.L.; Maris, J.M.; London, W.B.; Buxton, A.; Stram, D.; Castleberry, R.P.; Shimada, H.; Sandler, A.; et al. Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N. Engl. J. Med. 2010, 363, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, K.K.; London, W.B.; Ambros, P.F.; Nakagawara, A.; Berthold, F.; Schleiermacher, G.; Park, J.R.; et al. Advances in risk classification and treatment strategies for neuroblastoma. J. Clin. Oncol. 2015, 33, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Nuchtern, J.G.; London, W.B.; Barnewolt, C.E.; Naranjo, A.; McGrady, P.W.; Geiger, J.D.; Diller, L.; Schmidt, M.L.; Maris, J.M.; Cohn, S.L.; et al. A prospective study of expectant observation as primary therapy for neuroblastoma in young infants a children’s oncology group study. Ann. Surg. 2012, 256, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The international neuroblastoma risk group (INRG) classification system: An INRG task force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Monclair, T.; Brodeur, G.M.; Ambros, P.F.; Brisse, H.J.; Cecchetto, G.; Holmes, K.; Kaneko, M.; London, W.B.; Matthay, K.K.; Nuchtern, J.G.; et al. The international neuroblastoma risk group (INRG) staging system: An INRG task force report. J. Clin. Oncol. 2009, 27, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Look, A.T.; Hayes, F.A.; Shuster, J.J.; Douglass, E.C.; Castleberry, R.P.; Bowman, L.C.; Smith, E.I.; Brodeur, G.M. Clinical relevance of tumor cell ploidy and N-myc gene amplification in childhood neuroblastoma: A pediatric oncology group study. J. Clin. Oncol. 1991, 9, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Varmus, H.E.; Bishop, J.M. Human N-myc gene contributes to neoplastic transformation of mammalian cells in culture. Nature 1985, 316, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Alitalo, K.; Klempnauer, K.-H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Look, A.T.; Hayes, F.A.; Nitschke, R.; McWilliams, N.B.; Green, A.A. Cellular DNA content as a predictor of response to chemotherapy in infants with unresectable neuroblastoma. N. Engl. J. Med. 1984, 311, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Oppedal, B.R.; Storm-Mathisen, I.; Lie, S.O.; Brandtzaeg, P. Prognostic factors in neuroblastoma. Clinical, histopathologic, and immunohistochemical features and DNA ploidy in relation to prognosis. Cancer 1988, 62, 772–780. [Google Scholar] [CrossRef]
- Fong, C.T.; Dracopoli, N.C.; White, P.S.; Merrill, P.T.; Griffith, R.C.; Housman, D.E.; Brodeur, G.M. Loss of heterozygosity for the short arm of chromosome 1 in human neuroblastomas: Correlation with N-myc amplification. Proc. Natl. Acad. Sci. USA 1989, 86, 3753–3757. [Google Scholar] [CrossRef] [PubMed]
- Bown, N.; Cotterill, S.; Lastowska, M.; O’Neill, S.; Pearson, A.D.; Plantaz, D.; Meddeb, M.; Danglot, G.; Brinkschmidt, C.; Christiansen, H.; et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N. Engl. J. Med. 1999, 340, 1954–1961. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; White, P.S.; Weiss, M.J.; Hogarty, M.D.; Thompson, P.M.; Stram, D.O.; Gerbing, R.; Matthay, K.K.; Seeger, R.C.; Brodeur, G.M.; et al. Allelic deletion at 11q23 is common in MYCN single copy neuroblastomas. Oncogene 1999, 18, 4948–4957. [Google Scholar] [CrossRef] [PubMed]
- Kaat, D.; Frank, S. Epigenetic regulation of neuroblastoma development. Cell Tissue Res. 2018, 372, 309–324. [Google Scholar]
- Olsson, M.; Beck, S.; Kogner, P.; Martinsson, T.; Carén, H. Genome-wide methylation profiling identifies novel methylated genes in neuroblastoma tumors. Epigenetics 2016, 11, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Schleiermacher, G.; Mosseri, V.; London, W.B.; Maris, J.M.; Brodeur, G.M.; Attiyeh, E.; Haber, M.; Khan, J.; Nakagawara, A.; Speleman, F.; et al. Segmental chromosomal alterations have prognostic impact in neuroblastoma: A report from the INRG project. Br. J. Cancer 2012, 107, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Decock, A.; Ongenaert, M.; Cannoodt, R.; Verniers, K.; De Wilde, B.; Laureys, G.; Van Roy, N.; Berbegall, A.P.; Bienertova-Vasku, J.; Bown, N. Methyl-CpG-binding domain sequencing reveals a prognostic methylation signature in neuroblastoma. Oncotarget 2016, 7, 1960–1972. [Google Scholar] [CrossRef] [PubMed]
- Westermann, F.; Schwab, M. Genetic parameters of neuroblastomas. Cancer Lett. 2002, 184, 127–147. [Google Scholar] [CrossRef]
- Chatten, J.; Voorhess, M.L. Familial neuroblastoma. N. Engl. J. Med. 1967, 277, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Hanenson, I.B.; Lampkin, B.C. Familial neuroblastoma. Am. J. Dis. Child. 1971, 121, 415–416. [Google Scholar] [CrossRef] [PubMed]
- Hardy, P.C.; Nesbit, M.E., Jr. Familial neuroblastoma: Report of a kindred with a high incidence of infantile tumors. J. Pediatr. 1972, 80, 74–77. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr.; Strong, L.C. Mutation and cancer: Neuroblastoma and pheochromocytoma. Am. J. Hum. Genet. 1972, 24, 514–532. [Google Scholar] [PubMed]
- Maris, J.M.; Weiss, M.J.; Mosse, Y.; Hii, G.; Guo, C.; White, P.S.; Hogarty, M.D.; Mirensky, T.; Brodeur, G.M.; Rebbeck, T.R.; et al. Evidence for a hereditary neuroblastoma predisposition locus at chromosome 16p12-13. Cancer Res. 2002, 62, 6651–6658. [Google Scholar] [PubMed]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Trochet, D.; Bourdeaut, F.; Janoueix-Lerosey, I.; Deville, A.; de Pontual, L.; Schleiermacher, G.; Coze, C.; Philip, N.; Frebourg, T.; Munnich, A.; et al. Germline mutations of the paired-like homeobox 2b (PHOX2B) gene in neuroblastoma. Am. J. Hum. Genet. 2004, 74, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Bourdeaut, F.; Trochet, D.; Janoueix-Lerosey, I.; Ribeiro, A.; Deville, A.; Coz, C.; Michiels, J.F.; Lyonnet, S.; Amiel, J.; Delattre, O. Germline mutations of the paired-like homeobox 2b (PHOX2B) gene in neuroblastoma. Cancer Lett. 2005, 228, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, T.; Trachsel, D.; Engelcke, G.; Hammer, J. Congenital central hypoventilation syndrome associated with hirschsprung’s disease and neuroblastoma: Case of multiple neurocristopathies. Pediatr. Pulmonol. 2002, 33, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Trochet, D.; Hong, S.J.; Lim, J.K.; Brunet, J.F.; Munnich, A.; Kim, K.S.; Lyonnet, S.; Goridis, C.; Amiel, J. Molecular consequences of PHOX2B missense, frameshift and alanine expansion mutations leading to autonomic dysfunction. Hum. Mol. Genet. 2005, 14, 3697–3708. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Laudenslager, M.; Khazi, D.; Carlisle, A.J.; Winter, C.L.; Rappaport, E.; Maris, J.M. Germline PHOX2B mutation in hereditary neuroblastoma. Am. J. Hum. Genet. 2004, 75, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Pattyn, A.; Morin, X.; Cremer, H.; Goridis, C.; Brunet, J.F. The homeobox gene PHOX2B is essential for the development of autonomic neural crest derivatives. Nature 1999, 399, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Trochet, D.; O’Brien, L.M.; Gozal, D.; Trang, H.; Nordenskjold, A.; Laudier, B.; Svensson, P.J.; Uhrig, S.; Cole, T.; Niemann, S.; et al. PHOX2B genotype allows for prediction of tumor risk in congenital central hypoventilation syndrome. Am. J. Hum. Genet. 2005, 76, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.M.; Zhou, L.; Rand, C.M.; Weese-Mayer, D.E. Congenital central hypoventilation syndrome: PHOX2B mutations and phenotype. Am. J. Respir. Crit. Care Med. 2006, 174, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Raabe, E.H.; Laudenslager, M.; Winter, C.; Wasserman, N.; Cole, K.; LaQuaglia, M.; Maris, D.J.; Mosse, Y.P.; Maris, J.M. Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene 2008, 27, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Van Limpt, V.; Schramm, A.; van Lakeman, A.; Sluis, P.; Chan, A.; van Noesel, M.; Baas, F.; Caron, H.; Eggert, A.; Versteeg, R. The PHOX2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene 2004, 23, 9280–9288. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.; Haberle, B.; Konig, I.R.; Kappler, R.; Suttorp, M.; Schackert, H.K.; Roesner, D.; Fitze, G. Rare occurrence of PHOX2B mutations in sporadic neuroblastomas. J. Pediatr. Hematol. Oncol. 2008, 30, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef] [PubMed]
- George, R.E.; Sanda, T.; Hanna, M.; Frohling, S.; Luther, W., 2nd; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Azarova, A.M.; Gautam, G.; George, R.E. Emerging importance of ALK in neuroblastoma. Semin. Cancer Boil. 2011, 21, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Fransson, S.; Hansson, M.; Ruuth, K.; Djos, A.; Berbegall, A.; Javanmardi, N.; Abrahamsson, J.; Palmer, R.H.; Noguera, R.; Hallberg, B.; et al. Intragenic anaplastic lymphoma kinase (ALK) rearrangements: Translocations as a novel mechanism of ALK activation in neuroblastoma tumors. Genes Chromosom. Cancer 2015, 54, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Bachetti, T.; Di Paolo, D.; Di Lascio, S.; Mirisola, V.; Brignole, C.; Bellotti, M.; Caffa, I.; Ferraris, C.; Fiore, M.; Fornasari, D.; et al. Phox2b-mediated regulation of ALK expression: In vitro identification of a functional relationship between two genes involved in neuroblastoma. PLoS ONE 2010, 5, e13108. [Google Scholar] [CrossRef] [PubMed]
- Applebaum, M.A.; Desai, A.V.; Bender, J.L.G.; Cohn, S.L. Emerging and investigational therapies for neuroblastoma. Expert Opin. Orphan Drugs 2017, 5, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, V.P.; Coggins, G.E.; Maris, J.M. Genetic susceptibility to neuroblastoma. Curr. Opin. Genet. Dev. 2017, 42, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.C.; Laudenslager, M.; Haglund, E.A.; Attiyeh, E.F.; Pawel, B.; Courtright, J.; Plegaria, J.; Christensen, J.G.; Maris, J.M.; Mosse, Y.P. Inhibition of ALK mutated neuroblastomas by the selective inhibitor PF-02341066. J. Clin. Oncol. 2009, 27, 10008b. [Google Scholar] [CrossRef]
- Schlisio, S.; Kenchappa, R.S.; Vredeveld, L.C.; George, R.E.; Stewart, R.; Greulich, H.; Shahriari, K.; Nguyen, N.V.; Pigny, P.; Dahia, P.L.; et al. The kinesin KIF1Bβ acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 2008, 22, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Yeh, I.T.; Lenci, R.E.; Qin, Y.; Buddavarapu, K.; Ligon, A.H.; Leteurtre, E.; Cao, C.D.; Cardot-Bauters, C.; Pigny, P.; Dahia, P.L.M. A germline mutation of the KIF1Bβ gene on 1p36 in a family with neural and nonneural tumors. Hum. Genet. 2008, 124, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Kamihara, J.; Bourdeaut, F.; Foulkes, W.D.; Molenaar, J.J.; Mosse, Y.P.; Nakagawara, A.; Parareda, A.; Scollon, S.R.; Schneider, K.W.; Skalet, A.H.; et al. Retinoblastoma and neuroblastoma predisposition and surveillance. Clin. Cancer Res. 2017, 23, e98–e106. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Miranda, B.; Westra, S.J.; Boechat, M.I.; Yazdani, S. Noonan syndrome associated with neuroblastoma: A case report. Pediatr. Radiol. 1997, 27, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Cotton, J.L.; Williams, R.G. Noonan syndrome and neuroblastoma. Arch. Pediatr. Adolesc. Med. 1995, 149, 1280–1281. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.A.; Allanson, J.E.; Dahlgren, J.; Gelb, B.D.; Hall, B.; Pierpont, M.E.; Roberts, A.E.; Robinson, W.; Takemoto, C.M.; Noonan, J.A. Noonan syndrome: Clinical features, diagnosis, and management guidelines. Pediatrics 2010, 126, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Origone, P.; Defferrari, R.; Mazzocco, K.; Lo Cunsolo, C.; De Bernardi, B.; Tonini, G.P. Homozygous inactivation of nf1 gene in a patient with familial NF1 and disseminated neuroblastoma. Am. J. Med. Genet. A 2003, 118A, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Martinsson, T.; Sjöberg, R.-M.; Hedborg, F.; Kogner, P. Homozygous deletion of the neurofibromatosis-1 gene in the tumor of a patient with neuroblastoma. Cancer Genet. Cytogenet. 1997, 95, 183–189. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Primers 2017, 3, 17004. [Google Scholar] [CrossRef] [PubMed]
- Moroni, I.; Bedeschi, F.; Luksch, R.; Casanova, M.; D’Incerti, L.; Uziel, G.; Selicorni, A. Costello syndrome: A cancer predisposing syndrome? Clin. Dysmorphol. 2000, 9, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Gripp, K.W.; Lin, A.E. Costello syndrome: A Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet. Med. 2012, 14, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Tidyman, W.E.; Rauen, K.A. The rasopathies: Developmental syndromes of RAS/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 2009, 19, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Daage, L.C.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Seidinger, A.L.; Fortes, F.P.; Mastellaro, M.J.; Cardinalli, I.A.; Zambaldi, L.G.; Aguiar, S.S.; Yunes, J.A. Occurrence of neuroblastoma among TP53 p.R337H carriers. PLoS ONE 2015, 10, e0140356. [Google Scholar] [CrossRef] [PubMed]
- Diskin, S.J.; Capasso, M.; Diamond, M.; Oldridge, D.A.; Conkrite, K.; Bosse, K.R.; Russell, M.R.; Iolascon, A.; Hakonarson, H.; Devoto, M.; et al. Rare variants in TP53 and susceptibility to neuroblastoma. J. Natl. Cancer Inst. 2014, 106, dju047. [Google Scholar] [CrossRef] [PubMed]
- Sartori, S.; Priante, E.; Pettenazzo, A.; Marson, P.; Suppiej, A.; Benini, F.; Perilongo, G.; Toldo, I. Intrathecal synthesis of oligoclonal bands in rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation syndrome: New evidence supporting immunological pathogenesis. J. Child. Neurol. 2013, 29, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, N.; Berard, E.; Chastagner, P.; Feillet, F.; Wagner, K.; Sommelet, D. Hypothalamic dysfunction associated with neuroblastoma: Evidence for a new paraneoplastic syndrome? Med. Pediatr. Oncol. 2003, 40, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Bougneres, P.; Pantalone, L.; Linglart, A.; Rothenbuhler, A.; Le Stunff, C. Endocrine manifestations of the rapid-onset obesity with hypoventilation, hypothalamic, autonomic dysregulation, and neural tumor syndrome in childhood. J. Clin. Endocrinol. Metab. 2008, 93, 3971–3980. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.S.; McGrath, S.; Marcus, C.L. Late-onset central hypoventilation with hypothalamic dysfunction: A distinct clinical syndrome. Pediatr. Pulmonol. 2000, 29, 62–68. [Google Scholar] [CrossRef]
- Emery, L.G.; Shields, M.; Shah, N.R.; Garbes, A. Neuroblastoma associated with Beckwith-Wiedemann syndrome. Cancer 1983, 52, 176–179. [Google Scholar] [CrossRef]
- Chitayat, D.; Friedman, J.M.; Dimmick, J.E. Neuroblastoma in a child with Wiedemann-Beckwith syndrome. Am. J. Med. Genet. 1990, 35, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.M.; Vansenne, F.; Kadouch, D.J.; Ibrahim, A.; Bliek, J.; Hopman, S.; Mannens, M.M.; Merks, J.H.; Maher, E.R.; Hennekam, R.C. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am. J. Med. Genet. A 2016, 170, 2248–2260. [Google Scholar] [CrossRef] [PubMed]
- Mussa, A.; Molinatto, C.; Baldassarre, G.; Riberi, E.; Russo, S.; Larizza, L.; Riccio, A.; Ferrero, G.B. Cancer risk in Beckwith-Wiedemann syndrome: A systematic review and meta-analysis outlining a novel (epi)genotype specific histotype targeted screening protocol. J. Pediatr. 2016, 176, 142–149.e141. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Murray, A.; Hanks, S.; Douglas, J.; Armstrong, R.; Banka, S.; Bird, L.M.; Clericuzio, C.L.; Cormier-Daire, V.; Cushing, T.; et al. Weaver syndrome and EZH2 mutations: Clarifying the clinical phenotype. Am. J. Med. Genet. Part A 2013, 161, 2972–2980. [Google Scholar] [CrossRef] [PubMed]
- Schimke, R.N.; Collins, D.L.; Stolle, C.A. Paraganglioma, neuroblastoma, and a SDHB mutation: Resolution of a 30-year-old mystery. Am. J. Med. Genet. Part A 2010, 152A, 1531–1535. [Google Scholar] [CrossRef] [PubMed]
- Benn, D.E.; Gimenez-Roqueplo, A.P.; Reilly, J.R.; Bertherat, J.; Burgess, J.; Byth, K.; Croxson, M.; Dahia, P.L.; Elston, M.; Gimm, O.; et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J. Clin. Endocrinol. Metab. 2006, 91, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P. Fanconi’s anemia and malignancies. Am. J. Hematol. 1996, 53, 99–110. [Google Scholar] [CrossRef]
- Malric, A.; Defachelles, A.-S.; Leblanc, T.; Lescoeur, B.; Lacour, B.; Peuchmaur, M.; Maurage, C.-A.; Pierron, G.; Guillemot, D.; d’Enghien, C.D.; et al. Fanconi anemia and solid malignancies in childhood: A national retrospective study. Pediatr. Blood Cancer 2015, 62, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.; Schindler, D.; Hanenberg, H.; Barker, K.; Hanks, S.; Kalb, R.; Neveling, K.; Kelly, P.; Seal, S.; Freund, M.; et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 2006, 39, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Mathew, C.G. Fanconi anaemia genes and susceptibility to cancer. Oncogene 2006, 25, 5875–5884. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Ritenour, L.E.; Randall, M.P.; Bosse, K.R.; Diskin, S.J. Genetic susceptibility to neuroblastoma: Current knowledge and future directions. Cell Tissue Res. 2018, 372, 287–307. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M.; Mosse, Y.P.; Bradfield, J.P.; Hou, C.; Monni, S.; Scott, R.H.; Asgharzadeh, S.; Attiyeh, E.F.; Diskin, S.J.; Laudenslager, M.; et al. Chromosome 6p22 locus associated with clinically aggressive neuroblastoma. N. Engl. J. Med. 2008, 358, 2585–2593. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.R.; Penikis, A.; Oldridge, D.A.; Alvarez-Dominguez, J.R.; McDaniel, L.; Diamond, M.; Padovan, O.; Raman, P.; Li, Y.; Wei, J.S.; et al. CASC15-S is a tumor suppressor lncRNA at the 6p22 neuroblastoma susceptibility locus. Cancer Res. 2015, 75, 3155–3166. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.K.; Mitra, S.; Subhash, S.; Hertwig, F.; Kanduri, M.; Mishra, K.; Fransson, S.; Ganeshram, A.; Mondal, T.; Bandaru, S.; et al. The risk-associated long noncoding RNA NBAT-1 controls neuroblastoma progression by regulating cell proliferation and neuronal differentiation. Cancer Cell 2014, 26, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Mondal, T.; Juvvuna, P.K.; Kirkeby, A.; Mitra, S.; Kosalai, S.T.; Traxler, L.; Hertwig, F.; Wernig-Zorc, S.; Miranda, C.; Deland, L.; et al. Sense-antisense lncRNA pair encoded by locus 6p22.3 determines neuroblastoma susceptibility via the USP36-CHD7-SOX9 regulatory axis. Cancer Cell 2018, 33, 417–434.e417. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Devoto, M.; Hou, C.; Asgharzadeh, S.; Glessner, J.T.; Attiyeh, E.F.; Mosse, Y.P.; Kim, C.; Diskin, S.J.; Cole, K.A.; et al. Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nat. Genet. 2009, 41, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Diskin, S.J.; Totaro, F.; Longo, L.; De Mariano, M.; Russo, R.; Cimmino, F.; Hakonarson, H.; Tonini, G.P.; Devoto, M.; et al. Replication of GWAS-identified neuroblastoma risk loci strengthens the role of BARD1 and affirms the cumulative effect of genetic variations on disease susceptibility. Carcinogenesis 2013, 34, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Xu, X.L.; Yang, M.C.; Wei, F.; Ayi, T.C.; Bowcock, A.M.; Baer, R. Cell cycle-dependent colocalization of BARD1 and BRCA1 proteins in discrete nuclear domains. Proc. Natl. Acad. Sci. USA 1997, 94, 12075–12080. [Google Scholar] [CrossRef] [PubMed]
- Irminger-Finger, I.; Jefford, C.E. Is there more to BARD1 than BRCA1? Nat. Rev. Cancer 2006, 6, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.R.; Diskin, S.J.; Cole, K.A.; Wood, A.C.; Schnepp, R.W.; Norris, G.; Nguyen le, B.; Jagannathan, J.; Laquaglia, M.; Winter, C.; et al. Common variation at BARD1 results in the expression of an oncogenic isoform that influences neuroblastoma susceptibility and oncogenicity. Cancer Res. 2012, 72, 2068–2078. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schuttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc is a critical function of aurora a in human neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M.; Morton, C.L.; Gorlick, R.; Kolb, E.A.; Lock, R.; Carol, H.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Wu, J.; et al. Initial testing of the aurora kinase a inhibitor MLN8237 by the pediatric preclinical testing program (PPTP). Pediatr. Blood Cancer 2010, 55, 26–34. [Google Scholar] [CrossRef] [PubMed]
- DuBois, S.G.; Marachelian, A.; Fox, E.; Kudgus, R.A.; Reid, J.M.; Groshen, S.; Malvar, J.; Bagatell, R.; Wagner, L.; Maris, J.M.; et al. Phase I study of the aurora a kinase inhibitor alisertib in combination with irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma: A NANT (new approaches to neuroblastoma therapy) trial. J. Clin. Oncol. 2016, 34, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Diskin, S.J.; Zhang, H.; Attiyeh, E.F.; Winter, C.; Hou, C.; Schnepp, R.W.; Diamond, M.; Bosse, K.; Mayes, P.A.; et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature 2011, 469, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.M.; Lester, K.; Joseph, S.; Curtis, D.J. LIM-domain-only proteins in cancer. Nat. Rev. Cancer 2013, 13, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, X.; Weichert-Leahey, N.; Dong, Z.; Zhang, C.; Lopez, G.; Tao, T.; He, S.; Wood, A.C.; Oldridge, D.; et al. LMO1 synergizes with MYCN to promote neuroblastoma initiation and metastasis. Cancer Cell 2017, 32, 310–323.e315. [Google Scholar] [CrossRef] [PubMed]
- Oldridge, D.A.; Wood, A.C.; Weichert-Leahey, N.; Crimmins, I.; Sussman, R.; Winter, C.; McDaniel, L.D.; Diamond, M.; Hart, L.S.; Zhu, S.; et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature 2015, 528, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Nguyen le, B.; Diskin, S.J.; Capasso, M.; Wang, K.; Diamond, M.A.; Glessner, J.; Kim, C.; Attiyeh, E.F.; Mosse, Y.P.; Cole, K.; et al. Phenotype restricted genome-wide association study using a gene-centric approach identifies three low-risk neuroblastoma susceptibility loci. PLoS Genet. 2011, 7, e1002026. [Google Scholar] [CrossRef] [PubMed]
- Diskin, S.J.; Capasso, M.; Schnepp, R.W.; Cole, K.A.; Attiyeh, E.F.; Hou, C.; Diamond, M.; Carpenter, E.L.; Winter, C.; Lee, H.; et al. Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat. Genet. 2012, 44, 1126–1130. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project, C.; Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar]
- Zhang, L.; Anglesio, M.S.; O’Sullivan, M.; Zhang, F.; Yang, G.; Sarao, R.; Mai, P.N.; Cronin, S.; Hara, H.; Melnyk, N.; et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat. Med. 2007, 13, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, J.J.; Domingo-Fernandez, R.; Ebus, M.E.; Lindner, S.; Koster, J.; Drabek, K.; Mestdagh, P.; van Sluis, P.; Valentijn, L.J.; van Nes, J.; et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat. Genet. 2012, 44, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Schnepp, R.W.; Khurana, P.; Attiyeh, E.F.; Raman, P.; Chodosh, S.E.; Oldridge, D.A.; Gagliardi, M.E.; Conkrite, K.L.; Asgharzadeh, S.; Seeger, R.C.; et al. A LIN28B-RAN-AURKA signaling network promotes neuroblastoma tumorigenesis. Cancer Cell 2015, 28, 599–609. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, L.D.; Conkrite, K.L.; Chang, X.; Capasso, M.; Vaksman, Z.; Oldridge, D.A.; Zachariou, A.; Horn, M.; Diamond, M.; Hou, C.; et al. Common variants upstream of MLF1 at 3q25 and within CPZ at 4p16 associated with neuroblastoma. PLoS Genet. 2017, 13, e1006787. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Capasso, M.; Diskin, S.; Cimmino, F.; Acierno, G.; Totaro, F.; Petrosino, G.; Pezone, L.; Diamond, M.; McDaniel, L.; Hakonarson, H.; et al. Common genetic variants in NEFL influence gene expression and neuroblastoma risk. Cancer Res. 2014, 74, 6913–6924. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; McDaniel, L.D.; Cimmino, F.; Cirino, A.; Formicola, D.; Russell, M.R.; Raman, P.; Cole, K.A.; Diskin, S.J. The functional variant rs34330 of CDKN1B is associated with risk of neuroblastoma. J. Cell. Mol. Med. 2017, 21, 3224–3230. [Google Scholar] [CrossRef] [PubMed]
- Diskin, S.J.; Hou, C.; Glessner, J.T.; Attiyeh, E.F.; Laudenslager, M.; Bosse, K.; Cole, K.; Mosse, Y.P.; Wood, A.; Lynch, J.E.; et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature 2009, 459, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline mutations in predisposition genes in pediatric cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Roy, A.; Yang, Y.; Wang, T.; Scollon, S.; Bergstrom, K.; Kerstein, R.A.; Gutierrez, S.; Petersen, A.K.; Bavle, A.; et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hungate, E.A.; Applebaum, M.A.; Skol, A.D.; Vaksman, Z.; Diamond, M.; McDaniel, L.; Volchenboum, S.L.; Stranger, B.E.; Maris, J.M.; Diskin, S.J.; et al. Evaluation of genetic predisposition for MYCN-amplified neuroblastoma. J. Natl. Cancer Inst. 2017, 109, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Shohet, J.M.; Ghosh, R.; Coarfa, C.; Ludwig, A.; Benham, A.L.; Chen, Z.; Patterson, D.M.; Barbieri, E.; Mestdagh, P.; Sikorski, D.N.; et al. A genome-wide search for promoters that respond to increased MYCN reveals both new oncogenic and tumor suppressor microRNAs associated with aggressive neuroblastoma. Cancer Res. 2011, 71, 3841–3851. [Google Scholar] [CrossRef] [PubMed]
- Valentijn, L.J.; Koster, J.; Haneveld, F.; Aissa, R.A.; van Sluis, P.; Broekmans, M.E.; Molenaar, J.J.; van Nes, J.; Versteeg, R. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc. Natl. Acad. Sci. USA 2012, 109, 19190–19195. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Zhao, Y.; Hou, C.; Glessner, J.; McDaniel, L.; Diamond, M.A.; Thomas, K.; Li, J.; Wei, Z.; Liu, Y.; et al. Common variants in MMP20 at 11q22.2 predispose to 11q deletion and neuroblastoma risk. Nat. Commun. 2017, 8, 569. [Google Scholar] [CrossRef] [PubMed]
- Gamazon, E.R.; Pinto, N.; Konkashbaev, A.; Im, H.K.; Diskin, S.J.; London, W.B.; Maris, J.M.; Dolan, M.E.; Cox, N.J.; Cohn, S.L. Trans-population analysis of genetic mechanisms of ethnic disparities in neuroblastoma survival. J. Natl. Cancer Inst. 2013, 105, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Applebaum, M.A.; Henderson, T.O.; Lee, S.M.; Pinto, N.; Volchenboum, S.L.; Cohn, S.L. Second malignancies in patients with neuroblastoma: The effects of risk-based therapy. Pediatr. Blood Cancer 2015, 62, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, L.M.; Neglia, J.P.; Reulen, R.C.; Ronckers, C.M.; van Leeuwen, F.E.; Morton, L.M.; Hodgson, D.C.; Yasui, Y.; Oeffinger, K.C.; Henderson, T.O. Risk, risk factors, and surveillance of subsequent malignant neoplasms in survivors of childhood cancer: A review. J. Clin. Oncol. 2018, 36, 2145–2152. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S. Genetic variation as a modifier of association between therapeutic exposure and subsequent malignant neoplasms in cancer survivors. Cancer 2015, 121, 648–663. [Google Scholar] [CrossRef] [PubMed]
- Applebaum, M.A.; Vaksman, Z.; Lee, S.M.; Hungate, E.A.; Henderson, T.O.; London, W.B.; Pinto, N.; Volchenboum, S.L.; Park, J.R.; Naranjo, A.; et al. Neuroblastoma survivors are at increased risk for second malignancies: A report from the international neuroblastoma risk group project. Eur. J. Cancer 2017, 72, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Henderson, T.O.; Bhatia, S.; Pinto, N.; London, W.B.; McGrady, P.; Crotty, C.; Sun, C.L.; Cohn, S.L. Racial and ethnic disparities in risk and survival in children with neuroblastoma: A children’s oncology group study. J. Clin. Oncol. 2011, 29, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.D.; Burchard, E.G.; De la Vega, F.M. Genomics for the world. Nature 2011, 475, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Popejoy, A.B.; Fullerton, S.M. Genomics is failing on diversity. Nature 2016, 538, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, S.E.; French, B.; Kasner, S.E.; Johnson, J.A.; Anderson, J.L.; Gage, B.F.; Rosenberg, Y.D.; Eby, C.S.; Madigan, R.A.; McBane, R.B.; et al. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N. Engl. J. Med. 2013, 369, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Mapes, B.; El Charif, O.; Al-Sawwaf, S.; Dolan, M.E. Genome-wide association studies of chemotherapeutic toxicities: Genomics of inequality. Clin. Cancer Res. 2017, 23, 4010–4019. [Google Scholar] [CrossRef] [PubMed]
- Latorre, V.; Diskin, S.J.; Diamond, M.A.; Zhang, H.; Hakonarson, H.; Maris, J.M.; Devoto, M. Replication of neuroblastoma SNP association at the BARD1 locus in African-Americans. Cancer Epidemiol. Biomark. Prev. 2012, 21, 658–663. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zou, Y.; Wang, T.; Zhang, R.; Yang, T.; Zhu, J.; Wang, F.; Xia, H. Genetic variations of GWAS-identified genes and neuroblastoma susceptibility: A replication study in southern Chinese children. Transl. Oncol. 2017, 10, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]


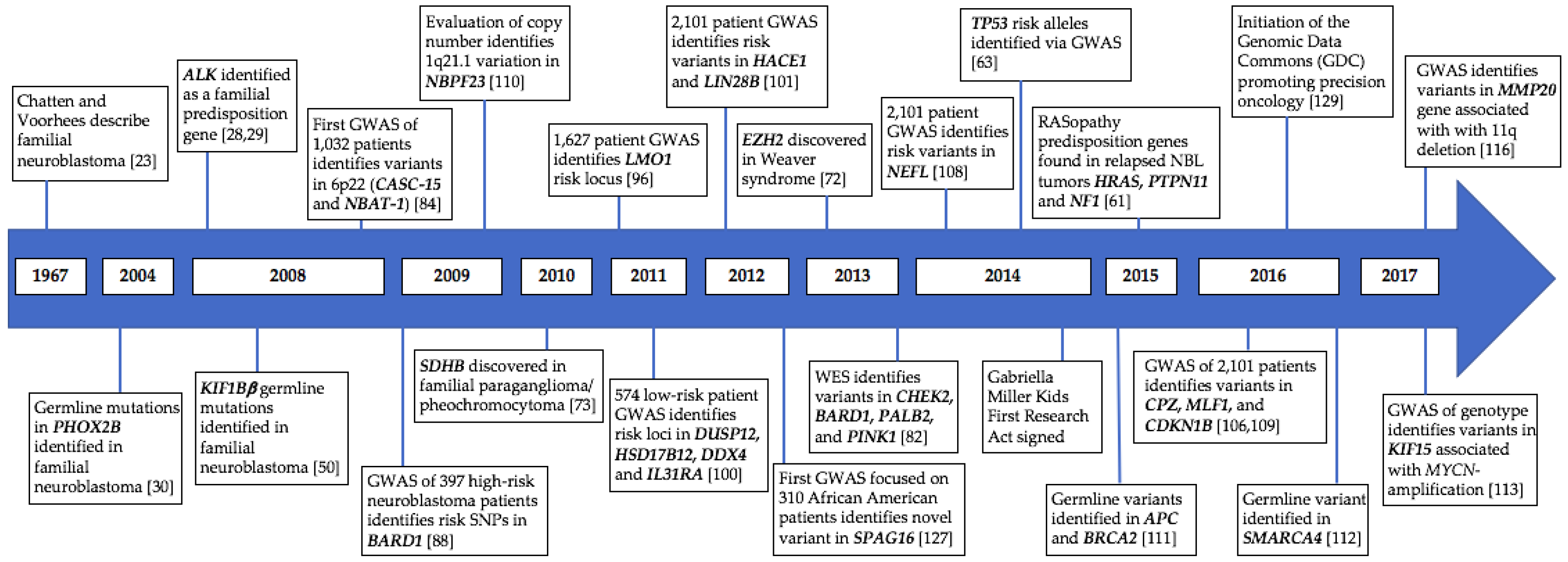{kind=link}
| Syndrome/Disease | Gene | Typical Genetic Alterations | Clinical Findings | Pre-Disposed Tumors |
|---|---|---|---|---|
| Congenital Central Hypoventilation Syndrome (CCHS) [32,33,36] | PHOX2B | Polyalanine and nonpolyalanine repeat expansion (frameshift or missense) | Respiratory dysfunction, autonomic dysfunction, Hirschsprung disease, neural crest tumors | Neuroblastoma, ganglioneuroma, ganglioneuroblastoma |
| ROHHAD [64,65,66,67] | Unknown | Unknown | Autonomic dysfunction, endocrinopathies, alveolar hypoventilation | Neuroblastoma, ganglioneuroma, ganglioneuroblastoma |
| Costello [58,59,60] | HRAS | Activating missense | Intellectual disability, coarse facial features, loose folds of skin, heart abnormalities, joint flexibility | Papilloma, rhabdomyosarcoma, neuroblastoma, transitional cell carcinoma |
| Noonan [52,53,54,60] | PTPN11, SOS1, RAF1, KRAS | Activating | Short stature, heat abnormalities, skeletal abnormalities, bleeding | Leukemia, neuroblastoma |
| Neurofibromatosis type 1 [55,56,57,60] | NF1 | Activating | Abnormal skin pigmentation, neurofibromas, scoliosis | Neurofibroma, MPNST, brain tumors, leukemia, optic glioma, neuroblastoma |
| Beckwith–Wiedemann [68,69,70,71] | CDKN1C *, H19, IGF2, KNBQOT1 | Abnormal methylation of chromosome 11 or uniparental disomy | Macrosomia, hemihypertrophy, abdominal wall defects, visceromegaly | Wilms tumor, hepatoblastoma, neuroblastoma |
| Li–Fraumeni [51,62,63] | TP53 | Missense | Increased cancer risk | Breast cancer, osteosarcoma, brain tumors, leukemia, neuroblastoma, adrenocortical carcinoma, soft tissue sarcoma |
| Weaver Syndrome [72] | EZH2 | Missense and truncating | Tall stature, intellectual disability, joint deformities, hypertelorism, micrognathia | Neuroblastoma |
| Familial Paraganglioma/Pheochromocytoma [73,74] | SDHB *, SDHAF2, SDHC, SDHD | Splice site, frameshift, nonsense | Growth of benign tumors in paraganglia | Paraganglioma, pheochromocytoma, neuroblastoma |
| Fanconi Anemia [75,76,77,78,79] | FANCA, FANCC, FANCG, BRCA1, BRCA2 *, PALB2 *, BRIP1 *, and many others | Truncating, frameshift, missense | Bone marrow failure, organ defects, skeletal abnormalities | Leukemia, Wilms tumor, medulloblastoma, neuroblastoma, embryonal tumors, sarcomas, nephroblastoma |
| Candidate Gene(s) | Variant * | Genomic Location |
|---|---|---|
| TP53 [63] | rs35850753 | 17p13.1 |
| CASC-15 and NBAT-1 [84] | rs6939340 | 6p22 |
| BARD1 [88] | rs6435862 | 2q35 |
| LMO1 [96] | rs2168101 | 11p15.4 |
| DUSP12 [100] | rs1027702 | 1q23.3 |
| DDX4 [100] | rs2619046 | 5q11.2 |
| IL31RA [100] | rs10055201 | 5q11.2 |
| HSD17B12 [100] | rs11037575 | 11p11.2 |
| HACE1 [101] | rs4336470 | 6q16 |
| LIN28B [101] | rs17065417 | 6q16 |
| CPZ [106] | rs3796727 | 4p16 |
| MLF1 [106] | rs6441201 | 3q25 |
| NEFL [108] | rs1059111 | 8q21 |
| CDKN1B [109] | rs34330 | 12p13 |
| KIF15 [113] | rs80059929 | 3p21.31 |
| MMP20 [116] | rs10895322 | 11q22.2 |
| SPAG16 [117] | rs1033069 | 2q34 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barr, E.K.; Applebaum, M.A. Genetic Predisposition to Neuroblastoma. Children 2018, 5, 119. https://doi.org/10.3390/children5090119
Barr EK, Applebaum MA. Genetic Predisposition to Neuroblastoma. Children. 2018; 5(9):119. https://doi.org/10.3390/children5090119
Chicago/Turabian StyleBarr, Erin K., and Mark A. Applebaum. 2018. "Genetic Predisposition to Neuroblastoma" Children 5, no. 9: 119. https://doi.org/10.3390/children5090119
APA StyleBarr, E. K., & Applebaum, M. A. (2018). Genetic Predisposition to Neuroblastoma. Children, 5(9), 119. https://doi.org/10.3390/children5090119