Sleep Disorders in Childhood Neurogenetic Disorders

Abstract
1. Introduction
2. Review of Disorders
2.1. Down Syndrome
2.1.1. Overview of Genetics
2.1.2. Diagnosis and Molecular Basis
2.1.3. Clinical Characteristics
Developmental Aspects
2.1.4. Sleep Disorders
Sleep Complaints
Sleep Architecture
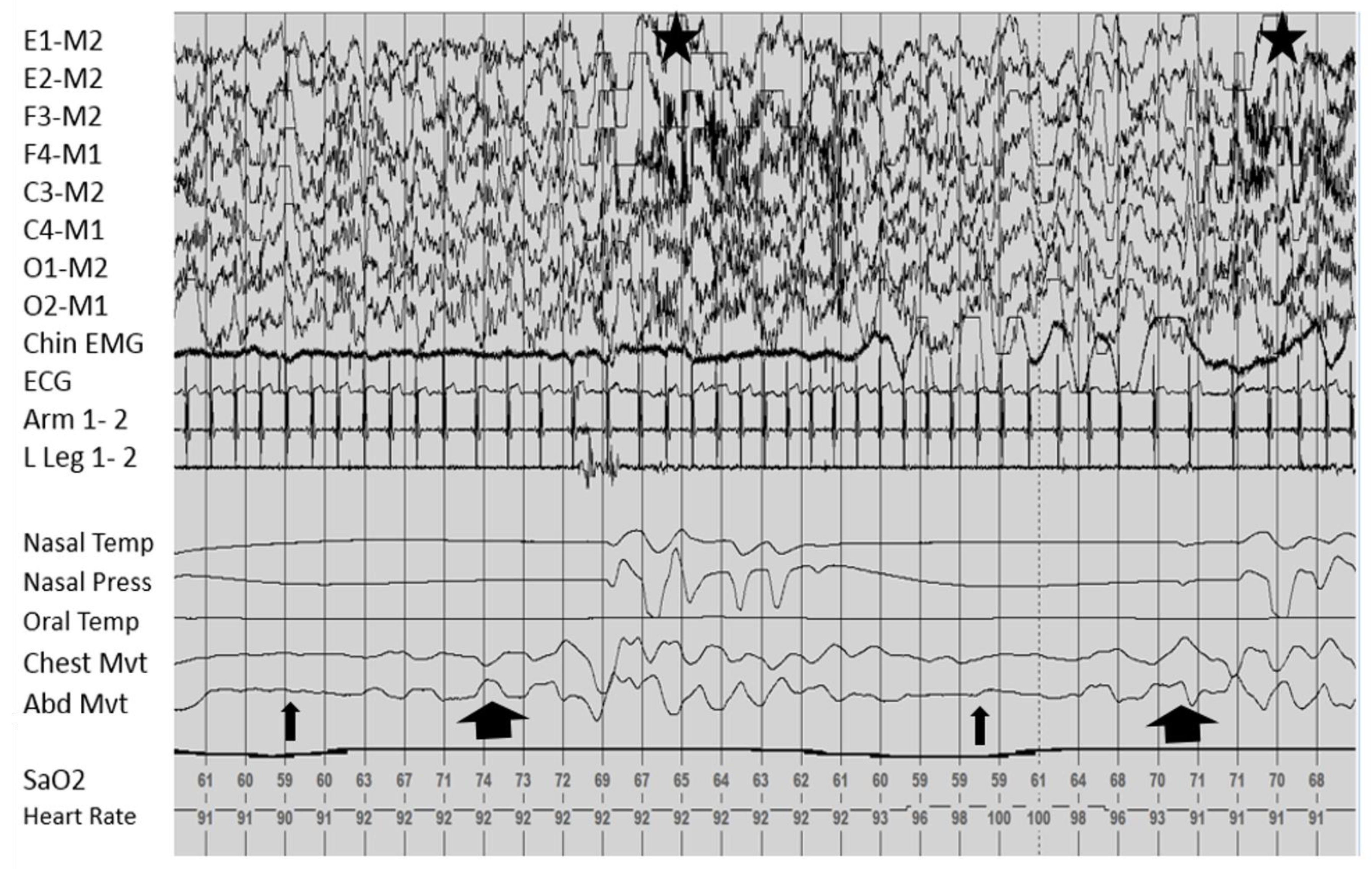
Sleep Related Breathing
Movements and Nocturnal Events
2.1.5. Management
2.1.6. Research and Future Directions
2.2. Angelman Syndrome
2.2.1. Overview of Genetics
2.2.2. Diagnosis and Molecular Genetics
2.2.3. Clinical Characteristics
Developmental Aspects
2.2.4. Sleep Disorders
Sleep Complaints
Sleep Architecture
Sleep Related Breathing
Movements and Nocturnal Events
2.2.5. Management
2.2.6. Research and Future Directions
2.3. Prader–Willi Syndrome
2.3.1. Overview of Genetics
2.3.2. Diagnosis and Molecular Genetics
2.3.3. Clinical Characteristics
Developmental Aspects
2.3.4. Sleep Disorders
Sleep Complaints
Sleep Architecture
Sleep Related Breathing
2.3.5. Management
2.3.6. Research and Future Directions
2.4. Smith–Magenis Syndrome
2.4.1. Overview of Genetics
2.4.2. Diagnosis and Molecular Genetics
2.4.3. Clinical Characteristics
Developmental Aspects
2.4.4. Sleep Disorders
Sleep Complaints
Sleep Architecture
Circadian Rhythm
2.4.5. Management
2.4.6. Research and Future Directions
2.5. Central Congenital Hypoventilation Syndrome
2.5.1. Overview of Genetics
2.5.2. Diagnosis and Molecular Genetics
2.5.3. Clinical Characteristics
Developmental Aspects
2.5.4. Sleep Disorders
Sleep Complaints
Sleep Architecture
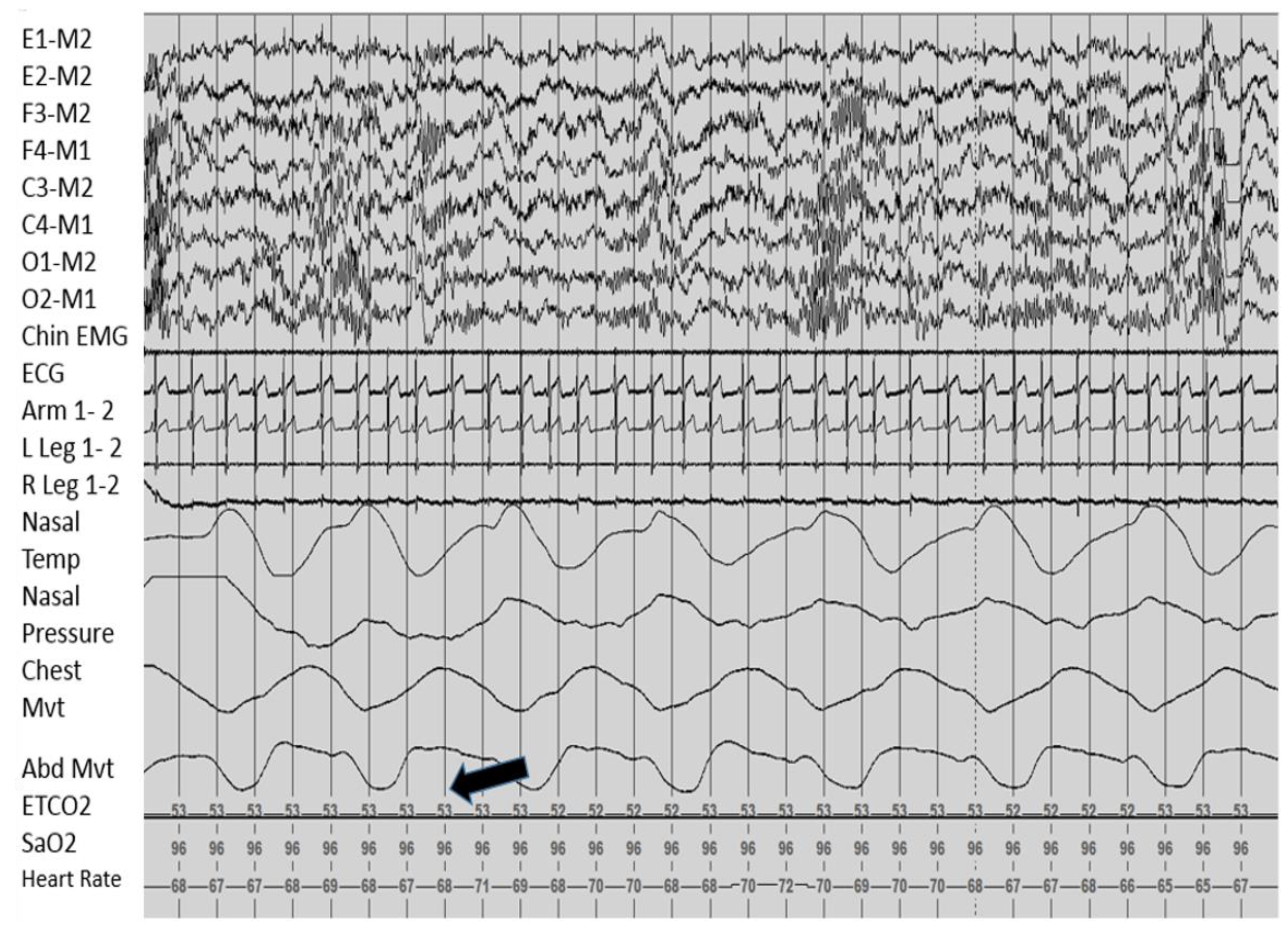
Sleep Related Breathing
2.5.5. Management
2.5.6. Research and Future Directions
2.6. Achondroplasia (Including Hypochondroplasia)
2.6.1. Overview of Genetics
2.6.2. Clinical Characteristics
Developmental Aspects
2.6.3. Sleep Disorders
Sleep Complaints
Sleep-Related Breathing
2.6.4. Management
2.6.5. Research and Future Directions
2.7. Mucopolysaccharidoses
2.7.1. Overview of Genetics
2.7.2. Diagnosis and Molecular Genetics
2.7.3. Clinical Characteristics
Developmental Aspects
2.7.4. Sleep Disorders
Sleep Complaints
Sleep Architecture
Circadian Rhythm
Sleep Related Breathing
Movements and Nocturnal Events
2.7.5. Management
2.7.6. Research and Future Directions
2.8. Duchenne Muscular Dystrophy
2.8.1. Overview of Genetics
2.8.2. Diagnosis and Molecular Genetics
2.8.3. Clinical Characteristics
Developmental Aspects
2.8.4. Sleep Disorders
Sleep Complaints
Sleep Architecture
Sleep Related Breathing
Movements and Nocturnal Events
2.8.5. Management
2.8.6. Research and Future Directions
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Muller, U.; Graeber, M.B.; Haberhausen, G.; Kohler, A. Molecular basis and diagnosis of neurogenetic disorders. J. Neurol. Sci. 1994, 124, 119–140. [Google Scholar] [CrossRef]
- Bargiela, D.; Yu-Wai-Man, P.; Keogh, M.; Horvath, R.; Chinnery, P.F. Prevalence of neurogenetic disorders in the North of England. Neurology 2015, 85, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Blackmer, A.B.; Feinstein, J.A. Management of sleep disorders in children with neurodevelopmental disorders: A review. Pharmacotherapy 2016, 36, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Robinson-Shelton, A.; Malow, B.A. Sleep Disturbances in neurodevelopmental disorders. Curr. Psychiatry Rep. 2016, 18, 6. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.D.; Kuhn, B.R.; DeHaai, K.A.; Wallace, D.P. Evaluation of a behavioral treatment package to reduce sleep problems in children with Angelman Syndrome. Res. Dev. Disabil. 2013, 34, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Griffith, G.M.; Hastings, R.P.; Oliver, C.; Howlin, P.; Moss, J.; Petty, J.; Tunnicliffe, P. Psychological well-being in parents of children with Angelman, Cornelia de Lange and Cri du Chat syndromes. J. Intellect. Disabil. Res. 2011, 55, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Mahon, L.V.; Lomax, M.; Grant, S.; Cross, E.; Hare, D.J.; Wraith, J.E.; Jones, S.; Bigger, B.; Langford-Smith, K.; Canal, M. Assessment of sleep in children with mucopolysaccharidosis type III. PLoS ONE 2014, 9, e84128. [Google Scholar] [CrossRef] [PubMed]
- Canal, M.M.; Wilkinson, F.L.; Cooper, J.D.; Wraith, J.E.; Wynn, R.; Bigger, B.W. Circadian rhythm and suprachiasmatic nucleus alterations in the mouse model of mucopolysaccharidosis IIIB. Behav. Brain Res. 2010, 209, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Maris, M.; Verhulst, S.; Wojciechowski, M.; Van de Heyning, P.; Boudewyns, A. Sleep problems and obstructive sleep apnea in children with down syndrome, an overwiew. Int. J. Pediatr. Otorhinolaryngol. 2016, 82, 12–15. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, F.J.; Camfferman, D.; Kennedy, J.D.; Martin, A.J.; Couper, T.; Lack, L.D.; Lushington, K.; McEvoy, R.D. Sleep-disordered breathing in Prader-Willi syndrome and its association with neurobehavioral abnormalities. J. Pediatr. 2005, 147, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Festen, D.A.; Wevers, M.; de Weerd, A.W.; van den Bossche, R.A.; Duivenvoorden, H.J.; Hokken-Koelega, A.C. Cognition and behavior in pre-pubertal children with Prader-Willi syndrome and associations with sleep-related breathing disorders. Am. J. Med. Genet. A 2008, 146, 3018–3025. [Google Scholar] [CrossRef] [PubMed]
- Festen, D.A.; Wevers, M.; de Weerd, A.W.; van den Bossche, R.A.; Duivenvoorden, H.J.; Otten, B.J.; Wit, J.M.; Hokken-Koelega, A.C. Psychomotor development in infants with Prader-Willi syndrome and associations with sleep-related breathing disorders. Pediatr. Res. 2007, 62, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Youssoufian, H.; Pyeritz, R.E. Mechanisms and consequences of somatic mosaicism in humans. Nat. Rev. Genet. 2002, 3, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Bull, M.J. Health supervision for children with Down syndrome. Pediatrics 2011, 128, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Fujiyama, A.; Taylor, T.D.; Watanabe, H.; Yada, T.; Park, H.S.; Toyoda, A.; Ishii, K.; Totoki, Y.; Choi, D.K.; et al. The DNA sequence of human chromosome 21. Nature 2000, 405, 311–319. [Google Scholar] [PubMed]
- Pelleri, M.C.; Cicchini, E.; Locatelli, C.; Vitale, L.; Caracausi, M.; Piovesan, A.; Rocca, A.; Poletti, G.; Seri, M.; Strippoli, P.; et al. Systematic reanalysis of partial trisomy 21 cases with or without Down syndrome suggests a small region on 21q22.13 as critical to the phenotype. Hum. Mol. Genet. 2016, 25, 2525–2538. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.K.; Suzuki, Y.; Yoshimura, S.; Togashi, T.; Hida, M.; Taylor, T.D.; Wang, Y.; Sugano, S.; Hattori, M.; Sakaki, Y. Molecular cloning and characterization of a gene expressed in mouse developing tongue, mDscr5 gene, a homolog of human DSCR5 (Down syndrome Critical Region gene 5). Mamm. Genome 2001, 12, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Maris, M.; Verhulst, S.; Wojciechowski, M.; Van de Heyning, P.; Boudewyns, A. Outcome of adenotonsillectomy in children with Down syndrome and obstructive sleep apnoea. Arch. Dis. Child. 2017, 102, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Mims, M.; Thottam, P.J.; Kitsko, D.; Shaffer, A.; Choi, S. Characterization of sleep architecture in down Syndrome patients pre and post airway surgery. Cureus 2017, 9, e983. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, L.C.; Phillips, N.N.; Hoban, T.F.; O’Brien, L.M. Characterization of a sleep architectural phenotype in children with Down syndrome. Sleep Breath. 2015, 19, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Ahn, M.; Roth, H.L.; Li, L.; Vaughn, B.V. Sleep apnea and hypoventilation in patients with Down syndrome: Analysis of 144 polysomnogram studies. Children 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Roland, P.S.; Rosenfeld, R.M.; Brooks, L.J.; Friedman, N.R.; Jones, J.; Kim, T.W.; Kuhar, S.; Mitchell, R.B.; Seidman, M.D.; Sheldon, S.H.; et al. Clinical practice guideline: Polysomnography for sleep-disordered breathing prior to tonsillectomy in children. Otolaryngol. Head Neck Surg. 2011, 145 (Suppl. 1), S1–S15. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.E.; Bichell, T.J.; Surdyka, K.; Malow, B.A. Sleep in children and adolescents with Angelman syndrome: Association with parent sleep and stress. J. Intellect. Disabil. Res. 2012, 56, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Pelc, K.; Cheron, G.; Boyd, S.G.; Dan, B. Are there distinctive sleep problems in Angelman syndrome? Sleep Med. 2008, 9, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Miano, S.; Bruni, O.; Leuzzi, V.; Elia, M.; Verrillo, E.; Ferri, R. Sleep polygraphy in Angelman syndrome. Clin. Neurophysiol. 2004, 115, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Judson, M.C.; Wallace, M.L.; Sidorov, M.S.; Burette, A.C.; Gu, B.; van Woerden, G.M.; King, I.F.; Han, J.E.; Zylka, M.J.; Elgersma, Y.; et al. GABAergic neuron-specific loss of ube3a causes Angelman syndrome-like EEG abnormalities and enhances seizure susceptibility. Neuron 2016, 90, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Laan, L.A.; Vein, A.A. Angelman syndrome: Is there a characteristic EEG? Brain Dev. 2005, 27, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Miano, S.; Bruni, O.; Elia, M.; Musumeci, S.A.; Verrillo, E.; Ferri, R. Sleep breathing and periodic leg movement pattern in Angelman Syndrome: A polysomnographic study. Clin. Neurophysiol. 2005, 116, 2685–2692. [Google Scholar] [CrossRef] [PubMed]
- Bruni, O.; Ferri, R.; D’Agostino, G.; Miano, S.; Roccella, M.; Elia, M. Sleep disturbances in Angelman syndrome: A questionnaire study. Brain Dev. 2004, 26, 233–240. [Google Scholar] [CrossRef]
- Vendrame, M.; Loddenkemper, T.; Zarowski, M.; Gregas, M.; Shuhaiber, H.; Sarco, D.P.; Morales, A.; Nespeca, M.; Sharpe, C.; Haas, K.; et al. Analysis of EEG patterns and genotypes in patients with Angelman syndrome. Epilepsy Behav. 2012, 23, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Takaesu, Y.; Komada, Y.; Inoue, Y. Melatonin profile and its relation to circadian rhythm sleep disorders in Angelman syndrome patients. Sleep Med. 2012, 13, 1164–1170. [Google Scholar] [CrossRef] [PubMed]
- Zhdanova, I.V.; Wurtman, R.J.; Wagstaff, J. Wagstaff, Effects of a low dose of melatonin on sleep in children with Angelman syndrome. J. Pediatr. Endocrinol. Metab. 1999, 12, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Braam, W.; Didden, R.; Smits, M.G.; Curfs, L.M. Melatonin for chronic insomnia in Angelman syndrome: A randomized placebo-controlled trial. J. Child Neurol. 2008, 23, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, A.P.; Holland, A.J.; Hauffa, B.P.; Hokken-Koelega, A.C.; Tauber, M. Recommendations for the diagnosis and management of Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 4183–4197. [Google Scholar] [CrossRef] [PubMed]
- Lan, M.C.; Hsu, Y.B.; Lan, M.Y.; Chiu, T.J.; Huang, T.T.; Wong, S.B.; Chen, Y.C.; Tsai, L.P. Drug-induced sleep endoscopy in children with Prader-Willi syndrome. Sleep Breath. 2016, 20, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Camfferman, D.; McEvoy, R.D.; O’Donoghue, F.; Lushington, K. Prader-Willi Syndrome and excessive daytime sleepiness. Sleep Med. Rev. 2008, 12, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Khayat, A.; Narang, I.; Bin-Hasan, S.; Amin, R.; Al-Saleh, S. Longitudinal evaluation of sleep disordered breathing in infants with Prader-Willi syndrome. Arch. Dis. Child. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pavone, M.; Caldarelli, V.; Khirani, S.; Colella, M.; Ramirez, A.; Aubertin, G.; Crino, A.; Brioude, F.; Gastaud, F.; Beydon, N.; et al. Sleep disordered breathing in patients with Prader-Willi syndrome: A multicenter study. Pediatr. Pulmonol. 2015, 50, 1354–1359. [Google Scholar] [CrossRef] [PubMed]
- Beauloye, V.; Dhondt, K.; Buysse, W.; Nyakasane, A.; Zech, F.; De Schepper, J.; Van Aken, S.; De Waele, K.; Craen, M.; Gies, I.; et al. Evaluation of the hypothalamic-pituitary-adrenal axis and its relationship with central respiratory dysfunction in children with Prader-Willi syndrome. Orphanet J. Rare Dis. 2015, 10, 106. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Lin, S.P.; Lin, C.C.; Tsai, L.P.; Chen, M.R.; Chuang, C.K.; Huang, C.Y. Polysomnographic characteristics in patients with Prader-Willi syndrome. Pediatr. Pulmonol. 2007, 42, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Manni, R.; Politini, L.; Nobili, L.; Ferrillo, F.; Livieri, C.; Veneselli, E.; Biancheri, R.; Martinetti, M.; Tartara, A. Hypersomnia in the Prader-Willi syndrome: Clinical-electrophysiological features and underlying factors. Clin. Neurophysiol. 2001, 112, 800–805. [Google Scholar] [CrossRef]
- Vgontzas, A.N.; Kales, A.; Seip, J.; Mascari, M.J.; Bixler, E.O.; Myers, D.C.; Vela-Bueno, A.V.; Rogan, P.K. Relationship of sleep abnormalities to patient genotypes in Prader-Willi syndrome. Am. J. Med. Genet. 1996, 67, 478–482. [Google Scholar] [CrossRef]
- Clift, S.; Dahlitz, M.; Parkes, J.D. Parkes, Sleep apnoea in the Prader-Willi syndrome. J. Sleep Res. 1994, 3, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Helbing-Zwanenburg, B.; Kamphuisen, H.A.; Mourtazaev, M.S. Mourtazaev, The origin of excessive daytime sleepiness in the Prader-Willi syndrome. J. Intellect. Disabil. Res. 1993, 37, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Hertz, G.; Cataletto, M.; Feinsilver, S.H.; Angulo, M. Sleep and breathing patterns in patients with Prader Willi syndrome (PWS): Effects of age and gender. Sleep 1993, 16, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Priano, L.; Grugni, G.; Miscio, G.; Guastamacchia, G.; Toffolet, L.; Sartorio, A.; Mauro, A. Sleep cycling alternating pattern (CAP) expression is associated with hypersomnia and GH secretory pattern in Prader-Willi syndrome. Sleep Med. 2006, 7, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Verrillo, E.; Bruni, O.; Franco, P.; Ferri, R.; Thiriez, G.; Pavone, M.; Petrone, A.; Paglietti, M.G.; Crino, A.; Cutrera, R. Analysis of NREM sleep in children with Prader-Willi syndrome and the effect of growth hormone treatment. Sleep Med. 2009, 10, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, D.S.; Gulliver, T.; Williams, G.; Harris, M.A.; Nyunt, O.; Suresh, S. Central sleep-disordered breathing and the effects of oxygen therapy in infants with Prader-Willi syndrome. Arch. Dis. Child. 2013, 98, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Vandeleur, M.; Davey, M.J.; Nixon, G.M. Nixon, Are sleep studies helpful in children with Prader-Willi syndrome prior to commencement of growth hormone therapy? J. Paediatr. Child Health 2013, 49, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.L.; Splaingard, M.; Repaske, D.R.; Zipf, W.; Atkins, J.; Jatana, K. Outcomes of adenotonsillectomy in patients with Prader-Willi syndrome. Arch. Otolaryngol. Head Neck Surg. 2012, 138, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- De Cock, V.C.; Diene, G.; Molinas, C.; Masson, V.D.; Kieffer, I.; Mimoun, E.; Tiberge, M.; Tauber, M. Efficacy of modafinil on excessive daytime sleepiness in Prader-Willi syndrome. Am. J. Med. Genet. A 2011, 155, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Spruyt, K.; Braam, W.; Smits, M.; Curfs, L.M. Sleep complaints and the 24-h melatonin level in individuals with Smith-Magenis syndrome: Assessment for effective intervention. CNS Neurosci. Ther. 2016, 22, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Poisson, A.; Nicolas, A.; Cochat, P.; Sanlaville, D.; Rigard, C.; de Leersnyder, H.; Franco, P.; Des Portes, V.; Edery, P.; Demily, C. Behavioral disturbance and treatment strategies in Smith-Magenis syndrome. Orphanet J. Rare Dis. 2015, 10, 111. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.R.; Zies, D.; Mullegama, S.V.; Grotewiel, M.S.; Elsea, S.H. Smith-Magenis syndrome results in disruption of CLOCK gene transcription and reveals an integral role for RAI1 in the maintenance of circadian rhythmicity. Am. J. Hum. Genet. 2012, 90, 941–949. [Google Scholar] [CrossRef] [PubMed]
- De Leersnyder, H.; Claustrat, B.; Munnich, A.; Verloes, A. Circadian rhythm disorder in a rare disease: Smith-Magenis syndrome. Mol. Cell. Endocrinol. 2006, 252, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Alaimo, J.T.; Barton, L.V.; Mullegama, S.V.; Wills, R.D.; Foster, R.H.; Elsea, S.H. Individuals with Smith-Magenis syndrome display profound neurodevelopmental behavioral deficiencies and exhibit food-related behaviors equivalent to Prader-Willi syndrome. Res. Dev. Disabil. 2015, 47, 27–38. [Google Scholar] [CrossRef] [PubMed]
- De Leersnyder, H.; de Blois, M.C.; Vekemans, M.; Sidi, D.; Villain, E.; Kindermans, C.; Munnich, A. beta(1)-adrenergic antagonists improve sleep and behavioural disturbances in a circadian disorder, Smith-Magenis syndrome. J. Med. Genet. 2001, 38, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Weese-Mayer, D.E.; Marazita, M.L.; Rand, C.M.; Berry-Kravis, E.M. Congenital Central Hypoventilation Syndrome; Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Weese-Mayer, D.E.; Berry-Kravis, E.M.; Ceccherini, I.; Keens, T.G.; Loghmanee, D.A.; Trang, H. An official ATS clinical policy statement: Congenital central hypoventilation syndrome: Genetic basis, diagnosis, and management. Am. J. Respir. Crit. Care Med. 2010, 181, 626–644. [Google Scholar] [CrossRef] [PubMed]
- Todd, E.S.; Weinberg, S.M.; Berry-Kravis, E.M.; Silvestri, J.M.; Kenny, A.S.; Rand, C.M.; Zhou, L.; Maher, B.S.; Marazita, M.L.; Weese-Mayer, D.E. Facial phenotype in children and young adults with PHOX2B-determined congenital central hypoventilation syndrome: Quantitative pattern of dysmorphology. Pediatr. Res. 2006, 59, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Charnay, A.J.; Antisdel-Lomaglio, J.E.; Zelko, F.A.; Rand, C.M.; Le, M.; Gordon, S.C.; Vitez, S.F.; Tse, J.W.; Brogadir, C.D.; Nelson, M.N.; et al. Congenital central hypoventilation syndrome: Neurocognition already reduced in preschool-aged children. Chest 2016, 149, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, J.M.; Weese-Mayer, D.E.; Nelson, M.N. Neuropsychologic abnormalities in children with congenital central hypoventilation syndrome. J. Pediatr. 1992, 120, 388–393. [Google Scholar] [CrossRef]
- Paddeu, E.M.; Giganti, F.; Piumelli, R.; De Masi, S.; Filippi, L.; Viggiano, M.P.; Donzelli, G. Sleeping problems in mothers and fathers of patients suffering from congenital central hypoventilation syndrome. Sleep Breath. 2015, 19, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Fleming, P.J.; Cade, D.; Bryan, M.H.; Bryan, A.C. Congenital central hypoventilation and sleep state. Pediatrics 1980, 66, 425–428. [Google Scholar] [PubMed]
- Guilleminault, C.; McQuitty, J.; Ariagno, R.L.; Challamel, M.J.; Korobkin, R.; McClead, R.E., Jr. Congenital central alveolar hypoventilation syndrome in six infants. Pediatrics 1982, 70, 684–694. [Google Scholar] [PubMed]
- Huang, J.; Colrain, I.M.; Panitch, H.B.; Tapia, I.E.; Schwartz, M.S.; Samuel, J.; Pepe, M.; Bandla, P.; Bradford, R.; Mosse, Y.P.; et al. Effect of sleep stage on breathing in children with central hypoventilation. J. Appl. Physiol. 2008, 105, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Gozal, D.; Marcus, C.L.; Ward, S.L.; Keens, T.G. Ventilatory responses to passive leg motion in children with congenital central hypoventilation syndrome. Am. J. Respir. Crit. Care Med. 1996, 153, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Weese-Mayer, D.E.; Silvestri, J.M.; Menzies, L.J.; Morrow-Kenny, A.S.; Hunt, C.E.; Hauptman, S.A. Congenital central hypoventilation syndrome: Diagnosis, management, and long-term outcome in thirty-two children. J. Pediatr. 1992, 120, 381–387. [Google Scholar] [CrossRef]
- Tenconi, R.; Khirani, S.; Amaddeo, A.; Michot, C.; Baujat, G.; Couloigner, V.; De Sanctis, L.; James, S.; Zerah, M.; Cormier-Daire, V.; et al. Sleep-disordered breathing and its management in children with achondroplasia. Am. J. Med. Genet. A 2017, 173, 868–878. [Google Scholar] [CrossRef] [PubMed]
- White, K.K.; Bompadre, V.; Goldberg, M.J.; Bober, M.B.; Campbell, J.W.; Cho, T.J.; Hoover-Fong, J.; Mackenzie, W.; Parnell, S.E.; Raggio, C.; et al. Best practices in the evaluation and treatment of foramen magnum stenosis in achondroplasia during infancy. Am. J. Med. Genet. A 2016, 170, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Afsharpaiman, S.; Saburi, A.; Waters, K.A. Respiratory difficulties and breathing disorders in achondroplasia. Paediatr. Respir. Rev. 2013, 14, 250–255. [Google Scholar]
- Zaffanello, M.; Cantalupo, G.; Piacentini, G.; Gasperi, E.; Nosetti, L.; Cavarzere, P.; Ramaroli, D.A.; Mittal, A.; Antoniazzi, F. Sleep disordered breathing in children with achondroplasia. World J. Pediatr. 2017, 13, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Onodera, K.; Sakata, H.; Niikuni, N.; Nonaka, T.; Kobayashi, K.; Nakajima, I. Survey of the present status of sleep-disordered breathing in children with achondroplasia Part I. A questionnaire survey. Int. J. Pediatr. Otorhinolaryngol. 2005, 69, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Zaffanello, M.; Lo Tartaro, P.; Piacentini, G.; Cantalupo, G.; Gasperi, E.; Antoniazzi, F. Sleep disordered breathing in a cohort of children with achondroplasia: Correlation between clinical and instrumental findings. Minerva Pediatr. 2015. [Google Scholar]
- White, K.K.; Parnell, S.E.; Kifle, Y.; Blackledge, M.; Bompadre, V. Is there a correlation between sleep disordered breathing and foramen magnum stenosis in children with achondroplasia? Am. J. Med. Genet. A 2016, 170, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.T.; Butler, I.J.; Horton, W.A. Foramen magnum decompression for homozygous achondroplasia. J. Neurosurg. 1989, 71, 300–301. [Google Scholar] [PubMed]
- Pereira, D.R.; Schweiger, C.; de Souza, C.F.; Fagondes, S.; Manica, D.; Giugliani, R.; Kuhl, G.; Marostica, P.J. Correlation between flexible fiberoptic laryngoscopic and polysomnographic findings in patients with mucopolysaccharidosis type VI. JIMD Rep. 2016, 29, 53–58. [Google Scholar] [PubMed]
- Wooten, W.I., 3rd; Muenzer, J.; Vaughn, B.V.; Muhlebach, M.S. Relationship of sleep to pulmonary function in mucopolysaccharidosis II. J. Pediatr. 2013, 162, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Moreira, G.A.; Kyosen, S.O.; Patti, C.L.; Martins, A.M.; Tufik, S. Prevalence of obstructive sleep apnea in patients with mucopolysaccharidosis types I, II, and VI in a reference center. Sleep Breath. 2014, 18, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Mumford, R.A.; Mahon, L.V.; Jones, S.; Bigger, B.; Canal, M.; Hare, D.J. Actigraphic investigation of circadian rhythm functioning and activity levels in children with mucopolysaccharidosis type III (Sanfilippo syndrome). J. Neurodev. Disord. 2015, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Muhlebach, M.S.; Wooten, W.; Muenzer, J. Muenzer, Respiratory manifestations in mucopolysaccharidoses. Paediatr. Respir. Rev. 2011, 12, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Averill, L.W.; Sawamoto, K.; Mackenzie, W.G.; Bober, M.B.; Pizarro, C.; Goff, C.J.; Xie, L.; Orii, T.; Theroux, M. Obstructive airway in Morquio A syndrome, the past, the present and the future. Mol. Genet. Metab. 2016, 117, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Montano, A.M.; Lock-Hock, N.; Steiner, R.D.; Graham, B.H.; Szlago, M.; Greenstein, R.; Pineda, M.; Gonzalez-Meneses, A.; Coker, M.; Bartholomew, D.; et al. Clinical course of sly syndrome (mucopolysaccharidosis type VII). J. Med. Genet. 2016, 53, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Kiykim, E.; Barut, K.; Cansever, M.S.; Zeybek, C.A.; Zubarioglu, T.; Aydin, A.; Kasapcopur, O. screening mucopolysaccharidosis type IX in patients with juvenile idiopathic arthritis. JIMD Rep. 2016, 25, 21–24. [Google Scholar] [PubMed]
- Arn, P.; Bruce, I.A.; Wraith, J.E.; Travers, H.; Fallet, S. Airway-related symptoms and surgeries in patients with mucopolysaccharidosis I. Ann. Otol. Rhinol. Laryngol. 2015, 124, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Gonuldas, B.; Yilmaz, T.; Sivri, H.S.; Gucer, K.S.; Kilinc, K.; Genc, G.A.; Kilic, M.; Coskun, T. Mucopolysaccharidosis: Otolaryngologic findings, obstructive sleep apnea and accumulation of glucosaminoglycans in lymphatic tissue of the upper airway. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chen, M.R.; Lin, C.C.; Chen, C.P.; Lin, D.S.; Chuang, C.K.; Niu, D.M.; Chang, J.H.; Lee, H.C.; Lin, S.P. Polysomnographic characteristics in patients with mucopolysaccharidoses. Pediatr. Pulmonol. 2010, 45, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Leighton, S.E.; Papsin, B.; Vellodi, A.; Dinwiddie, R.; Lane, R. Disordered breathing during sleep in patients with mucopolysaccharidoses. Int. J. Pediatr. Otorhinolaryngol. 2001, 58, 127–138. [Google Scholar] [CrossRef]
- Nashed, A.; Al-Saleh, S.; Gibbons, J.; MacLusky, I.; MacFarlane, J.; Riekstins, A.; Clarke, J.; Narang, I. Sleep-related breathing in children with mucopolysaccharidosis. J. Inherit. Metab. Dis. 2009, 32, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, F.; Andreucci, M.V.; Parenti, G.; Polverino, M.; Viggiano, D.; Montella, S.; Cesaro, A.; Ciccarelli, R.; Capaldo, B.; Andria, G. Upper airway obstructive disease in mucopolysaccharidoses: Polysomnography, computed tomography and nasal endoscopy findings. J. Inherit. Metab. Dis. 2007, 30, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Hoque, R. Sleep-Disordered Breathing in Duchenne Muscular Dystrophy: An Assessment of the Literature. J. Clin. Sleep Med. 2016, 12, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Jeronimo, G.; Nozoe, K.T.; Polesel, D.N.; Moreira, G.A.; Tufik, S.; Andersen, M.L. Impact of corticotherapy, nutrition, and sleep disorder on quality of life of patients with Duchenne muscular dystrophy. Nutrition 2016, 32, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Nozoe, K.T.; Moreira, G.A.; Tolino, J.R.; Pradella-Hallinan, M.; Tufik, S.; Andersen, M.L. The sleep characteristics in symptomatic patients with Duchenne muscular dystrophy. Sleep Breath. 2015, 19, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.P.; O’Halloran, K.D. Evidence of hypoxic tolerance in weak upper airway muscle from young mdx mice. Respir. Physiol. Neurobiol. 2016, 226, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Ricotti, V.; Mandy, W.P.; Scoto, M.; Pane, M.; Deconinck, N.; Messina, S.; Mercuri, E.; Skuse, D.H.; Muntoni, F. Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev. Med. Child Neurol. 2016, 58, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Wales, P.; Dakin, C.; Harris, M.A.; Cooper, D.G. Sleep-related breathing disorder in Duchenne muscular dystrophy: Disease spectrum in the paediatric population. J. Paediatr. Child Health 2005, 41, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Sawnani, H.; Thampratankul, L.; Szczesniak, R.D.; Fenchel, M.C.; Simakajornboon, N. Sleep disordered breathing in young boys with Duchenne muscular dystrophy. J. Pediatr. 2015, 166, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Bloetzer, C.; Jeannet, P.Y.; Lynch, B.; Newman, C.J. Sleep disorders in boys with Duchenne muscular dystrophy. Acta Paediatr. 2012, 101, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Kirk, V.G.; Flemons, W.W.; Adams, C.; Rimmer, K.P.; Montgomery, M.D. Sleep-disordered breathing in Duchenne muscular dystrophy: A preliminary study of the role of portable monitoring. Pediatr. Pulmonol. 2000, 29, 135–140. [Google Scholar] [CrossRef]
- Finder, J.D.; Birnkrant, D.; Carl, J.; Farber, H.J.; Gozal, D.; Iannaccone, S.T.; Kovesi, T.; Kravitz, R.M.; Panitch, H.; Schramm, C.; et al. Respiratory care of the patient with Duchenne muscular dystrophy: ATS consensus statement. Am. J. Respir. Crit. Care Med. 2004, 170, 456–465. [Google Scholar] [PubMed]
- Toussaint, M.; Steens, M.; Soudon, P. Soudon, Lung function accurately predicts hypercapnia in patients with Duchenne muscular dystrophy. Chest 2007, 131, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Farkas, G.A.; McCormick, K.M.; Gosselin, L.E. Gosselin, Episodic hypoxia exacerbates respiratory muscle dysfunction in DMD (mdx) mice. Muscle Nerve 2007, 36, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, M.R.; Fallavollita, J.A.; Farkas, G.A. The effects of experimental sleep apnea on cardiac and respiratory functions in 6 and 18 month old dystrophic (mdx) mice. PLoS ONE 2016, 11, e0147640. [Google Scholar] [CrossRef] [PubMed]
- Polat, M.; Sakinci, O.; Ersoy, B.; Sezer, R.G.; Yilmaz, H. Assessment of sleep-related breathing disorders in patients with duchenne muscular dystrophy. J. Clin. Med. Res. 2012, 4, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Hukins, C.A.; Hillman, D.R. Daytime predictors of sleep hypoventilation in Duchenne muscular dystrophy. Am. J. Respir. Crit. Care Med. 2000, 161, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Villanova, M.; Brancalion, B.; Mehta, A.D. Duchenne muscular dystrophy: Life prolongation by noninvasive ventilatory support. Am. J. Phys. Med. Rehabil. 2014, 93, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.E.; Calverley, P.M.; Edwards, R.H. Hypoxemia during sleep in Duchenne muscular dystrophy. Am. Rev. Respir. Dis. 1988, 137, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Krieg, A.M. FDA approves eteplirsen for Duchenne Muscular dystrophy: The next chapter in the eteplirsen saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Suspected Localization of Pathogenesis of Sleep Disorder | Neurogenetic Disorders |
|---|---|
| Multi-system Brainstem/orofacial structure/airway/chest | Down syndrome (DS) |
| Central nervous system Brain Brain (hypothalamus) Brain Brainstem/autonomic system | Angelman syndrome (AS) Prader–Willi syndrome (PWS) Smith–Magenis syndrome (SMS) Congenital central hypoventilation syndrome (CCHS) |
| Skeletal/spinal cord Medullary-cervical cord Upper airway/cervical cord | Achondroplasia/hypochondroplasia Mucopolysaccharidoses (MPS) |
| Peripheral nervous system Muscles | Duchenne muscular dystrophy (DMD) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dosier, L.B.M.; Vaughn, B.V.; Fan, Z. Sleep Disorders in Childhood Neurogenetic Disorders. Children 2017, 4, 82. https://doi.org/10.3390/children4090082
Dosier LBM, Vaughn BV, Fan Z. Sleep Disorders in Childhood Neurogenetic Disorders. Children. 2017; 4(9):82. https://doi.org/10.3390/children4090082
Chicago/Turabian StyleDosier, Laura Beth Mann, Bradley V. Vaughn, and Zheng Fan. 2017. "Sleep Disorders in Childhood Neurogenetic Disorders" Children 4, no. 9: 82. https://doi.org/10.3390/children4090082
APA StyleDosier, L. B. M., Vaughn, B. V., & Fan, Z. (2017). Sleep Disorders in Childhood Neurogenetic Disorders. Children, 4(9), 82. https://doi.org/10.3390/children4090082