
Congenital Malignant Ectomesenchymoma Presenting as a Neck Mass in a Newborn

 , , and
, , and
Abstract
1. Introduction
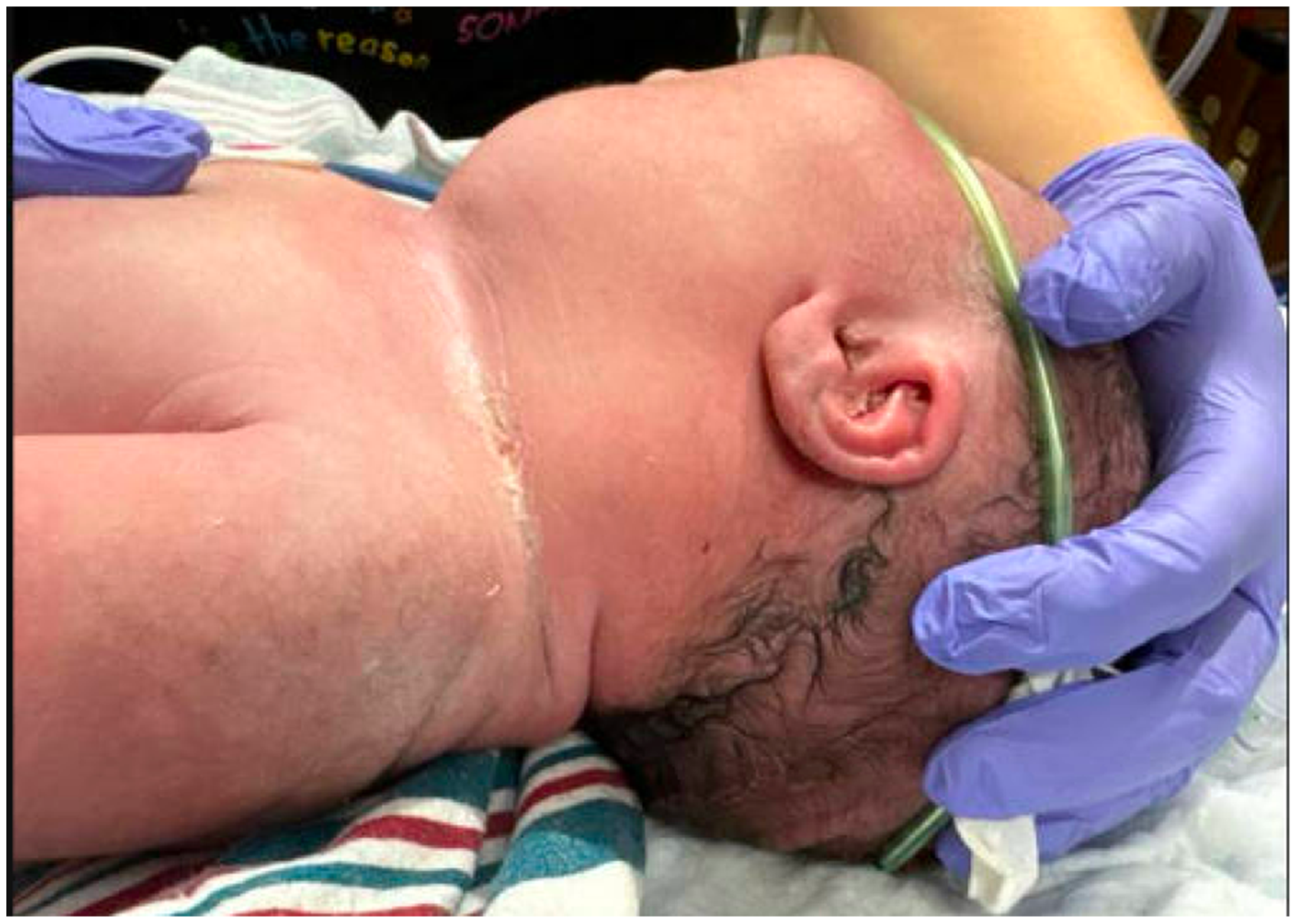
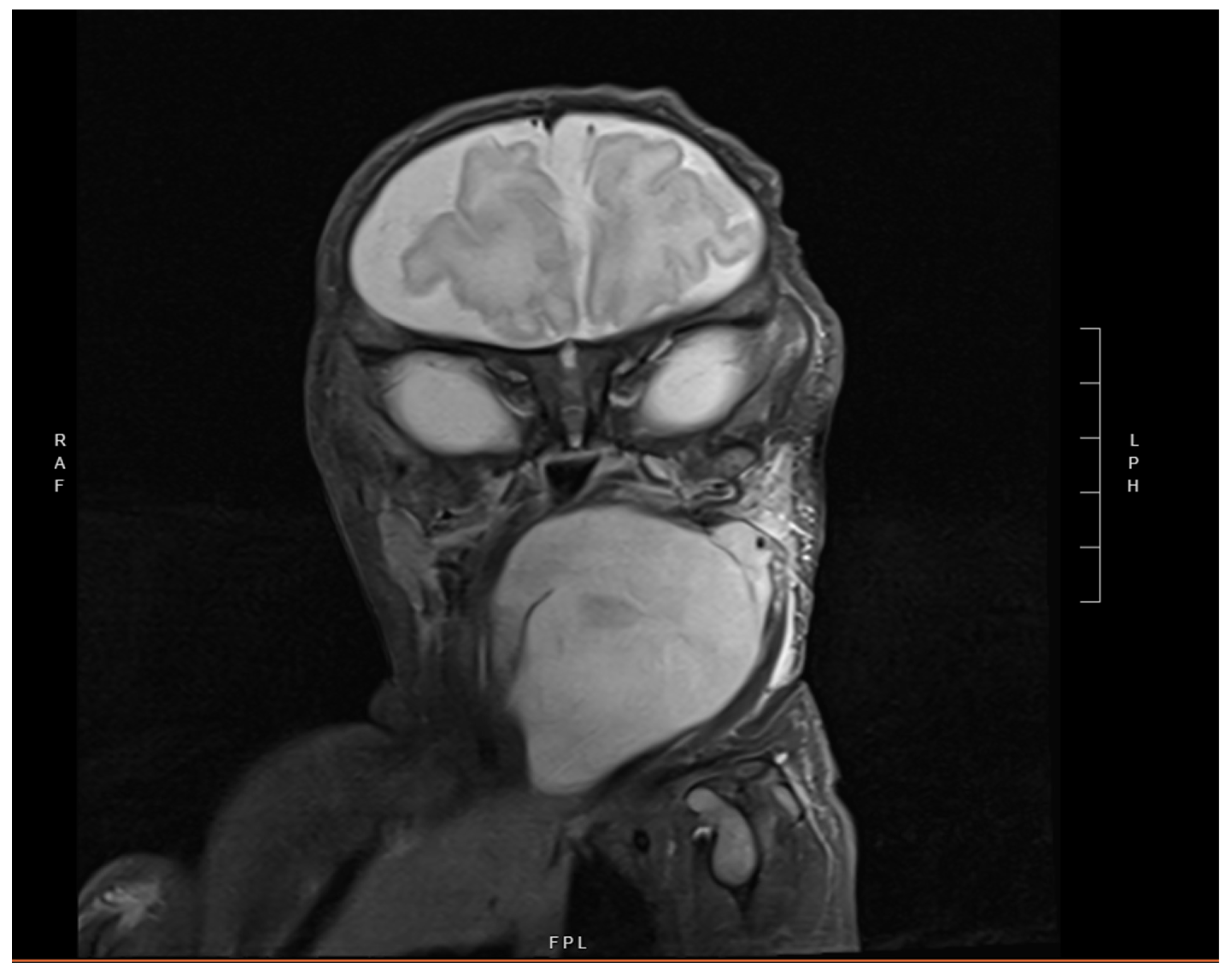
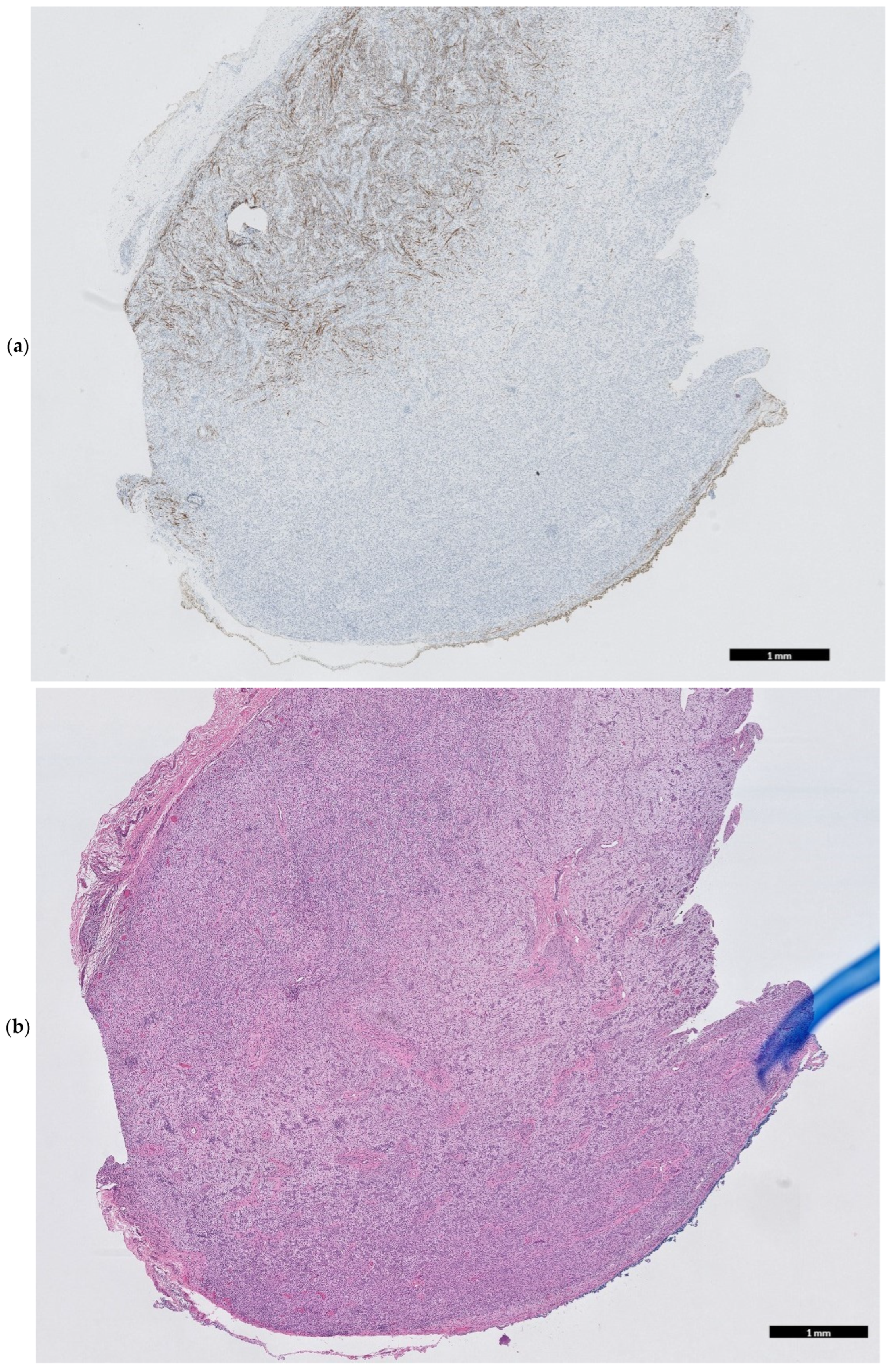
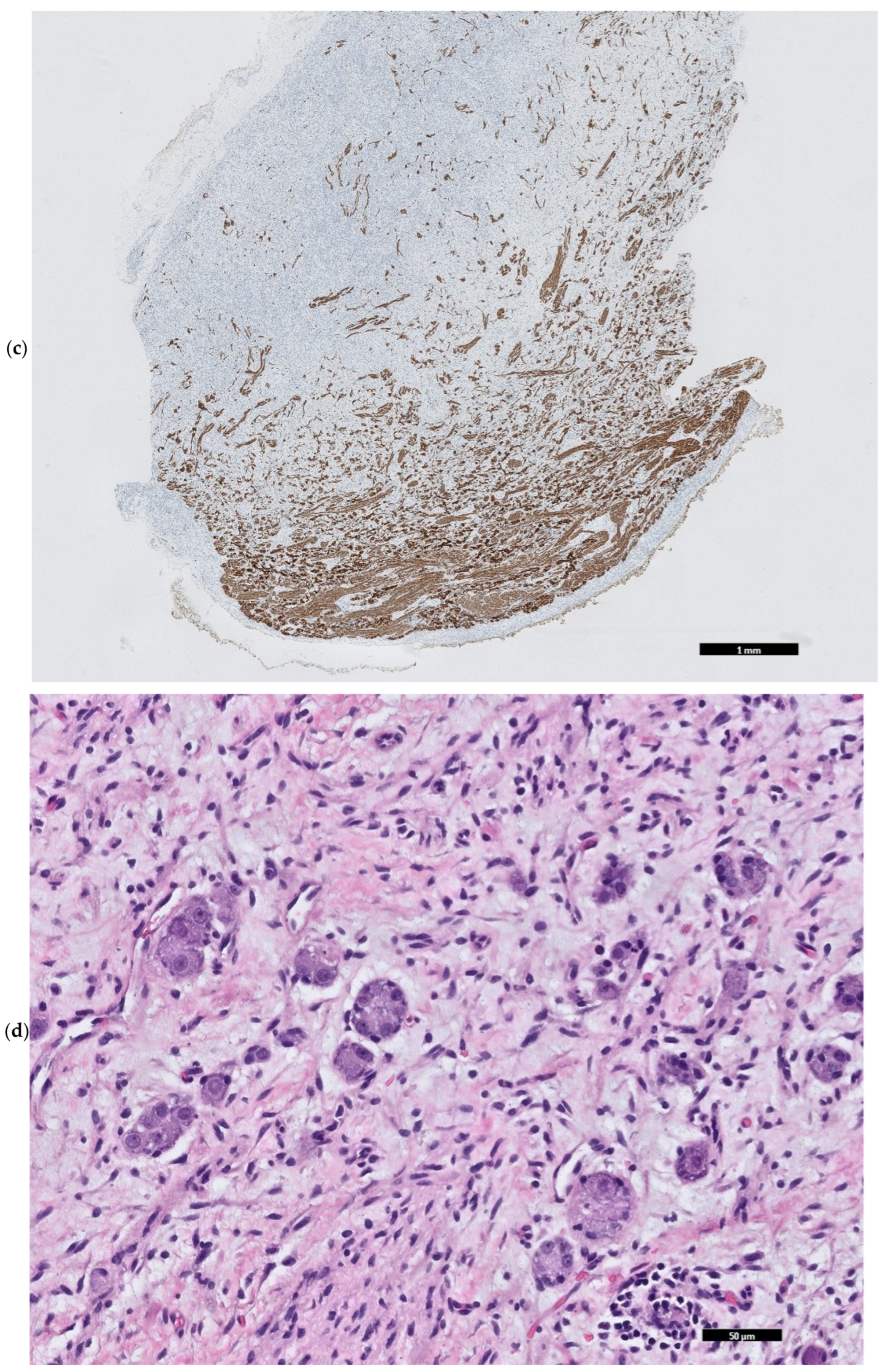
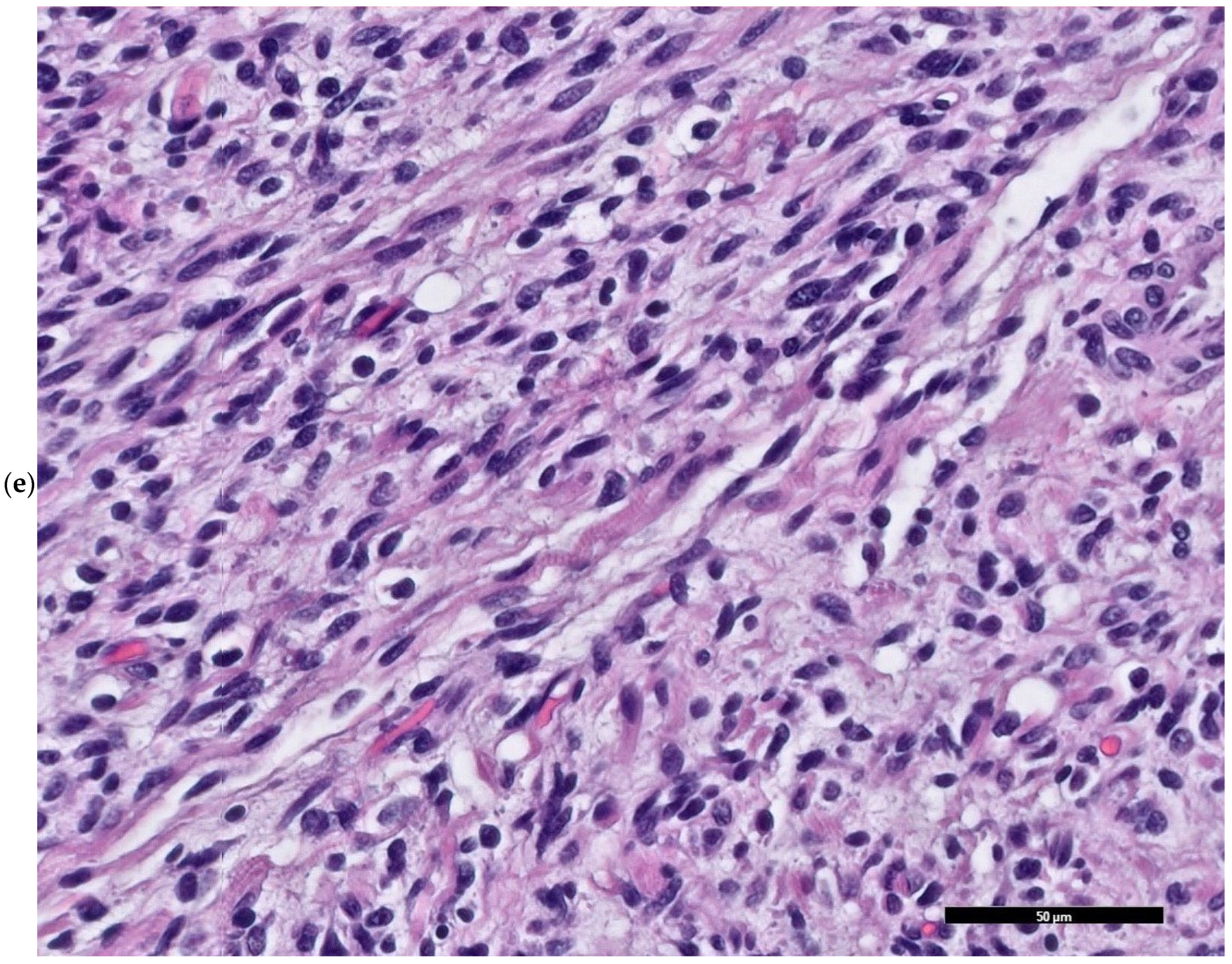
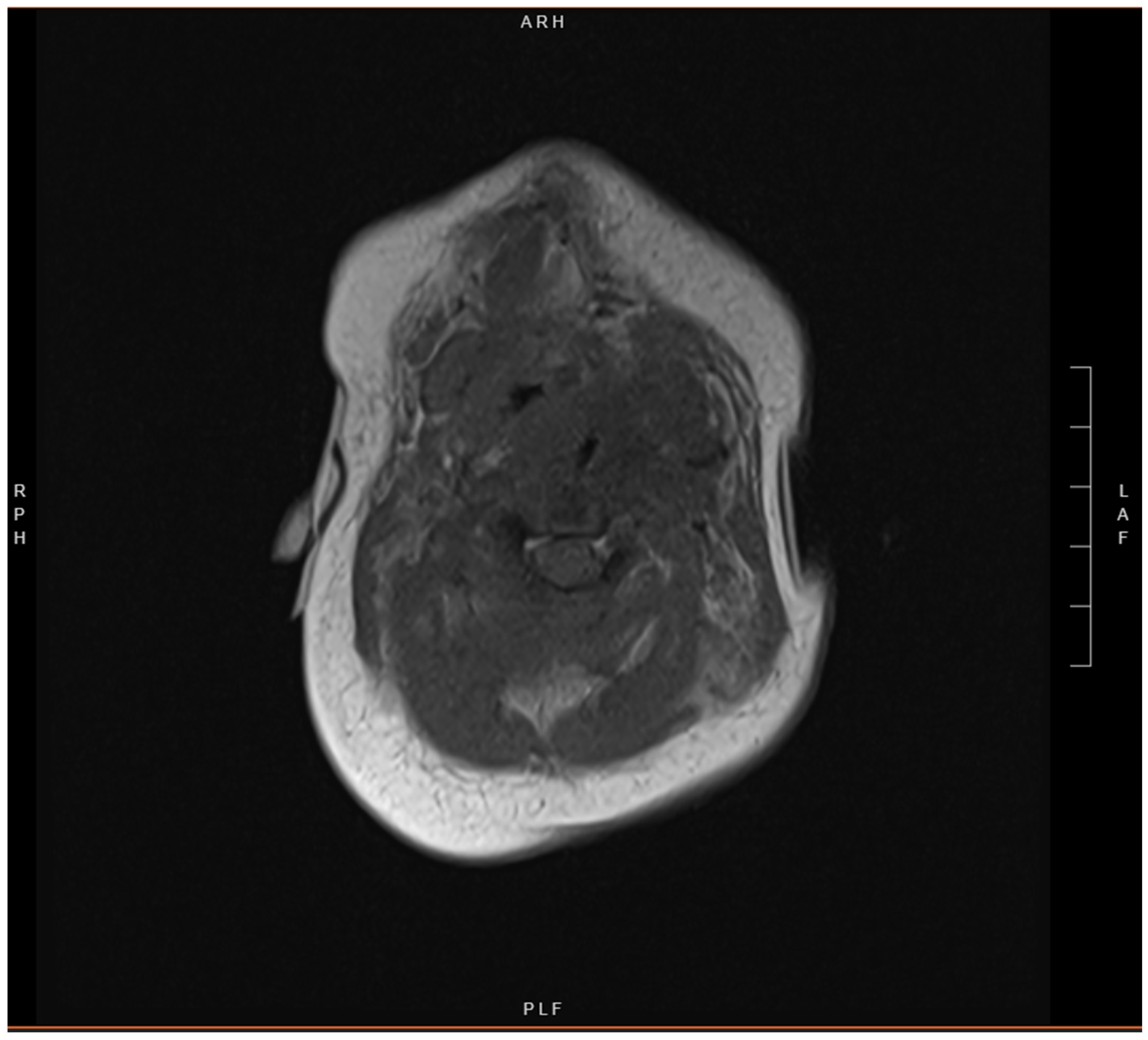
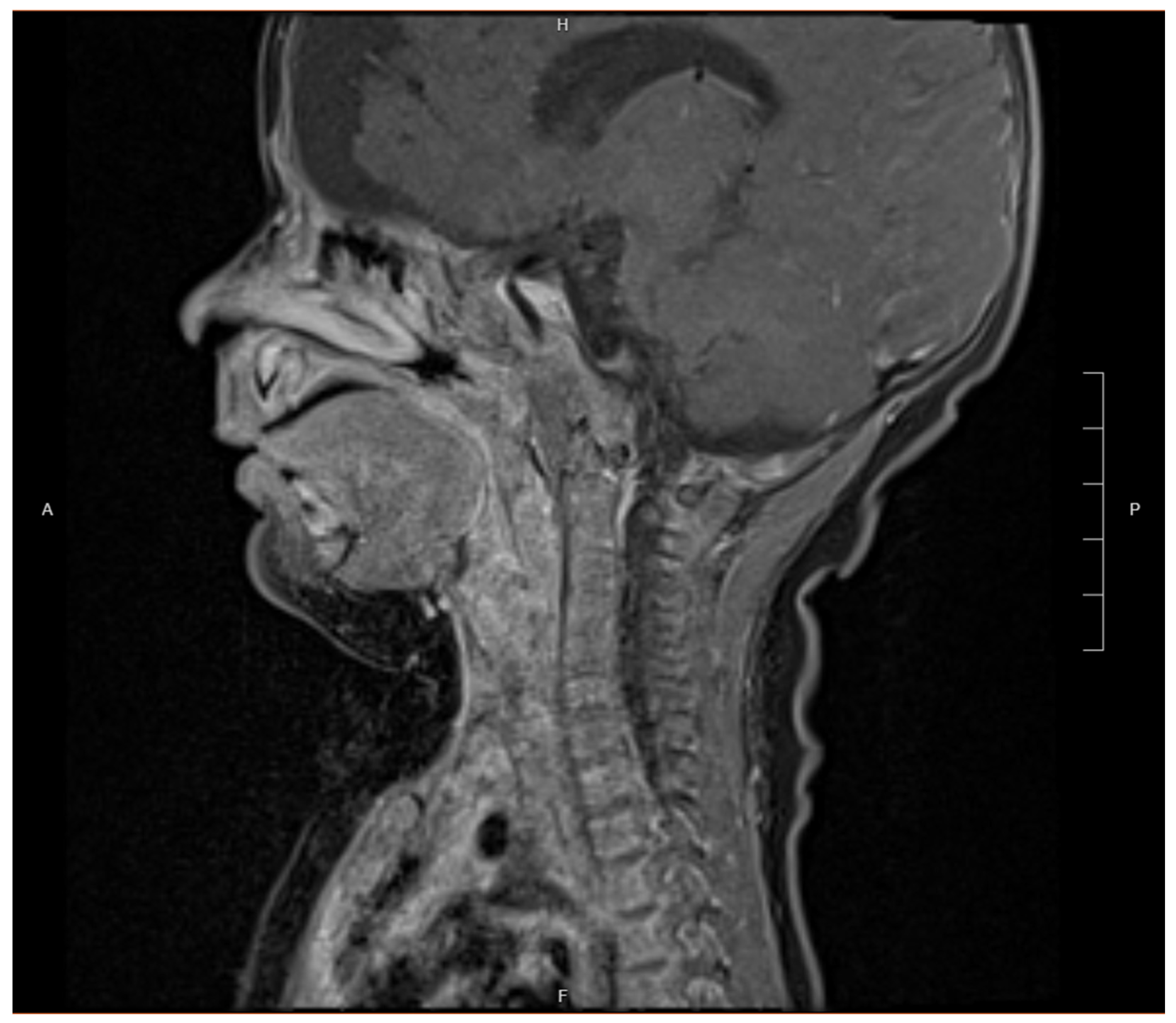
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MEM | Malignant Ectomesenchymoma |
| NICU | Neonatal Intensive Care Unit |
| CPAP | Continuous Positive Airway Pressure |
| HVA | Homovanillic Acid |
| VMA | Vanillylmandelic Acid |
| MRI | Magnetic Resonance Imaging |
| PET | Positron Emission Tomography |
| CT | Computed Tomography |
| VAC | Vincristine, Actinomycin, and Cyclophosphamide |
| VIT | Vincristine, Irinotecan, and Temozolomide |
| H&E | Hematoxylin and Eosin |
| STIR | Short Tau Inversion Recovery |
| RMS | Rhabdomyosarcoma |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | |||||||||
| ABL1 | ABL2 | ABRAXAS1 * | ACVR1 | ACVR1B * | ADGRA2 * | AIP | AKT1 | AKT2 | AKT3 |
| ALK | AMER1 | APC | AR | ARAF | ARFRP1 * | ARID1A | ARID1B $ | ARID2 | ASXL1 |
| ATM | ATR | ATRX | AURKA | AURKB | AXIN1 | AXIN2 | AXL | B2M | BAP1 |
| BARD1 | BCL10 * | BCL11B * | BCL2 | BCL2L1 * | BCL2L2 * | BCL6 | BCL7A | BCOR ^ | BCORL1 |
| BIRC3 | BLM | BMPR1A | BRAF | BRCA1 | BRCA2 | BRCC3 * | BRD3 * | BRD4 | BRINP3 * |
| BRIP1 | BTG1 | BTK | BUB1B * | C19MC | CALR | CARD11 | CASP8 * | CBFB | CBL |
| CBLB | CBLC | CCN6 | CCND1 | CCND2 | CCND3 | CCNE1 | CD19 * | CD274 | CD28 * |
| CD40 * | CD58 | CD79A | CD79B | CDC73 | CDH1 | CDK12 | CDK4 | CDK6 | CDK8 |
| CDKN1A | CDKN1B | CDKN1C | CDKN2A | CDKN2B | CDKN2C | CEBPA | CHD2 | CHD4 | CHEK1 |
| CHEK2 | CIC | CIITA * | COL1A1 | CREBBP | CRKL | CRLF2 | CSF1R | CSF3R | CSNK1A1 * |
| CTCF | CTLA4 * | CTNNA1 * | CTNNB1 | CUL3 | CUX1 | CXCR4 | CYLD | DAXX | DDR2 |
| DDX3X | DDX41 | DGCR8 | DICER1 | DIS3L2 | DKC1 | DNM2 | DNMT3A | DOT1L | DROSHA |
| EBF1 | EED | EFNB2 * | EGFR | EGR2 * | EIF1AX | ELOC | EMSY * | EP300 | EPCAM |
| EPHA3 | EPHA5 | EPHA7 * | EPHB1 | EPHB4 * | ERBB2 | ERBB3 | ERBB4 | ERCC1 * | ERCC2 |
| ERCC3 * | ERCC4 * | ERCC5 * | ERG * | ERRFI1 * | ESR1 | ETNK1 | ETS1 * | ETV1 * | ETV4 * |
| ETV5 * | ETV6 | EWSR1 * | EXT1 | EXT2 | EZH2 | FANCA | FANCB | FANCC | FANCD2 * |
| FANCE * | FANCF * | FANCG * | FANCI | FANCL * | FANCM * | FAS | FAT1 | FAT3 * | FBXO11 |
| FBXW7 | FGF10 * | FGF14 * | FGF19 | FGF23 * | FGF3 | FGF4 | FGF6 * | FGFR1 | FGFR2 |
| FGFR3 | FGFR4 | FH | FLCN | FLT1 | FLT3 ^ | FLT4 | FOXA1 | FOXL2 | FOXO1 * |
| FOXP1 | FOXR2 * | FRK | FRS2 * | FUBP1 | FUS * | GABRA6 * | GATA1 | GATA2 | GATA3 |
| GATA4 * | GATA6 * | GEN1 * | GID4 * | GLI1 | GLI2 | GNA11 | GNA13 | GNA14 | GNAQ |
| GNAS | GNB1 | GPC3 * | GREM1 * | GRIN2A | GRM3 * | GSK3B | H1-4 | H3-3A | H3-3B |
| H3C2 | H3C3 | HDAC4 | HDAC9 | HGF | HIF1A | HMGA2 * | HNF1A | HOXB13 * | HRAS |
| HSD3B1 | HSP90AA1 * | ID3 | IDH1 | IDH2 | IGF1R | IGF2 * | IKBKE | IKZF1 | IKZF2 |
| IKZF3 | IL2RG * | IL6ST | IL7R | INHBA * | INPP4B | IRF2 | IRF4 | IRF8 * | IRS2 * |
| ITK * | ITPKB * | JAK1 | JAK2 | JAK3 | JAZF1 * | JUN * | KAT6A * | KBTBD4 | KCNJ5 |
| KDM4C | KDM5A | KDM5C | KDM6A | KDR | KEAP1 | KEL * | KIF1B * | KIF5B * | KIT |
| KLF2 | KLF4 | KLHL6 * | KMT2A | KMT2B | KMT2C | KMT2D | KRAS | LEF1 | LIFR * |
| LMO1 | LRP1B * | LYN * | LYST * | LZTR1 | MAGI2 * | MAML2 * | MAP2K1 | MAP2K2 | MAP2K4 |
| MAP3K1 | MAP3K14 | MAP3K3 * | MAP3K7 * | MAPK1 | MAPK3 | MAX | MCL1 | MDM2 | MDM4 |
| MECOM | MED12 | MEF2B | MEN1 | MET | MITF | MLH1 | MLH3 | MLLT1 | MN1 |
| MPL | MRE11 | MSH2 | MSH3 | MSH6 | MTOR | MUTYH | MYB | MYBL1 | MYC |
| MYCL * | MYCN | MYD88 | MYH9 * | MYOD1 | NBN | NCOA2 * | NCOR1 | NCOR2 | NF1 |
| NF2 | NFE2 | NFE2L2 | NFKBIA | NFKBIE | NKX2-1 | NOTCH1 | NOTCH2 | NOTCH3 | NPM1 |
| NRAS | NSD1 | NSD2 | NT5C2 | NTHL1 | NTRK1 | NTRK2 | NTRK3 | NUP214 * | NUP93 * |
| NUTM1 * | OTX2 | PAK3 | PALB2 | PAX3 * | PAX5 | PAX7 * | PAX8 * | PBRM1 | PDCD1 |
| PDCD1LG2 * | PDGFB * | PDGFRA | PDGFRB | PDK1 * | PHF6 | PHOX2B | PIGA | PIK3C2B * | PIK3CA |
| PIK3CB | PIK3CD | PIK3CG | PIK3R1 | PIK3R2 | PIM1 | PLAG1 * | PLCG1 | PLCG2 | PML * |
| PMS1 | PMS2 | POLD1 * | POLE | POLR2A | POT1 | PPARG * | PPM1D | PPP2R1A | PRDM1 |
| PREX2 | PRKAR1A | PRKCA | PRKCI * | PRKDC | PRKN | PRPF40B | PRPF8 | PRSS8 * | PTCH1 |
| PTEN | PTPN11 | PTPRD | PTPRT | QKI * | RAB35 * | RAC1 | RAD21 | RAD50 | RAD51 |
| RAD51B | RAD51C | RAD51D | RAF1 | RANBP2 * | RARA * | RASA1 | RB1 | RBM10 | RECQL * |
| RECQL4 * | REL * | RELA * | RET | RHOA | RICTOR | RINT1 * | RIT1 | RNF43 | ROS1 * |
| RPS15 | RPS19 | RPS20 * | RPS6KA3 | RPTOR | RRAGC | RSPO2 * | RSPO3 * | RUNX1 | RUNX1T1 * |
| SAMD9 | SAMD9L | SBDS * | SDHA | SDHAF2 | SDHB | SDHC | SDHD | SETBP1 | SETD2 |
| SF1 | SF3A1 | SF3B1 | SGK1 | SH2B3 | SIX1 | SIX2 | SLIT2 * | SLX4 | SMAD2 |
| SMAD3 | SMAD4 | SMARCA4 | SMARCB1 | SMARCE1 | SMC1A | SMC3 | SMO | SNCAIP * | SOCS1 |
| SOS1 | SOX10 * | SOX2 | SOX9 * | SPEN | SPOP | SPTA1 | SRC | SRP72 | SRSF2 |
| SS18 * | STAG2 | STAT3 | STAT4 * | STAT5B | STAT6 | STK11 | SUFU | SUZ12 | SYK * |
| TAF1 | TBL1XR1 | TBX3 | TBXT * | TCF12 | TCF3 | TCF7L2 | TEK | TENT5C * | TERC |
| TERT | TET1 * | TET2 | TFE3 * | TFEB * | TGFBR2 | TMEM127 | TMPRSS2 * | TNFAIP3 | TNFRSF14 |
| TOP1 | TOP2A * | TP53 | TP63 | TPMT * | TRAF3 | TRAF7 | TRRAP | TSC1 | TSC2 |
| TSHR | TYK2 | U2AF1 | U2AF2 | USP6 * | USP7 | VAV1 | VEGFA | VHL | WRN |
| WT1 | XPC * | XPO1 | XRCC2 * | YAP1 * | YWHAE * | ZBTB2 * | ZFTA * | ZMYM3 | ZNF217 * |
| ZNF703 * | ZNF750 | ZRSR2 | |||||||
| (b) | |||||||||
| ABL1 | ABL2 | AKT3 | ALK | ARHGAP26 | AXL | BCL10 | BCL11B | BCL2 | BCL6 |
| BCOR | BCORL1 | BCR | BIRC3 | BRAF | BRD3 | BRD4 | BTG1 | C11ORF95 | CAMTA1 |
| CARD11 | CBFB | CBL | CCND1 | CCND3 | CDK6 | CHIC2 | CIC | CIITA | COL1A1 |
| CREBBP | CRLF2 | CSF1R | CTNNB1 * | DEK | DNAJB1 | DUSP22 | EBF1 | EGFR ^ | EP300 |
| EPC1 | EPOR | ERBB2 | ERG | ETV1 | ETV4 | ETV5 | ETV6 | EWSR1 | FER |
| FGFR1 | FGFR2 | FGFR3 | FGR | FLT3 | FOS | FOSB | FOXO1 | FOXP1 | FOXR2 |
| FUS | GLI1 | GLI2 | GLIS2 | HLF | HMGA2 | HTRA1 | IKZF1 | IKZF2 | IKZF3 |
| IRF4 | ITK | JAK1 | JAK2 | JAK3 | JAZF1 | KAT6A | KIF5B | KLF2 | KMT2A |
| LMO1 | LYN | MALT1 | MAML2 | MAST1 | MAST2 | MEAF6 | MECOM | MEF2D | MET $ |
| MKL1 | MKL2 | MLLT10 | MLLT4 | MN1 | MSMB | MYB | MYBL1 | MYC | MYH11 |
| MYH9 | NCOA1 | NCOA2 | NCOA3 | NF1 | NF2 | NFKB2 | NOTCH1 | NOTCH2 | NPM1 |
| NR4A3 | NRG1 | NSD1 | NTRK1 | NTRK2 | NTRK3 | NUMBL | NUP214 | NUP98 | NUTM1 |
| PAG1 | PAX3 | PAX5 | PAX7 | PAX8 | PDCD1LG2 | PDGFB | PDGFRA | PDGFRB | PHF1 |
| PIK3CA | PKN1 | PLAG1 | PML | PPARG | PRDM1 | PRDM16 | PRKACA | PRKCA | PRKCB |
| PTCH1 | PTK2B | PVT1 | QKI | RAF1 | RARA | RELA | RET | ROS1 | RSPO2 |
| RSPO3 | RUNX1 | RUNX1T1 | SETD2 | SS18 | SSX1 | SSX2 | SSX4 | STAT6 | SUZ12 |
| SYK | TAF15 | TAL1 | TBL1XR1 | TCF12 | TCF3 | TERT | TET1 | TFE3 | TFEB |
| TFG | THADA | TMPRSS2 | TOP1 | TP63 | TYK2 | USP6 | VGLL2 | WHSC1 | YAP1 |
| YWHAE | ZCCHC7 | ZNF384 | |||||||
References
- Patrichi, A.I.; Gurzu, S. Pathogenetic and molecular classifications of soft tissue and bone tumors: A 2024 update. Pathol. Res. Pract. 2024, 260, 155406. [Google Scholar] [CrossRef] [PubMed]
- Boué, D.R.; Parham, D.M.; Webber, B.; Crist, W.M.; Qualman, S.J. Clinicopathologic study of ectomesenchymomas from intergroup rhabdomyosarcoma study groups III and IV. Pediatr. Dev. Pathol. 2000, 3, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, F.; Tirtei, E.; Divincenzo, F.; Campello, A.; Rubino, C.; Augustoni, E.; Linari, A.; Asaftei, S.D.; Fagioli, F. An integrative morpho-molecular approach in malignant ectomesenchymoma diagnosis: Report of a new paediatric case and a review of the literature. Front. Oncol. 2024, 14, 1320541. [Google Scholar] [CrossRef] [PubMed]
- Milano, G.M.; Orbach, D.; Casanova, M.; Berlanga, P.; Schoot, R.A.; Corradini, N.; Brennan, B.; Ramirez-Villar, G.L.; Hjalgrim, L.L.; van Noesel, M.M.; et al. Malignant ectomesenchymoma in children: The European pediatric Soft tissue sarcoma Study Group experience. Pediatr. Blood Cancer 2023, 70, e30116. [Google Scholar] [PubMed]
- Nael, A.; Siaghani, P.; Wu, W.W.; Nael, K.; Shane, L.; Romansky, S.G. Metastatic Malignant Ectomesenchymoma Initially Presenting as a Pelvic Mass: Report of a Case and Review of Literature. Case Rep. Pediatr. 2014, 2014, 792925. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-C.; Alaggio, R.; Sung, Y.-S.M.; Chen, C.-L.M.; Zhang, L.; Kao, Y.-C.; Agaram, N.P.; Wexler, L.H.; Antonescu, C.R. Frequent HRAS mutations in malignant ectomesenchymoma. Am. J. Surg. Pathol. 2016, 40, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.B.; Chou, P.M.; George, D.; Jennings, L.J.; Arva, N.C. Malignant Ectomesenchymoma: Series Analysis of a His-tologically and Genetically Heterogeneous Tumor. Int. J. Surg. Pathol. 2018, 26, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Dantonello, T.M.; Leuschner, I.; Vokuhl, C.; Gfroerer, S.; Schuck, A.; Kube, S.; Nathrath, M.; Bernbeck, B.; Kaatsch, P.; Pal, N.; et al. Malignant ectomesenchymoma in children and adolescents: Report from the Cooperative Weichteil-sarkom Studiengruppe (CWS). Pediatr. Blood Cancer 2013, 60, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Altenburger, D.L.; Wagner, A.S.; Eslin, D.E.; Pearl, G.S.; Pattisapu, J.V. A rare case of malignant pediatric ectomesenchymoma arising from the falx cerebri. J. Neurosurg. Pediatr. 2011, 7, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.C.; Abraham, J.A. Upper Extremity Considerations for Oncologic Surgery. Orthop. Clin. N. Am. 2014, 45, 541–564. [Google Scholar] [CrossRef]
- University of Kentucky. Project Inherited Cancer Risk Panel. 2024. Available online: https://ukhealthcare.uky.edu/kentucky-childrens-hospital/services/hematology-oncology/cancer-predisposition-clinic/project-inherited-cancer-risk (accessed on 21 March 2025).
- Cincinnati Children’s. CinCSeq Comprehensive Cancer Panel. 2024. Available online: https://www.testmenu.com/cincinnatichildrens/Tests/1169861 (accessed on 21 March 2025).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanchard, I.S.C.; Bhavsar, R.C.; Olszewski, A.M.; Shelman, N.R.; D’Orazio, J.A.; Bhandary, P.; Sithisarn, T. Congenital Malignant Ectomesenchymoma Presenting as a Neck Mass in a Newborn. Children 2025, 12, 480. https://doi.org/10.3390/children12040480
Blanchard ISC, Bhavsar RC, Olszewski AM, Shelman NR, D’Orazio JA, Bhandary P, Sithisarn T. Congenital Malignant Ectomesenchymoma Presenting as a Neck Mass in a Newborn. Children. 2025; 12(4):480. https://doi.org/10.3390/children12040480
Chicago/Turabian StyleBlanchard, Ianna S. C., Ravi C. Bhavsar, Ashley M. Olszewski, Nathan R. Shelman, John A. D’Orazio, Prasad Bhandary, and Thitinart Sithisarn. 2025. "Congenital Malignant Ectomesenchymoma Presenting as a Neck Mass in a Newborn" Children 12, no. 4: 480. https://doi.org/10.3390/children12040480
APA StyleBlanchard, I. S. C., Bhavsar, R. C., Olszewski, A. M., Shelman, N. R., D’Orazio, J. A., Bhandary, P., & Sithisarn, T. (2025). Congenital Malignant Ectomesenchymoma Presenting as a Neck Mass in a Newborn. Children, 12(4), 480. https://doi.org/10.3390/children12040480