
Syndromic and Monogenic Obesity: New Opportunities Due to Genetic-Based Pharmacological Treatment

 , , and
, , and
Abstract
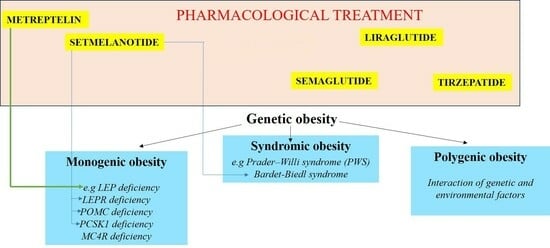
1. Introduction
2. Syndromic Obesity
2.1. Prader–Willi Syndrome (PWS)
2.2. Bardet–Biedl Syndrome (BBS)
2.3. Pseudohypoparathyroidism (PHP) Type 1a
2.4. Alström Syndrome (ALMS)
2.5. 16p11.2 Deletion Syndrome
2.6. WAGR Syndrome
2.7. Smith–Magenis Syndrome (SMS)
2.8. Cohen Syndrome (CS)
2.9. MYT1L-Variants Syndrome
2.10. Börjeson–Forssman–Lehmann Syndrome (BFLS)
2.11. Carpenter Syndrome (CRPT1)
2.12. Down Syndrome (DS)
2.13. Kallmann Syndrome (KS)
3. Monogenic Obesity
3.1. Congenital Leptin Deficiency
3.2. Congenital Leptin Receptor Deficiency
3.3. POMC Deficiency
3.4. PCSK1 Deficiency
3.5. MC4R Deficiency
3.6. SH2B1 Deficiency
3.7. CPE Deficiency
3.8. SRC1 Deficiency
4. Genetic-Based Pharmacological Treatment of Obesity
5. Other Current and Promising Therapeutic Approaches
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Obesity and Overweight. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 9 June 2021).
- Hampl, S.E.; Hassink, S.G.; Skinner, A.C.; Armstrong, S.C.; Barlow, S.E.; Bolling, C.F.; Avila Edwards, K.C.; Eneli, I.; Hamre, R.; Joseph, M.M.; et al. Clinical Practice Guideline for the Evaluation and Treatment of Children and Adolescents with Obesity. Pediatrics 2023, 151, e2022060640. [Google Scholar] [CrossRef]
- Faccioli, N.; Poitou, C.; Clément, K.; Dubern, B. Current Treatments for Patients with Genetic Obesity. J. Clin. Res. Pediatr. Endocrinol. 2023, 15, 108–119. [Google Scholar] [CrossRef]
- Sohn, Y.B. Genetic obesity: An update with emerging therapeutic approaches. Ann. Pediatr. Endocrinol. Metab. 2022, 27, 169–175. [Google Scholar] [CrossRef]
- Koves, I.H.; Roth, C. Genetic and Syndromic Causes of Obesity and Its Management. Indian J. Pediatr. 2018, 85, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Haqq, A.M.; Muehlbauer, M.J.; Newgard, C.B.; Grambow, S.; Freemark, M. The metabolic phenotype of Prader-Willi syndrome (PWS) in childhood: Heightened insulin sensitivity relative to body mass index. J. Clin. Endocrinol. Metab. 2011, 96, E225–E232. [Google Scholar] [CrossRef] [PubMed]
- Bardet, G. On congenital obesity syndrome with polydactyly and retinitis pigmentosa (a contribution to the study of clinical forms of hypophyseal obesity). Obes. Res. 1995, 3, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Biedl, A. A pair of siblings with adiposo-genital dystrophy. Obes. Res. 1995, 3, 404. [Google Scholar] [CrossRef] [PubMed]
- Schachat, A.P.; Maumenee, I.H. Bardet Biedl syndrome and related disorders. Arch. Ophthalmol. 1982, 100, 285–288. [Google Scholar] [CrossRef] [PubMed]
- M’hamdi, O.; Ouertani, I.; Chaabouni-Bouhamed, H. Update on the genetics of bardet-biedl syndrome. Mol. Syndromol. 2014, 5, 51–56. [Google Scholar] [CrossRef]
- Beales, P.L.; Badano, J.L.; Ross, A.J.; Ansley, S.J.; Hoskins, B.E.; Kirsten, B.; Mein, C.A.; Froguel, P.; Scambler, P.; Lewis, R.A.; et al. Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl syndrome. Am. J. Hum. Genet. 2003, 72, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Albright, F.; Burnett, C.H.; Smith, P.H.; Parson, W. Pseudohypoparathyroidism—An example of “Seabright–Bantam Syndrome”. Endocrinology 1942, 30, 922–932. [Google Scholar]
- Jüppner, H. Molecular Definition of Pseudohypoparathyroidism Variants. J. Clin. Endocrinol. Metab. 2021, 106, 1541–1552. [Google Scholar] [CrossRef]
- Levine, M.A. An update on the clinical and molecular characteristics of pseudohypoparathyroidism. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Alstrom, C.H.; Hallgren, B.; Nilsson, L.B.; Asander, H. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: A specific syndrome (not hitherto described) distinct from the Laurence-Moon-Bardet-Biedl syndrome: A clinical, endocrinological and genetic examination based on a large pedigree. Acta Psychiatr. Neurol. Scand. Suppl. 1959, 129, 1–35. [Google Scholar]
- Marshall, J.D.; Maffei, P.; Beck, S.; Barrett, T.G.; Paisey, R.; Naggert, J.K. Clinical utility gene card for: Alstrom Syndrome—Update 2013. Eur. J. Hum. Genet. 2013, 21, 3–4. [Google Scholar] [CrossRef]
- Álvarez-Satta, M.; Castro-Sánchez, S.; Valverde, D. Alström syndrome: Current perspectives. Appl. Clin. Genet. 2015, 8, 171–179. [Google Scholar]
- Szelest, M.; Stefaniak, M.; Ręka, G.; Jaszczuk, I.; Lejman, M. Three case reports of patients indicating the diversity of molecular and clinical features of 16p11.2 microdeletion anomaly. BMC Med. Genom. 2021, 14, 76. [Google Scholar] [CrossRef]
- Finelli, P.; Natacci, F.; Bonati, M.T.; Gottardi, G.; Engelen, J.J.; de Die-Smulders, C.E.; Sala, M.; Giardino, D.; Larizza, L. FISH characterisation of an identical (16)(p11.2p12.2) tandem duplication in two unrelated patients with autistic behaviour. J. Med. Genet. 2004, 41, e90. [Google Scholar] [CrossRef]
- Zufferey, F.; Sherr, E.H.; Beckmann, N.D.; Hanson, E.; Maillard, A.M.; Hippolyte, L.; Mace, A.; Ferrari, C.; Kutalik, J.; Andrieux, J.; et al. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J. Med. Genet. 2012, 49, 660. [Google Scholar] [CrossRef]
- Marakhonov, A.V.; Vasilyeva, T.A.; Voskresenskaya, A.A.; Sukhanova, N.V.; Kadyshev, V.V.; Kutsev, S.I.; Zinchenko, R.A. LMO2 gene deletions significantly worsen the prognosis of Wilms’ tumor development in patients with WAGR syndrome. Hum. Mol. Genet. 2019, 28, 3323–3326. [Google Scholar] [CrossRef]
- Turleau, C.; de Grouchy, J.; Dufier, J.L.; Phuc, L.H.; Schmelck, P.H.; Rappaport, R.; Nihoul-Fekete, C.; Diebold, N. Aniridia, male pseudohermaphroditism, gonadoblastoma, mental retardation, and del 11p13. Hum. Genet. 1981, 57, 300–306. [Google Scholar] [CrossRef]
- Rodriguez-Lopez, R.; Perez, J.M.; Balsera, A.M.; Rodriguez, G.G.; Moreno, T.H.; Garcia de Caceres, M.; Serrano, M.G.; Freijo, F.C.; Ruiz, J.R.; Angueira, F.B.; et al. The modifier effect of the BDNF gene in the phenotype of the WAGRO syndrome. Gene 2013, 516, 285–290. [Google Scholar] [CrossRef]
- Han, J.C.; Liu, Q.R.; Jones, M.; Levinn, R.L.; Menzie, C.M.; Jefferson-George, K.S.; Adler-Wailes, D.C.; Sanford, E.L.; Lacbawan, F.L.; Uhl, G.R.; et al. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N. Engl. J. Med. 2008, 359, 918–927. [Google Scholar] [CrossRef]
- Smith, A.C.M.; Magenis, R.E.; Elsea, S.H. Overview of Smith-Magenis Syndrome. J. Assoc. Genet. Technol. 2005, 31, 163–167. [Google Scholar] [PubMed]
- Slager, R.E.; Newton, T.L.; Vlangos, C.N.; Finucane, B.; Elsea, S.H. Mutations in RAI1 Associated with Smith-Magenis Syndrome. Nat. Genet. 2003, 33, 466–468. [Google Scholar] [CrossRef]
- Cohen, M.M.; Hall, B.D.; Smith, D.W.; Graham, C.B.; Lampert, K.J. A new syndrome with hypotonia, obesity, mental deficiency, and facial, oral, ocular, and limb anomalies. J. Pediatr. 1973, 83, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Balikova, I.; Lehesjoki, A.E.; de Ravel, T.J.; Thienpont, B.; Chandler, K.E.; Clayton-Smith, J.; Traskelin, A.L.; Fryns, J.P.; Vermeesch, J.R. Deletions in the VPS13B (COH1) gene as a cause of Cohen syndrome. Hum. Mutat. 2009, 30, E845–E854. [Google Scholar] [CrossRef] [PubMed]
- Parri, V.; Katzaki, E.; Uliana, V.; Scionti, F.; Tita, R.; Artuso, R.; Longo, I.; Boschloo, R.; Vijzelaar, R.; Selicorni, A.; et al. High frequency of COH1 intragenic deletions and duplications detected by MLPA in patients with Cohen syndrome. Eur. J. Hum. Genet. 2010, 18, 1133–1140. [Google Scholar] [CrossRef]
- Duplomb, L.; Duvet, S.; Picot, D.; Jego, G.; El Chehadeh-Djebbar, S.; Marle, N.; Gigot, N.; Aral, B.; Carmignac, V.; Thevenon, J.; et al. Cohen syndrome is associated with major glycosylation defects. Hum. Mol. Genet. 2014, 23, 2391–2399. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, P.; Bebin, M.; Bruet, S.; Cooper, G.M.; Thompson, M.L.; Duban-Bedu, B.; Gerard, B.; Piton, A.; Suckno, S.; Deshpando, C.; et al. MYT1L mutations cause intellectual disability and variable obesity by dysregulating gene expression and development of the neuroendocrine hypothalamus. PLoS Genet. 2017, 13, e1006957. [Google Scholar] [CrossRef]
- Börjeson, M.; Forssman, H.; Lehmann, O. An X-linked, Recessively Inherited Syndrome Characterized by Grave Mental Deficiency, Epilepsy, and Endocrine Disorder. Acta Medica Scand. 1962, 171, 13–22. [Google Scholar] [CrossRef]
- Jahani-Asl, A.; Cheng, C.; Zhang, C.; Bonni, A. Pathogenesis of Börjeson-Forssman-Lehmann syndrome: Insights from PHF6 function. Neurobiol. Dis. 2016, 96, 227–235. [Google Scholar] [CrossRef]
- Carpenter, G. Acrocephaly, with other congenital malformations-autopsy. Proc. R. Soc. Med. 1909, 2, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.; Seelow, D.; Jehee, F.S.; Perlyn, C.A.; Alonso, L.G.; Bueno, D.F.; Donnai, D.; Josifova, D.; Mathijssen, I.M.; Morton, J.E.; et al. RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am. J. Hum. Genet. 2007, 80, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Havercamp, S.M.; Tasse, M.J.; Navas, P.; Benson, B.A.; Allain, D.; Manickam, K. Exploring the weight and health status of adults with Down syndrome. J. Educ. Train. Stud. 2017, 5, 97–108. [Google Scholar] [CrossRef]
- Artioli, T. Understanding obesity in Down’s syndrome children. J. Obes. Metab. 2017, 1, 1–3. [Google Scholar]
- Stamou, M.I.; Georgopoulos, N.A. Kallmann syndrome: Phenotype and genotype of hypogonadotropic hypogonadism. Metabolism 2018, 86, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Dubern, B.; Clement, K. Leptin and leptin receptor-related monogenic obesity. Biochimie 2012, 94, 2111–2115. [Google Scholar] [CrossRef]
- Funcke, J.B.; von Schnurbein, J.; Lennerz, B.; Lahr, G.; Debatin, K.M.; Fischer-Posovszky, P.; Wabitsch, M. Monogenic forms of childhood obesity due to mutations in the leptin gene. Mol. Cell. Pediatr. 2014, 1, 3. [Google Scholar] [CrossRef]
- Cottrell, E.C.; Mercer, J.G. Leptin receptors. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2012; pp. 3–21. [Google Scholar]
- Farooqi, I.S.; Wangensteen, T.; Collins, S.; Kimber, W.; Matarese, G.; Keogh, J.M.; Lank, E.; Bottomley, B.; Lopez-Fernandez, J.; Ferraz-Amaro, I.; et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N. Engl. J. Med. 2007, 356, 237–247. [Google Scholar] [CrossRef]
- Cetinkaya, S.; Guran, T.; Kurnaz, E.; Keskin, M.; Sagsak, E.; Savas, E.S.; Suntharalingham, J.P.; Buonocore, F.; Achermann, J.C.; Aycan, Z. A patient with proopiomelanocortin deficiency: An increasingly important diagnosis to make. J. Clin. Res. Pediatr. Endocrinol. 2018, 10, 68–73. [Google Scholar] [CrossRef]
- Cone, R.D. Studies on the physiological functions of the melanocortin system. Endocr. Rev. 2006, 27, 736–749. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Volders, K.; Stanhope, R.; Heuschkel, R.; White, A.; Lank, E.; Keogh, J.; O’Rahilly, S.; Creemers, J.W.M. Hyperphagia and early-onset obesity due to a novel homozygous missense mutation in prohormone convertase 1/3. J. Clin. Endocrinol. Metab. 2007, 92, 3369–3373. [Google Scholar] [CrossRef]
- Jackson, R.S.; Creemers, J.W.; Ohagi, S.; Raffin-Sanson, M.L.; Sanders, L.; Montague, C.T.; Hutton, J.C.; O’Rahilly, S. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat. Genet. 1997, 16, 303–306. [Google Scholar] [CrossRef]
- Doche, M.E.; Bochukova, E.G.; Su, H.W.; Pearce, L.R.; Keogh, J.M.; Henning, E.; Cline, J.M.; Saeed, S.; Dale, A.; Cheetman, T.; et al. Human SH2B1 mutations are associated with maladaptive behaviors and obesity. J. Clin. Investig. 2012, 122, 4732–4736. [Google Scholar] [CrossRef]
- Ji, L.; Wu, H.T.; Qin, X.Y.; Lan, R. Dissecting carboxypeptidase E: Properties, functions and pathophysiological roles in disease. Endocr. Connect. 2017, 6, R18–R38. [Google Scholar] [CrossRef]
- Alsters, S.I.; Goldstone, A.P.; Buxton, J.L.; Zekavati, A.; Sosinsky, A.; Yiorkas, S.; Holder, S.; Klaber, R.E.; Bridges, N.; van Haelst, M.M.; et al. Truncating homozygous mutation of carboxypeptidase E (CPE) in a morbidly obese female with type 2 diabetes mellitus, intellectual disability and hypogonadotrophic hypogonadism. PLoS ONE 2015, 10, e0131417. [Google Scholar] [CrossRef] [PubMed]
- Bosch, E.; Hebebrand, M.; Popp, B.; Penger, T.; Behring, B.; Cox, H.; Towner, S.; Kraus, C.; Wilson, W.G.; Khan, S.; et al. BDV syndrome: An emerging syndrome with profound obesity and neurodevelopmental delay resembling Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2021, 106, 3413–3427. [Google Scholar] [CrossRef] [PubMed]
- York, B.; O’Malley, B.W. Steroid receptor coactivator (SRC) family: Masters of systems biology. J. Biol. Chem. 2010, 285, 38743–38750. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, A.E.; Bochukova, E.G.; Marenne, G.; Koegh, J.M.; Atanassova, N.; Bounds, R.; Wheeler, E.; Mistry, V.; Henning, E.; Korner, A.; et al. Rare variant analysis of human and rodent obesity genes in individuals with severe childhood obesity. Sci. Rep. 2017, 7, 4394. [Google Scholar] [CrossRef]
- Cacciottolo, T.M.; Henning, E.; Keogh, J.M.; Bel Lassen, P.; Lawler, K.; Bounds, R.; Ahmed, R.; Perdikari, A.; Mendes de Oliveira, E.; Smith, M.; et al. Obesity Due to Steroid Re-ceptor Coactivator-1 Deficiency Is Associated with Endocrine and Metabolic Abnormalities. J. Clin. Endocrinol. Metab. 2022, 107, e2532–e2544. [Google Scholar] [CrossRef]
- Picard, F.; Gehin, M.; Annicotte, J.; Rocchi, S.; Champy, M.F.; O’Malley, B.W.; Chambon, P.; Auwerx, J. SRC-1 and TIF2 control energy balance between white and brown adipose tissues. Cell 2002, 111, 931–941. [Google Scholar] [CrossRef]
- Yang, Y.; van der Klaauw, A.A.; Zhu, L.; Cacciottolo, T.S.; He, Y.; Stadler, L.K.J.; Wang, C.; Xu, P.; Saito, K.; Hinton, A.; et al. Steroid receptor coactivator-1 modulates the function of Pomc neurons and energy homeostasis. Nat. Commun. 2019, 10, 1718. [Google Scholar] [CrossRef]
- Ogden, C.L.; Carroll, M.D.; Lawman, H.G. Trends in obesity prevalence among children and adolescents in the United States, 1988–1994 through 2013–2014. JAMA 2016, 315, 2292–2299. [Google Scholar] [CrossRef]
- Thaker, V.V. Genetic and epigenetic causes of obesity. Adolesc. Med. State Art Rev. 2017, 28, 379–405. [Google Scholar]
- Farooqi, S. Insights from the genetics of severe childhood obesity. Horm. Res. 2007, 68, 5–7. [Google Scholar] [CrossRef]
- Vollbach, H.; Brandt, S.; Lahr, G. Prevalence and phenotypic characterization of MC4R variants in a large pediatric cohort. Int. J. Obes. 2017, 41, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Cuda, S.; Censani, M. Progress in pediatric obesity: New and advanced therapies. Curr. Opin. Pediatr. 2022, 34, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; Jebb, S.A.; Langmack, G.E.; Lawrence, E.C.; Cheetham, C.H.; Prentice, A.M.; Hughes, I.A.; McCamish, M.A.; O’Rahilly, S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N. Engl. J. Med. 1999, 341, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Wabitsch, M.; Funcke, J.B.; Lennerz, B.; Kuhnle-Krahl, U.; Lahr, G.; Debatin, K.M.; Vatter, P.; Gierschic, P.; Moepps, B.; Fischer-Posovszky, O. Biologically inactive leptin and early- onset extreme obesity. N. Engl. J. Med. 2015, 372, 48–54. [Google Scholar] [CrossRef]
- Kόhnen, P.; Clement, K.; Wiegand, S.; Blankenstein, O.; Gottesdiener, K.; Martini, L.L.; Mai, K.; Blume-Petavi, U.; Gruters, A.; Krude, H. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N. Engl. J. Med. 2016, 375, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Clement, K.; van den Akker, E.; Argente, J.; Bahm, A.; Chung, W.K.; Connors, H.; De Waele, K.; Farooqi, A.; Gonneau-Lejeune, J.; Gordon, G.; et al. Setmelanotide POMC and LEPR Phase 3 Trial investigators. Efficacy and safety of setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency: Single- arm, open- label, multicentre, phase 3 trials. Lancet Diabetes Endocrinol. 2020, 8, 960–970. [Google Scholar] [CrossRef]
- Markham, A. Setmelanotide: First approval. Drugs 2021, 81, 397–403. [Google Scholar] [CrossRef]
- Kühnen, P.; Clement, K. Long-term MC4R agonist treatment in POMC-deficient patients. N. Engl. J. Med. 2022, 387, 852–854. [Google Scholar] [CrossRef]
- Collet, T.H.; Dubern, B.; Mokrosinski, J.; Connors, J.; Keogh, J.M.; de Oliveira, E.M.; Henning, E.; Poitou-Bernet, C.; Oppert, J.M.; Tounian, P.; et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (setmelanotide) in MC4R deficiency. Mol. Metab. 2017, 6, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Haws, R.; Brady, S.; Davis, E.; Fletty, K.; Yuan, G.; Gordon, G.; Stewart, M.; Yanovski, J. Effect of setmelanotide, a melanocortin-4 receptor agonist, on obesity in Bardet–Biedl syndrome. Diabetes Obes. Metab. 2020, 22, 2133–2140. [Google Scholar] [CrossRef] [PubMed]
- Haqq, A.M.; Chung, W.K.; Dollfus, H.; Haws, R.M.; Martos-Moreno, G.A.; Poitou, C.; Yanovski, J.A.; Mittleman, R.S.; Yang, G.; Forsythe, E.; et al. Efficacy and safety of setmelanotide, a melanocortin-4 receptor agonist, in patients with Bardet-Biedl syndrome and Alstrom syndrome: A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial with an open-label period. Lancet Diabetes Endocrinol. 2022, 10, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, E.; Haws, R.M.; Argente, J.; Beales, P.; Martos-Moreno, G.A.; Dollfus, A.; Chirila, C.; Gnanasakthy, A.; Buckley, B.; Mallya, U.; et al. Quality of life improvements following one year of setmelanotide in children and adult patients with Bardet-Biedl syndrome: Phase 3 trial results. Orphanet J. Rare Dis. 2023, 18, 12. [Google Scholar] [CrossRef] [PubMed]
- Dubern, B.; Faccioli, N.; Poitou, C.; Clément, K. Novel therapeutics in rare genetic obesities: A narrative review. Pharmacol. Res. 2023, 191, 106763. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Wang, Q.W.; Yang, X.Y.; Yang, W.; Li, D.R.; Jin, J.Y.; Zhang, H.C.; Zhang, X.F. GLP-1 receptor agonists for the treatment of obesity: Role as a promising approach. Front. Endocrinol. 2023, 14, 1085799. [Google Scholar] [CrossRef]
- Wilding, J.P.H.; Batterham, R.L.; Calanna, S.; Davies, M.; Van Gaal, L.F.; Lingvay, I.; McGowan, B.M.; Rosenstock, J.; Tran, M.T.D.; Wadden, T.A.; et al. Once-weekly semaglutide in adults with overweight or obesity. N. Engl. J. Med. 2021, 384, 989–1002. [Google Scholar] [CrossRef]
- Weghuber, D.; Barrett, T.; Barrientos-Perez, M.; Gies, I.; Hesse, D.; Jeppesen, O.K.; Kelly, A.S.; Mastrandrea, L.D.; Sorrig, R.; Arslanian, S.; et al. Once-weekly semaglutide in adolescents with obesity. N. Engl. J. Med. 2022, 387, 2245–2257. [Google Scholar] [CrossRef]
- Iepsen, E.W.; Zhang, J.; Thomsen, H.S.; Hansen, E.L.; Hollensted, M.; Madsbad, S.; Hansen, T.; Holst, J.J.; Holm, J.C.; Torekov, S.S. Patients with obesity caused by melanocortin-4 receptor mutations can be treated with a glucagon-like peptide-1 receptor agonist. Cell Metab. 2018, 28, 23–32.e3. [Google Scholar] [CrossRef]
- Iepsen, E.W.; Have, C.T.; Veedfald, S.; Madsbad, S.; Holst, J.J.; Grarup, N.; Pedersen, O.; Brandslund, I.; Holm, J.C.; Hansen, T.; et al. GLP-1 receptor agonist treatment in morbid obesity and type 2 diabetes due to pathogenic homozygous melanocortin-4 receptor mutation: A case report. Cell Rep. Med. 2020, 1, 100006. [Google Scholar] [CrossRef]
- Samms, R.J.; Zhang, G.; He, W.; Ilkayeva, O.; Droz, B.A.; Bauer, S.M.; Stutsman, C.; Pirro, V.; Collins, K.W.; Furber, E.C.; et al. Tirzepatide induces a thermogenic-like amino acid signature in brown adipose tissue. Mol. Metab. 2022, 64, 101550. [Google Scholar] [CrossRef] [PubMed]
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 2010, 1, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Boer, G.A.; Hay, D.L.; Tups, A. Obesity pharmacotherapy: Incretin action in the central nervous system. Trends Pharm. Sci. 2023, 44, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Jastreboff, A.M.; Aronne, L.J.; Ahmad, N.N.; Wharton, S.; Connery, L.; Alves, B.; Kiyosue, A.; Zhang, S.; Liu, B.; Bunck, M.C.; et al. Tirzepatide once weekly for the treatment of obesity. N. Engl. J. Med. 2022, 387, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Coskun, T.; Urva, S.; Roell, W.C.; Qu, H.; Loghin, C.; Moyers, J.S.; O’Farrell, L.S.; Briere, D.A.; Sloop, K.W.; Thomas, M.K.; et al. LY3437943, a novel triple glucagon, GIP, and GLP-1 receptor agonist for glycemic control and weight loss: From discovery to clinical proof of concept. Cell Metab. 2022, 34, 1234–1247.e9. [Google Scholar] [CrossRef] [PubMed]
- Tahani, N.; Maffei, P.; Dollfus, H.; Paisey, R.; Valverde, D.; Milan, G.; Han, J.C.; Favaretto, F.; Madathil, S.C.; Dawson, C.; et al. Consensus clinical management guidelines for Alstrom syndrome. Orphanet J. Rare Dis. 2020, 15, 253. [Google Scholar] [CrossRef]
- Feuillan, P.P.; Ng, D.; Han, J.C.; Sapp, J.C.; Wetsch, K.; Spaulding, E.; Zheng, Y.C.; Caruso, R.C.; Brooks, B.P.; Johnston, J.J.; et al. Patients with Bardet-Biedl syndrome have hyperleptinemia suggestive of leptin resistance. J. Clin. Endocrinol. Metab. 2011, 96, E528–E535. [Google Scholar] [CrossRef] [PubMed]
- Pedemonti, B.; Ceccomancini, R.; D’Acunti, A.; Stegmann, J. Effectiveness of a transdisciplinary approach on hyperphagia management among patients with Prader Willi syndrome. Endocrinol. Diabetes Nutr. 2023, 70, 347–351. [Google Scholar] [CrossRef]
- Irizarry, K.A.; Miller, M.; Freemark, M.; Haqq, A.M. Prader Willi Syndrome: Genetics, Metabolomics, Hormonal Function, and New Approaches to Therapy. Adv. Pediatr. 2016, 63, 47–77. [Google Scholar] [CrossRef] [PubMed]
- Ng, N.B.H.; Low, Y.W.; Rajgor, D.D.; Low, J.M.; Lim, Y.Y.; Loke, K.Y.; Lee, Y.S. The effects of glucagon-like peptide (GLP)-1 receptor agonists on weight and glycaemic control in Prader-Willi syndrome: A systematic review. Clin. Endocrinol. 2022, 96, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Diene, G.; Angulo, M.; Hale, P.M.; Jepsen, C.H.; Hofman, P.L.; Hokken-Koelega, A.; Ramesh, C.; Turan, S.; Tauber, M. Liraglutide for Weight Management in Children and Adolescents with Prader-Willi Syndrome and Obesity. J. Clin. Endocrinol. Metab. 2022, 108, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Han, J.C.; Rasmussen, M.C.; Forte, A.R.; Schrage, S.B.; Zafar, S.K.; Haqq, A.M. Management of Monogenic and Syndromic Obesity. Gastroenterol. Clin. N. Am. 2023, 52, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Roof, E.; Deal, C.L.; McCandless, S.E.; Cowan, R.L.; Miller, J.L.; Hamilton, J.K.; Roeder, E.R.; McCormack, S.E.; Roshan Lal, T.R.; Abdul-Latif, H.D.; et al. Intranasal Carbetocin Reduces Hyperphagia, Anxiousness and Distress in Prader-Willi Syndrome: CARE-PWS Phase 3 Trial. J. Clin. Endocrinol. Metab. 2023, 108, 1696–1708. [Google Scholar] [CrossRef]
- Miller, J.L.; Gevers, E.; Bridges, N.; Yanovski, J.A.; Salehi, P.; Obrynba, K.S.; Felner, E.I.; Bird, L.M.; Shoemaker, A.H.; Angulo, M.; et al. Diazoxide Choline Extended-Release Tablet in People with Prader-Willi Syndrome: A Double-Blind, Placebo-Controlled Trial. J. Clin. Endocrinol. Metab. 2023, 108, 1676–1685. [Google Scholar] [CrossRef]
- McCandless, S.E.; Yanovski, J.A.; Miller, J.; Fu, C.; Bird, L.M.; Salehi, P.; Chan, C.L.; Stafford, D.; Abuzzahab, M.J.; Viskochil, D.; et al. Effects of MetAP2 inhibition on hyperphagia and body weight in Prader-Willi syndrome: A randomized, double-blind, placebo-controlled trial. Diabetes Obes. Metab. 2017, 19, 1751–1761. [Google Scholar] [CrossRef]
- Huynh, K.; Klose, M.; Krogsgaard, K.; Drejer, J.; Byberg, S.; Madsbad, S.; Magkos, F.; Aharaz, A.; Edsberg, B.; Tfelt-Hansen, J.; et al. Randomized controlled trial of Tesomet for weight loss in hypothalamic obesity. Eur. J. Endocrinol. 2022, 186, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Nolan, B.J.; Proietto, J.; Sumithran, P. Intensive management of obesity in people with Prader-Willi syndrome. Endocrine 2022, 77, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, G.; Salehi, V.; Browne, A.; Riddle, R.; Hall, E.; Fam, J.; Tichansky, D.; Myers, S. Metabolic and bariatric surgery for obesity in Prader Willi syndrome: Systematic review and meta-analysis. Surg. Obes. Relat. Dis. 2023, 19, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Stratigopoulos, G.; Panigrahi, S.; Sui, L.; Zhang, Y.; Leduc, C.A.; Glover, H.J.; De Rosa, M.C.; Burnett, L.C.; et al. Bardet-Biedl syndrome proteins regulate intracellular signaling and neuronal function in patient-specific iPSC-derived neurons. J. Clin. Investig. 2021, 131, e146287. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Kim, H.; Hamann, C.A.; Rhea, E.M.; Brunger, J.M.; Lippmann, E.S. Nuclear receptor ligand screening in an iPSC-derived in vitro blood-brain barrier model identifies new contributors to leptin transport. Fluids Barriers CNS 2022, 19, 77. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Dani, V.; Dani, C. Human Pluripotent Stem Cells: A Relevant Model to Identify Pathways Governing Thermogenic Adipocyte Generation. Front. Endocrinol. 2020, 10, 932. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Liu, Y.; Qiu, Z.; Kumar, S.; Curran, J.E.; Blangero, J.; Chen, Y.; Lehman, D.M. Molecular Profiling of Human Induced Pluripotent Stem Cell-Derived Hypothalamic Neurones Provides Developmental Insights into Genetic Loci for Body Weight Regulation. J. Neuroendocrinol. 2017, 29. [Google Scholar] [CrossRef]
- Stelzer, Y.; Sagi, I.; Yanuka, O.; Eiges, R.; Benvenisty, N. The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome. Nat. Genet. 2014, 46, 551–557. [Google Scholar] [CrossRef]
- Soeda, S.; Saito, R.; Fujii, A.; Tojo, S.; Tokumura, Y.; Taniura, H. Abnormal DNA methylation in pluripotent stem cells from a patient with Prader-Willi syndrome results in neuronal differentiation defects. Stem Cell Res. 2021, 53, 102351. [Google Scholar] [CrossRef]
- Sledziowska, M.; Winczura, K.; Jones, M.; Almaghrabi, R.; Mischo, H.; Hebenstreit, D.; Garcia, P.; Grzechnik, P. Non-coding RNAs associated with Prader-Willi syndrome regulate transcription of neurodevelopmental genes in human induced pluripotent stem cells. Hum. Mol. Genet. 2023, 32, 608–620. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, X.; Li, J.; Jia, X.; Bai, X.; Zhao, Y.; Cheng, W.; Shu, M.; Zhu, Y.; Jin, S. Leptin gene-targeted editing in ob/ob mouse adipose tissue based on the CRISPR/Cas9 system. J. Genet. Genom. 2021, 48, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, L.; Qu, S.; Zhang, C. CRISPR-mediated gene editing to rescue haploinsufficient obesity syndrome. Protein Cell 2019, 10, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Milbank, E.; Dragano, N.; Vidal-Gómez, X.; Rivas-Limeres, V.; Garrido-Gil, P.; Wertheimer, M.; Recoquillon, S.; Pata, M.P.; Labandeira-Garcia, J.L.; Diéguez, C.; et al. Small extracellular vesicle targeting of hypothalamic AMPKα1 promotes weight loss in leptin receptor deficient mice. Metabolism 2023, 139, 155350. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Drug Class | Dose | Route | Approved for | Most Common Side Effects | |
|---|---|---|---|---|---|
| Metreleptin | Recombinant analog of leptin | 0.03 mg/kg | subcutaneous | LEP deficiency | production of anti-leptin antibodies, increased risk of lymphomas |
| Setmelanotide | MC4R agonist | Max 3 mg | subcutaneous | POMC deficiency PCSK1 deficiency LEPR deficiency BBS | hyperpigmentation, nausea, vomiting, and injection site reactions |
| Semaglutide | GLP-1 receptor agonist | 2.4 mg | subcutaneous | chronic weight management BMI ≥ 27 kg/m2, at least one weight-related ailment or BMI of ≥30 kg/m2. Age limit 12 years. | Nausea, vomiting, diarrhea, constipation |
| Liraglutide | GLP-1 receptor agonist | 3 mg | subcutaneous | chronic weight management among pediatric patients aged ≥ 12 who are obese | nausea, vomiting, diarrhea, dizziness fever |
| Tirzepatide | GIP receptor and GLP-1 receptor agonist | 2.5 mg | subcutaneous | chronic weight management BMI ≥ 27 kg/m2, at least one weight-related ailment or BMI of ≥30 kg/m2. It is only approved for adults. | nausea, diarrhea, vomiting, constipation, abdominal discomfort and pain, injection site reactions |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalinderi, K.; Goula, V.; Sapountzi, E.; Tsinopoulou, V.R.; Fidani, L. Syndromic and Monogenic Obesity: New Opportunities Due to Genetic-Based Pharmacological Treatment. Children 2024, 11, 153. https://doi.org/10.3390/children11020153
Kalinderi K, Goula V, Sapountzi E, Tsinopoulou VR, Fidani L. Syndromic and Monogenic Obesity: New Opportunities Due to Genetic-Based Pharmacological Treatment. Children. 2024; 11(2):153. https://doi.org/10.3390/children11020153
Chicago/Turabian StyleKalinderi, Kallirhoe, Vasiliki Goula, Evdoxia Sapountzi, Vasiliki Rengina Tsinopoulou, and Liana Fidani. 2024. "Syndromic and Monogenic Obesity: New Opportunities Due to Genetic-Based Pharmacological Treatment" Children 11, no. 2: 153. https://doi.org/10.3390/children11020153
APA StyleKalinderi, K., Goula, V., Sapountzi, E., Tsinopoulou, V. R., & Fidani, L. (2024). Syndromic and Monogenic Obesity: New Opportunities Due to Genetic-Based Pharmacological Treatment. Children, 11(2), 153. https://doi.org/10.3390/children11020153