
Oral Rehabilitation as Part of a Multidisciplinary Treatment in a Case Study of Pigmentary Incontinence

 , ,
, , 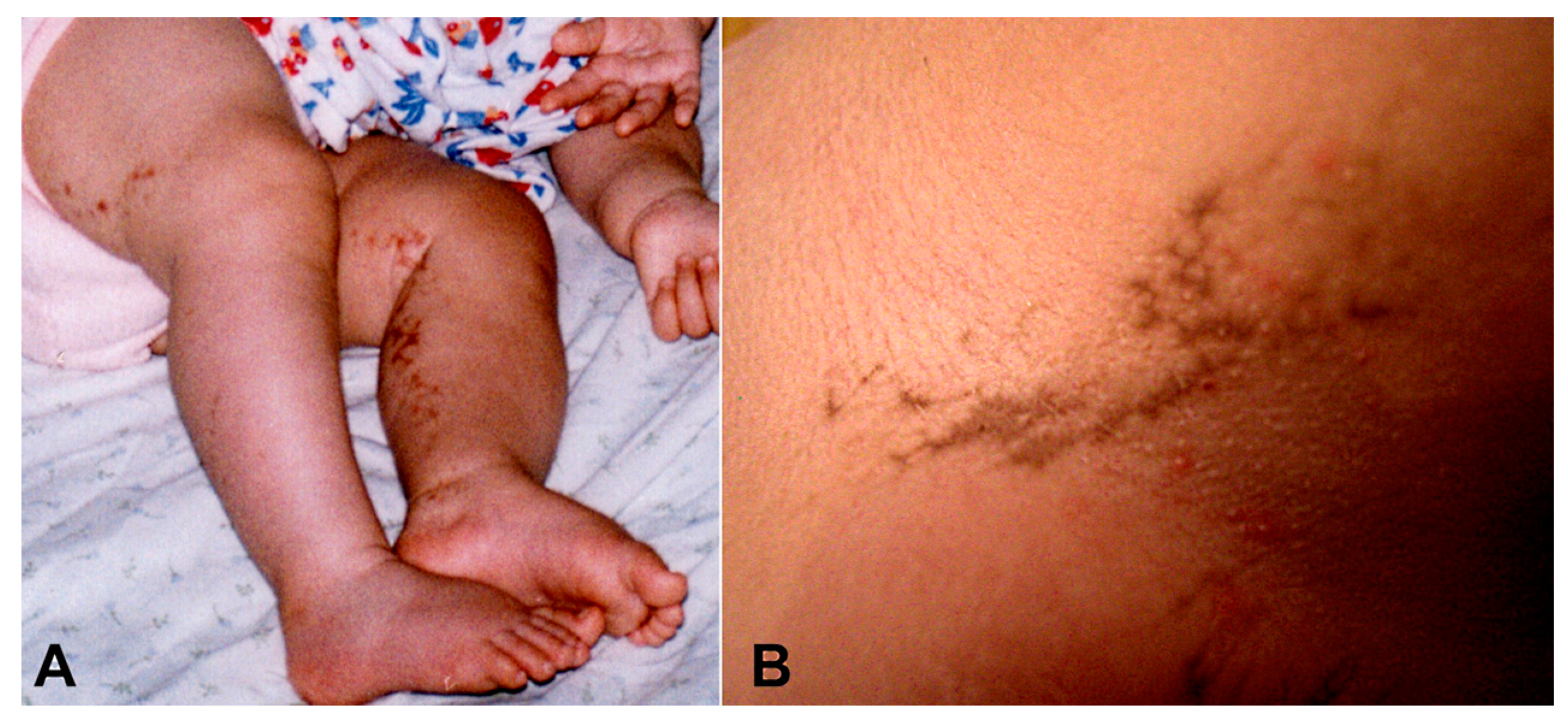{kind=link}
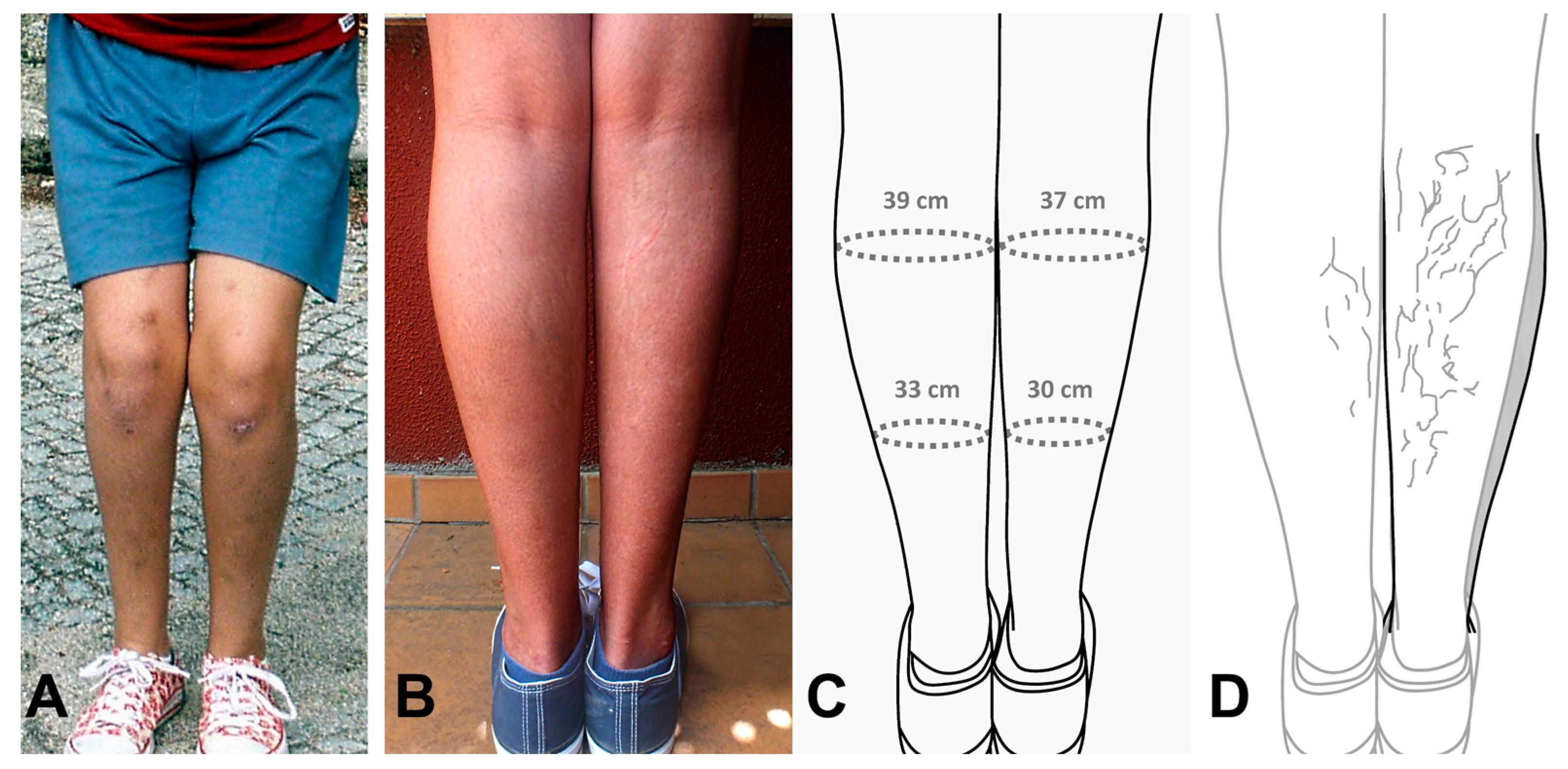{kind=link}
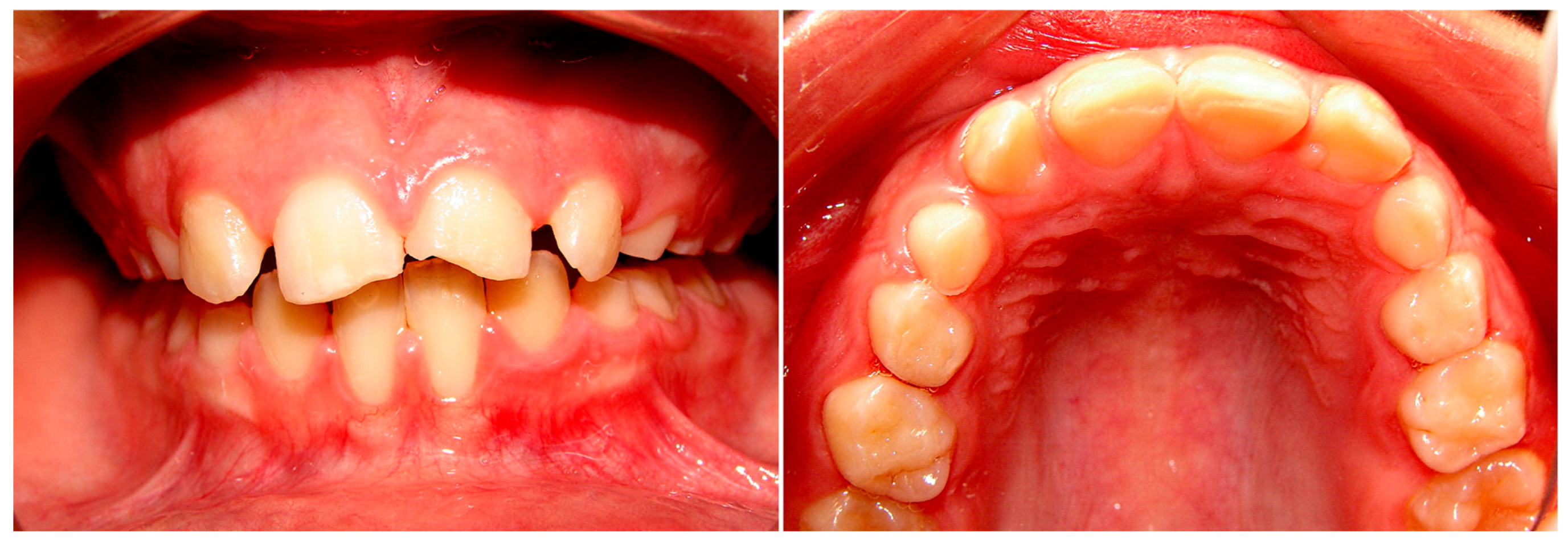{kind=link}
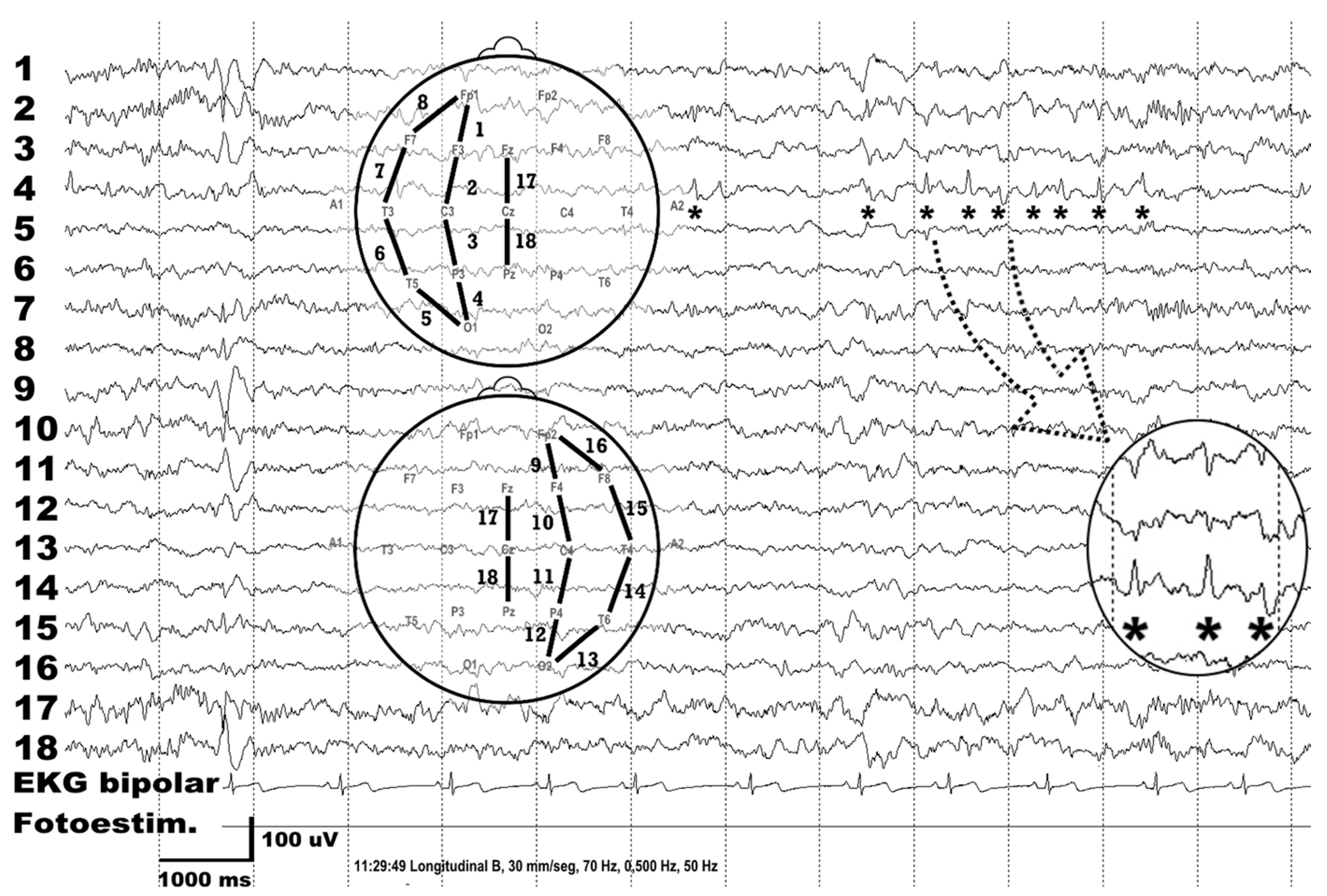{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
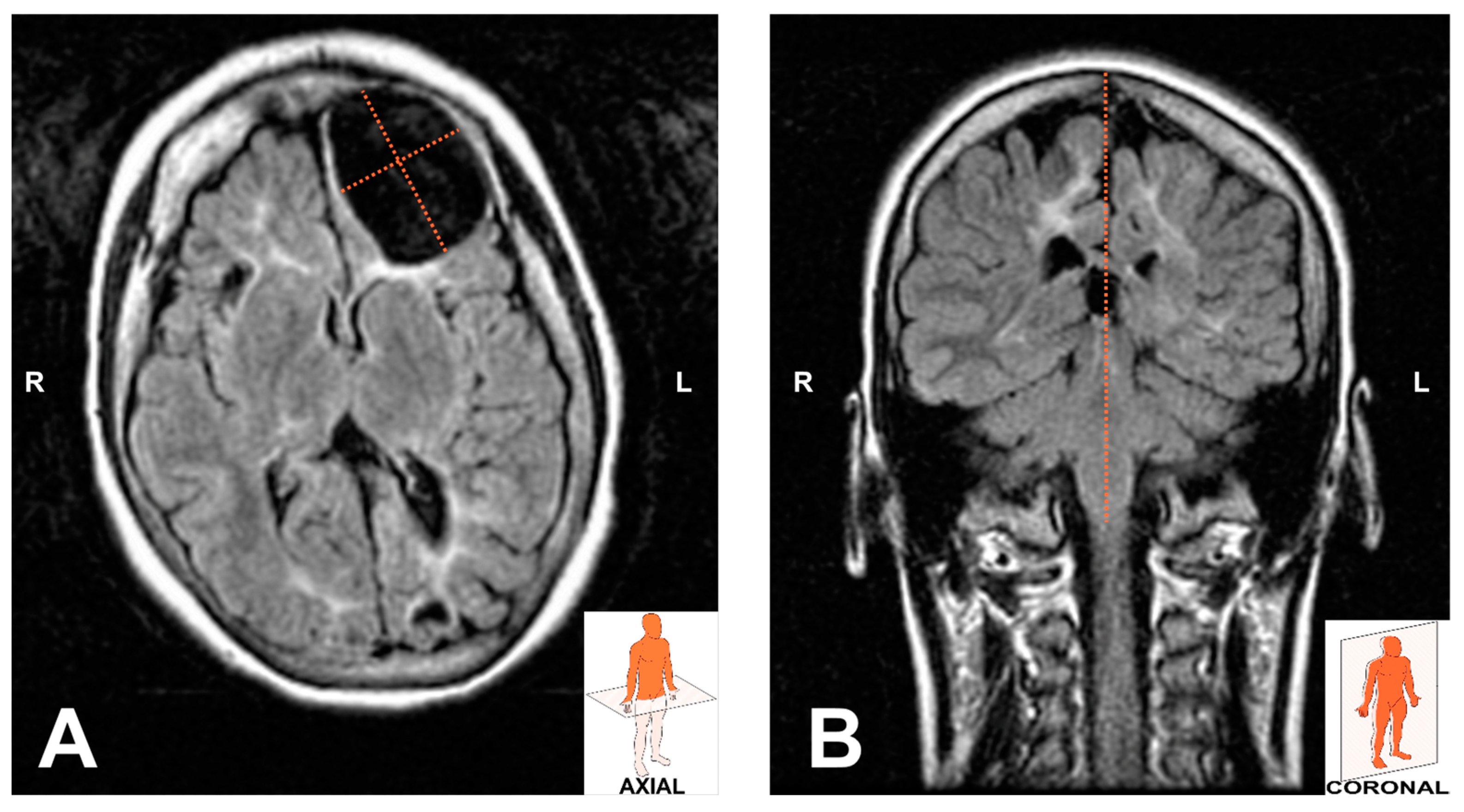
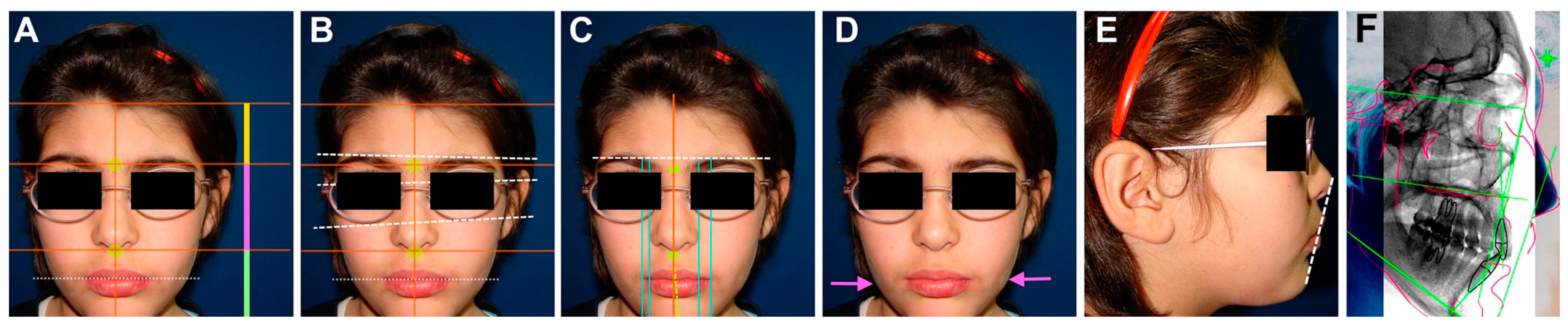
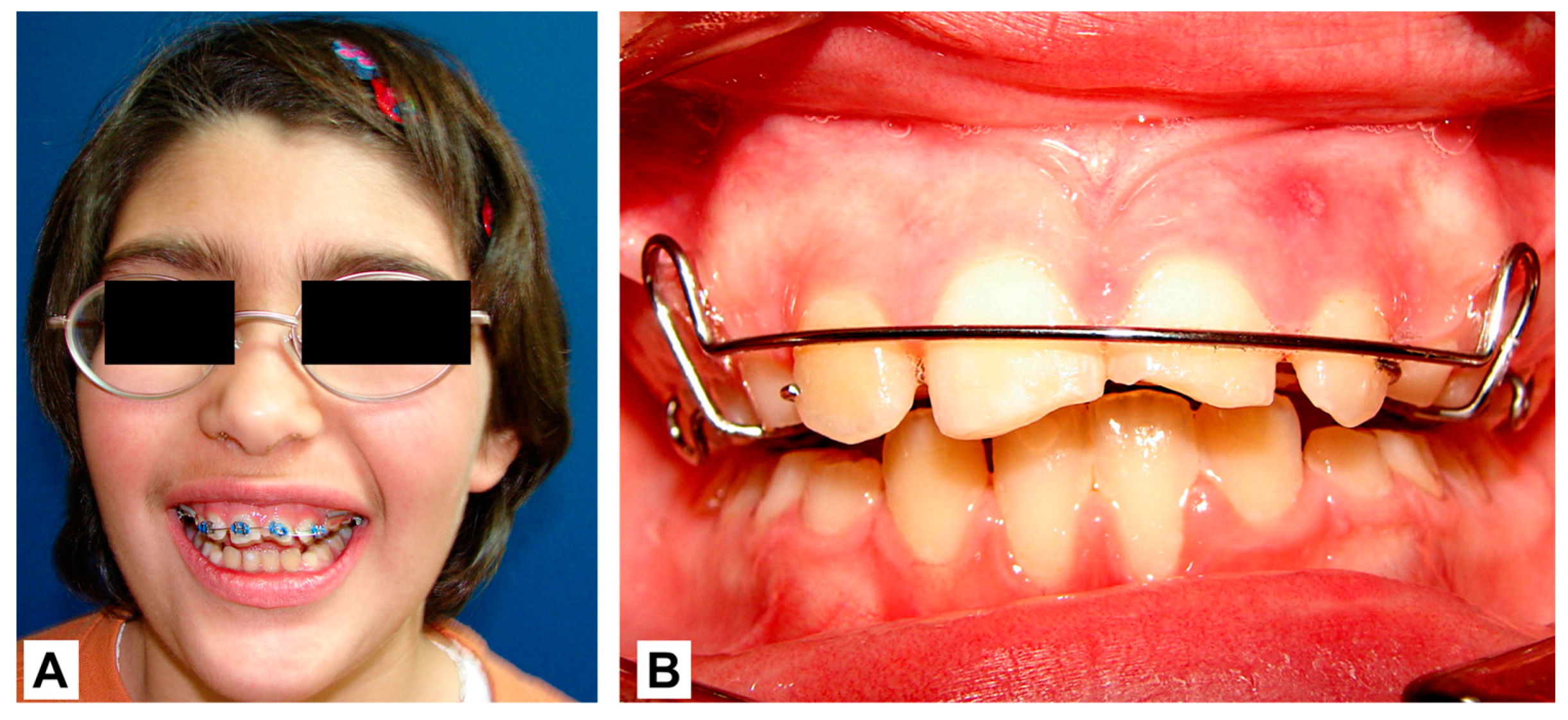
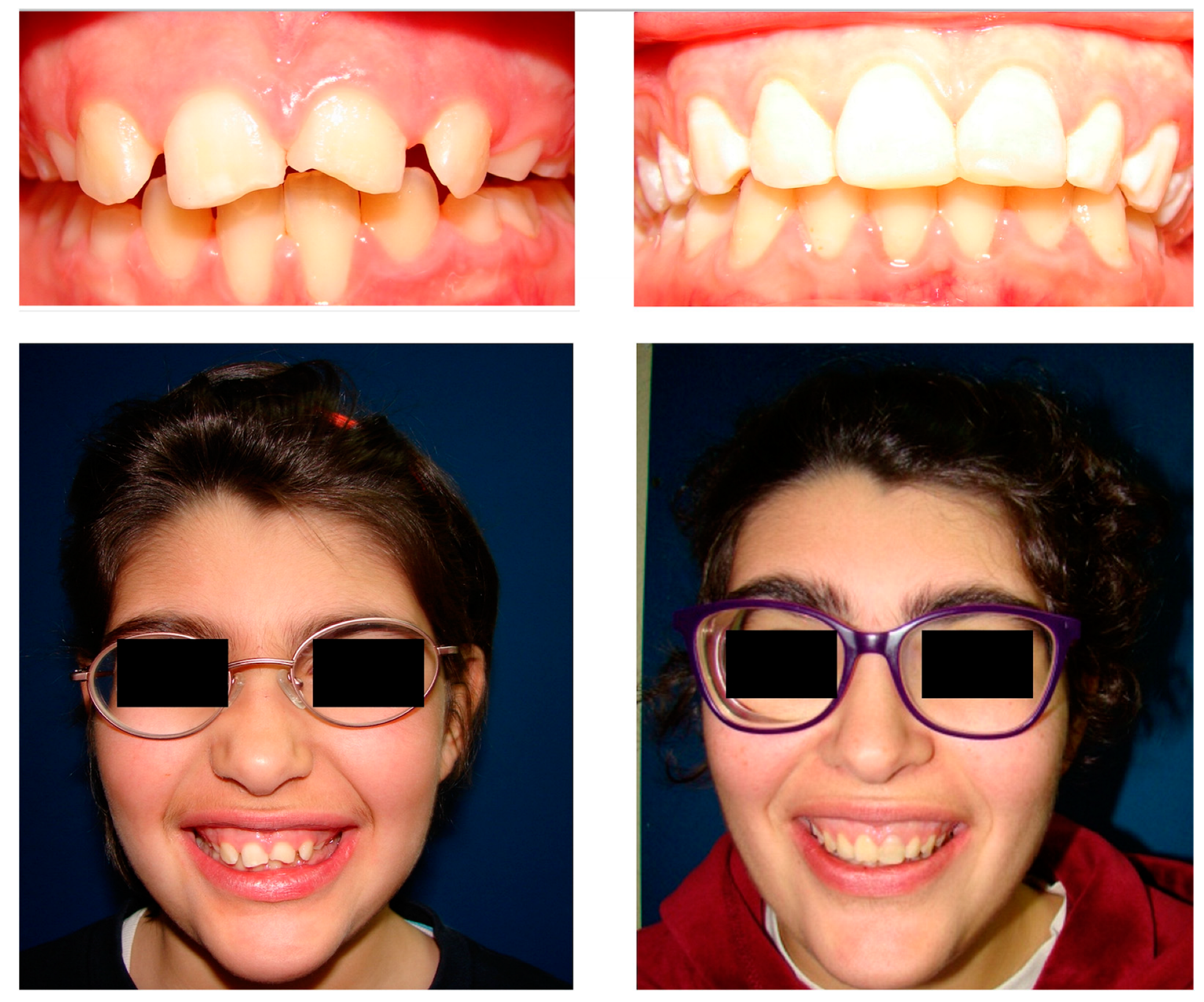
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Masu, S.; Seiji, M. Pigmentary incontinence in fixed drug eruptions. Histologic and electron microscopic findings. J. Am. Acad. Dermatol. 1983, 8, 525–532. [Google Scholar] [CrossRef]
- Narayanan, M.J.; Rangasamy, S.; Narayanan, V. Incontinentia pigmenti (Bloch-Sulzberger syndrome). Handb. Clin. Neurol. 2015, 132, 271–280. [Google Scholar]
- Smahi, A.; Courtois, G.; Vabres, P.; Yamaoka, S.; Heuertz, S.; Munnich, A.; Israël, A.; Heiss, N.S.; Klauck, S.M.; Kioschis, P.; et al. Genomic rearrangement in NEMO impairs NF-κB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature 2000, 405, 466–472. [Google Scholar]
- Buinauskiene, J.; Buinauskaite, E.; Valiukeviciene, S. Incontinentia pigmenti (Bloch-Sulzberger syndrome) in neonates. Medicina 2005, 41, 496–499. [Google Scholar] [PubMed]
- Kenwrick, S.; Woffendin, H.; Jakins, T.; Shuttleworth, S.G.; Mayer, E.; Greenhalgh, L.; Whittaker, J.; Rugolotto, S.; Bardaro, T.; Esposito, T.; et al. Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter syndrome. Am. J. Hum. Genet. 2001, 69, 1210–1217. [Google Scholar]
- Ardelean, D.; Pope, E. Incontinentia pigmenti in boys: A series and review of the literature. Pediatr. Dermatol. 2006, 23, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Franco, L.M.; Goldstein, J.; Prose, N.S.; Selim, M.A.; Tirado, C.A.; Coale, M.M.; McDonald, M.T. Incontinentia pigmenti in a boy with XXY mosaicism detected by fluorescence in situ hybridization. J. Am. Acad. Dermatol. 2006, 55, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Traupe, H.; Vehring, K.H. Unstable pre-mutation may explain mosaic disease expression of incontinentia pigmenti in males. Am. J. Med. Genet. 1994, 49, 397–398. [Google Scholar] [CrossRef] [PubMed]
- Berlin, A.L.; Paller, A.S.; Chan, L.S. Incontinentia pigmenti: A review and update on the molecular basis of pathophysiology. J. Am. Acad. Dermatol. 2002, 47, 169–187. [Google Scholar] [CrossRef]
- Doruk, C.; Bicakci, A.A.; Babacan, H. Orthodontic and orthopedic treatment of a patient with incontinentia pigmenti. Angle Orthod. 2003, 73, 763–778. [Google Scholar] [PubMed]
- Shankar, Y.U.; Fatima, N.; Kumar, M.A.; Prakash, K.S. Bloch Sulzberger syndrome (Incontinentia Pigmenti): A rare case report with dental defects. J. Indian Soc. Pedod. Prev. Dent. 2015, 33, 74–77. [Google Scholar] [PubMed]
- Chen, A.Y.; Chen, K. Dental treatment considerations for a pediatric patient with incontinentia pigmenti (Bloch-Sulzberger syndrome). Eur. J. Dent. 2017, 11, 264–267. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maahs, M.A.; Kiszewski, A.E.; Rosa, R.F.; Maria, F.D.; Prates, F.B.; Zen, P.R. Cephalometric skeletal evaluation of patients with Incontinentia Pigmenti. J. Oral Biol. Craniofac. Res. 2014, 4, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Steiner, C.C. Cephalometrics in Clinical Practice. Angle Orthod. 1959, 29, 8–29. [Google Scholar]
- Landy, S.J.; Donnai, D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J. Med. Genet. 1993, 30, 53–59. [Google Scholar] [CrossRef]
- Minić, S.; Trpinac, D.; Obradović, M. Systematic review of central nervous system anomalies in incontinentia pigmenti. Orphanet J. Rare Dis. 2013, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Carrascosa Romero, M.C.; Ruiz Cano, R.; Medina Monzón, C.; Perez García, L.; Martínez Gutiérrez, A.; Tebar Gil, R. Neonatal convulsions caused by incontinentia pigmenti with left opercular dysgenesia. Rev. Neurol. 2003, 36, 36–39. [Google Scholar] [PubMed]
- Swinney, C.C.; Han, D.P.; Karth, P.A. Incontinentia Pigmenti: A Comprehensive Review and Update. Ophthalmic Surg. Lasers Imaging Retin. 2015, 46, 650–657. [Google Scholar] [CrossRef]
- Holmström, G.; Bergendal, B.; Hallberg, G.; Marcus, S.; Hallén, A.; Dahl, N. Incontinentia pigmenti. En ovanlig sjukdom med många symtom [Incontinentia pigmenti. A rare disease with many symptoms]. Lakartidningen 2002, 99, 1345–1350. [Google Scholar] [PubMed]
- Santa-Maria, F.D.; Mariath, L.M.; Poziomczyk, C.S.; Maahs, M.A.P.; Rosa, R.F.M.; Zen, P.R.G.; Schüller-Faccini, L.; Kiszewski, A.E. Dental anomalies in 14 patients with IP: Clinical and radiological analysis and review. Clin. Oral Investig. 2017, 21, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Reyes, A.; Aznar-Martin, T.; Cabrera-Suarea, E. General and dental characteristics of Bloch-Sulzberger syndrome. Review of literature and presentation of a case report. Med. Oral 2002, 7, 293–297. [Google Scholar]
- Rafatjoo, R.; Taghdisi Kashani, A. Incontinentia Pigmenti; a Rare Multisystem Disorder: Case Report of a 10-Year-Old Girl. J. Dent. 2016, 17, 233–237. [Google Scholar]
- Wolf, D.S.; Golden, W.C.; Hoover-Fong, J.; Applegate, C.; Cohen, B.A.; Germain-Lee, E.L.; Goldberg, M.F.; Crawford, T.O.; Gauda, E.B. High-dose glucocorticoid therapy in the management of seizures in neonatal incontinentia pigmenti: A case report. J. Child Neurol. 2015, 30, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Delaporte, E.; Janin, A.; Blondel, V.; Copin, M.C.; Piette, F.; de Martinville, B.; Bergoend, H. Linear and whorled nevoid hypermelanosis versus incontinentia pigmenti: Is pigmentary incontinence really a distinctive feature? Dermatology. 1996, 192, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Llano-Rivas, I.; Soler-Sánchez, T.; Málaga-Diéguez, I.; Fernández-Toral, J. Incontinencia pigmenti. Cuatro pacientes con diferentes manifestaciones clínicas [Incontinentia pigmenti. Four patients with different clinical manifestations]. An. Pediatr. 2012, 76, 156–160. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cano-Rosás, M.; Vicente-Jiménez, J.d.; Diosdado-Cano, J.M.; Suárez-Quintanilla, D.; González-Sarmiento, R.; Curto, D.; Curto, A. Oral Rehabilitation as Part of a Multidisciplinary Treatment in a Case Study of Pigmentary Incontinence. Children 2023, 10, 1505. https://doi.org/10.3390/children10091505
Cano-Rosás M, Vicente-Jiménez Jd, Diosdado-Cano JM, Suárez-Quintanilla D, González-Sarmiento R, Curto D, Curto A. Oral Rehabilitation as Part of a Multidisciplinary Treatment in a Case Study of Pigmentary Incontinence. Children. 2023; 10(9):1505. https://doi.org/10.3390/children10091505
Chicago/Turabian StyleCano-Rosás, Mónica, Joaquín de Vicente-Jiménez, José María Diosdado-Cano, David Suárez-Quintanilla, Rogelio González-Sarmiento, Daniel Curto, and Adrián Curto. 2023. "Oral Rehabilitation as Part of a Multidisciplinary Treatment in a Case Study of Pigmentary Incontinence" Children 10, no. 9: 1505. https://doi.org/10.3390/children10091505
APA StyleCano-Rosás, M., Vicente-Jiménez, J. d., Diosdado-Cano, J. M., Suárez-Quintanilla, D., González-Sarmiento, R., Curto, D., & Curto, A. (2023). Oral Rehabilitation as Part of a Multidisciplinary Treatment in a Case Study of Pigmentary Incontinence. Children, 10(9), 1505. https://doi.org/10.3390/children10091505