


Cerebral Myelination in a Bronchopulmonary Dysplasia Murine Model


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Animals and Hyperoxia Intervention
2.2. Hematoxylin-Eosin (HE) Staining and Lung Morphological Assessment
2.3. Immunofluorescence of Cerebral Myelination
2.4. Western Blot of MBP and GFAP
2.5. Statistical Analysis
3. Results
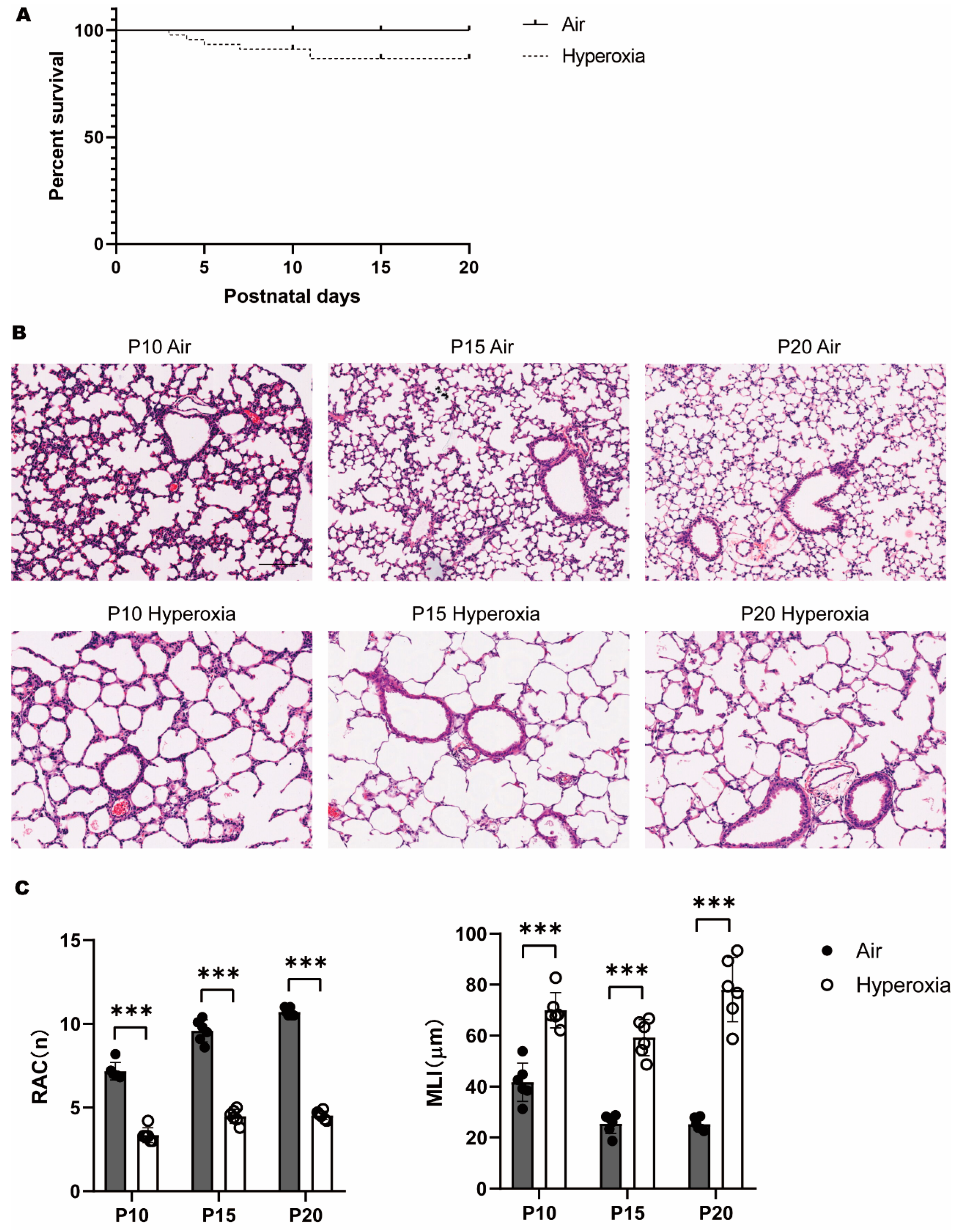
3.1. Persistent Alveolar Arrest during Neonatal Hyperoxia Exposure
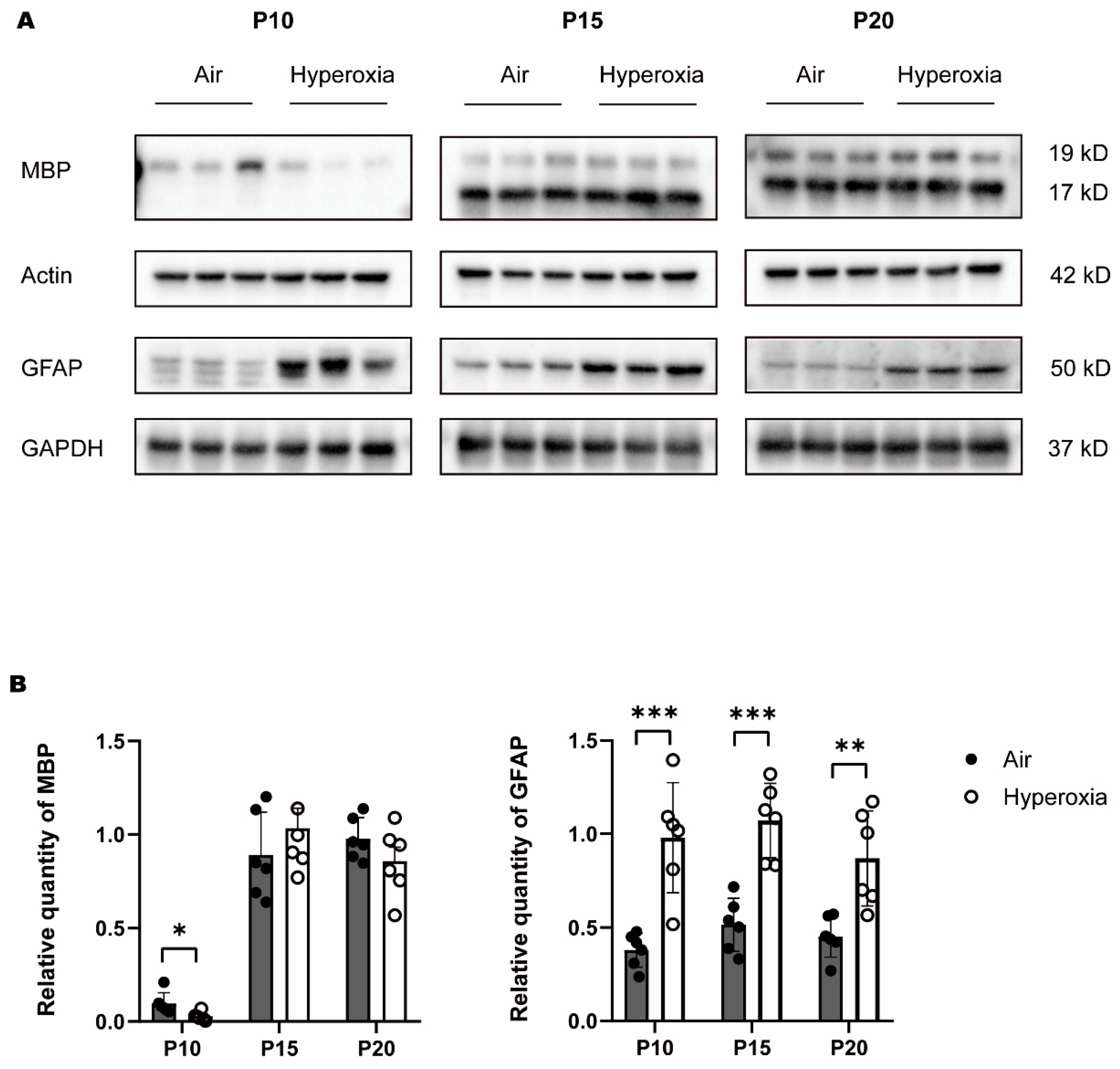
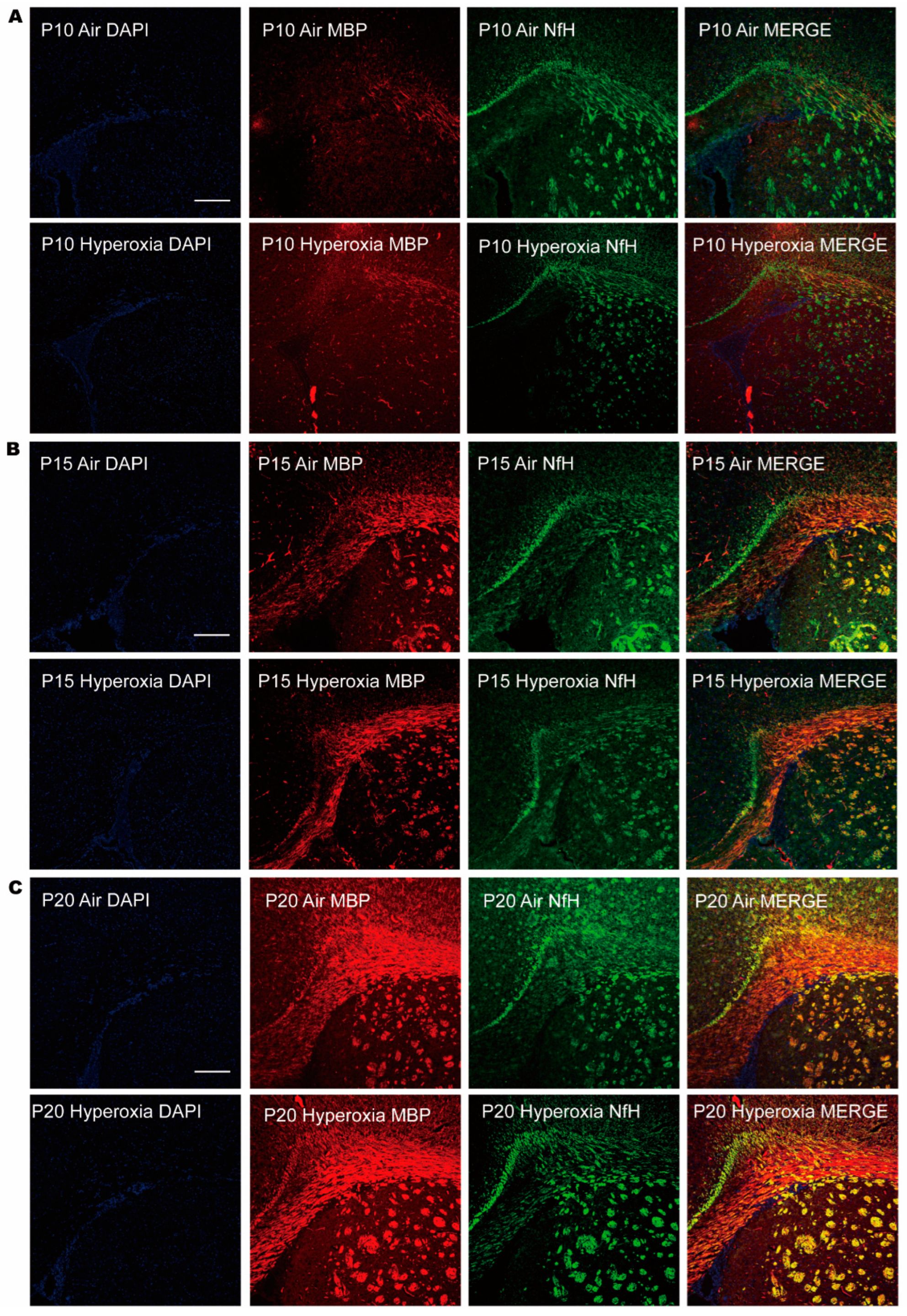
3.2. Transient Myelination Impairment during Neonatal Hyperoxia Exposure
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Silva, L.V.; De Araújo, L.B.; Azevedo, V.M.G.D.O. Assessment of the neuropsychomotor development in the first year of life of premature infants with and without bronchopulmonary dysplasia. Rev. Bras. De Ter. Intensiv. 2018, 30, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.K.; Shin, S.H.; Kim, E.-K.; Kim, H.-S. Developmental outcomes of preterm infants with bronchopulmonary dysplasia-associated pulmonary hypertension at 18–24 months of corrected age. BMC Pediatr. 2019, 19, 26. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.; Van Steenwinckel, J.; Fleiss, B.; Scheuer, T.; Bührer, C.; Faivre, V.; Lemoine, S.; Blugeon, C.; Schwendimann, L.; Csaba, Z.; et al. A unique cerebellar pattern of microglia activation in a mouse model of encephalopathy of prematurity. Glia 2022, 70, 1699–1719. [Google Scholar] [CrossRef] [PubMed]
- Jantzie, L.L.; Winer, J.L.; Maxwell, J.R.; Chan, L.; Robinson, S. Modeling Encephalopathy of Prematurity Using Prenatal Hypoxia-ischemia with Intra-amniotic Lipopolysaccharide in Rats. J. Vis. Exp. 2015, 105, 53196. [Google Scholar]
- Ten, V.S. Mitochondrial dysfunction in alveolar and white matter developmental failure in premature infants. Pediatr. Res. 2016, 81, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Jungner, S.; Kvist, S.V.; Romantsik, O.; Bruschettini, M.; Ekström, C.; Bendix, I.; Herz, J.; Felderhoff-Mueser, U.; Bibic, A.; Zandt, R.I.; et al. White Matter Brain Development after Exposure to Circulating Cell-Free Hemoglobin and Hyperoxia in a Rat Pup Model. Dev. Neurosci. 2020, 41, 1–13. [Google Scholar] [CrossRef]
- Lithopoulos, M.A.; Toussay, X.; Zhong, S.; Xu, L.; Mustafa, S.B.; Ouellette, J.; Freitas-Andrade, M.; Comin, C.H.; Bassam, H.A.; Baker, A.N.; et al. Neonatal hyperoxia in mice triggers long-term cognitive deficits via impairments in cerebrovascular function and neurogenesis. J. Clin. Investig. 2022, 132, e146095. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef]
- Nardiello, C.; Mižíková, I.; Silva, D.M.; Ruiz-Camp, J.; Mayer, K.; Vadász, I.; Herold, S.; Seeger, W.; Morty, R.E. Standardisation of oxygen exposure in the development of mouse models for bronchopulmonary dysplasia. Dis. Model. Mech. 2017, 10, 185–196. [Google Scholar] [CrossRef]
- Cooney, T.P.; Thurlbeck, W.M. The radial alveolar count method of Emery and Mithal: A reappraisal 1--postnatal lung growth. Thorax 1982, 37, 572–579. [Google Scholar] [CrossRef]
- Knudsen, L.; Weibel, E.R.; Gundersen, H.J.G.; Weinstein, F.V.; Ochs, M. Assessment of air space size characteristics by intercept (chord) measurement: An accurate and efficient stereological approach. J. Appl. Physiol. 2010, 108, 412–421. [Google Scholar] [CrossRef]
- Bohnert, S.; Wirth, C.; Schmitz, W.; Trella, S.; Monoranu, C.-M.; Ondruschka, B.; Bohnert, M. Myelin basic protein and neurofilament H in postmortem cerebrospinal fluid as surrogate markers of fatal traumatic brain injury. Int. J. Leg. Med. 2021, 135, 1525–1535. [Google Scholar] [CrossRef]
- Brenner, M.; Messing, A. Regulation of GFAP Expression. ASN Neuro 2021, 13, 1759091420981206. [Google Scholar] [CrossRef] [PubMed]
- Colombo, J.A.; Yáñez, A.; Lipina, S.J. Interlaminar astroglial processes in the cerebral cortex of non human primates: Response to injury. J. Fur Hirnforsch. 1997, 38, 503–512. [Google Scholar]
- Lebel, C.; Deoni, S. The development of brain white matter microstructure. Neuroimage 2018, 182, 207–218. [Google Scholar] [CrossRef]
- Fields, R.D. Change in the Brain’s White Matter. Science 2010, 330, 768–769. [Google Scholar] [CrossRef] [PubMed]
- Buyanova, I.S.; Arsalidou, M. Cerebral White Matter Myelination and Relations to Age, Gender, and Cognition: A Selective Review. Front. Hum. Neurosci. 2021, 15, 662031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Y.; Xu, J.; Kim, B.; Deng, W.; Guo, F. HIFα Regulates Developmental Myelination Independent of Autocrine Wnt Signaling. J. Neurosci. 2021, 41, 251–268. [Google Scholar] [CrossRef] [PubMed]
- Rokem, A.; Takemura, H.; Bock, A.S.; Scherf, K.S.; Behrmann, M.; Wandell, B.A.; Fine, I.; Bridge, H.; Pestilli, F. The visual white matter: The application of diffusion MRI and fiber tractography to vision science. J. Vis. 2017, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, O.; Catani, M.; Johansen-Berg, H.; Clark, C.; Thompson, A. Diffusion-based tractography in neurological disorders: Concepts, applications, and future developments. Lancet Neurol. 2008, 7, 715–727. [Google Scholar] [CrossRef]
- Benear, S.; Ngo, C.T.; Olson, I.R. Dissecting the Fornix in Basic Memory Processes and Neuropsychiatric Disease: A Review. Brain Connect. 2020, 10, 331–354. [Google Scholar] [CrossRef]
- Friederici, A.D. White-matter pathways for speech and language processing. Handb. Clin. Neurol. 2015, 129, 177–186. [Google Scholar] [CrossRef]
- Spaas, J.; van Veggel, L.; Schepers, M.; Tiane, A.; van Horssen, J.; Wilson, D.M.; Moya, P.R.; Piccart, E.; Hellings, N.; Eijnde, B.O.; et al. Oxidative stress and impaired oligodendrocyte precursor cell differentiation in neurological disorders. Cell Mol Life Sci. 2021, 78, 4615–4637. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef]
- Bacmeister, C.M.; Barr, H.J.; McClain, C.R.; Thornton, M.A.; Nettles, D.; Welle, C.G.; Hughes, E.G. Motor learning promotes remyelination via new and surviving oligodendrocytes. Nat. Neurosci. 2020, 23, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Hooft, J.V.; van der Lee, J.H.; Opmeer, B.C.; Aarnoudse-Moens, C.S.H.; Leenders, A.G.E.; Mol, B.W.J.; de Haan, T.R. Predicting developmental outcomes in premature infants by term equivalent MRI: Systematic review and meta-analysis. Syst. Rev. 2015, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Zuchero, J.B.; Fu, M.-M.; Sloan, S.A.; Ibrahim, A.; Olson, A.; Zaremba, A.; Dugas, J.C.; Wienbar, S.; Caprariello, A.V.; Kantor, C.; et al. CNS Myelin Wrapping Is Driven by Actin Disassembly. Dev. Cell 2015, 34, 152–167. [Google Scholar] [CrossRef]
- Brady, S.T.; Witt, A.S.; Kirkpatrick, L.L.; De Waegh, S.M.; Readhead, C.; Tu, P.-H.; Lee, V.M.-Y. Formation of Compact Myelin Is Required for Maturation of the Axonal Cytoskeleton. J. Neurosci. 1999, 19, 7278–7288. [Google Scholar] [CrossRef]
- Meschkat, M.; Steyer, A.M.; Weil, M.-T.; Kusch, K.; Jahn, O.; Piepkorn, L.; Agüi-Gonzalez, P.; Phan, N.T.N.; Ruhwedel, T.; Sadowski, B.; et al. White matter integrity in mice requires continuous myelin synthesis at the inner tongue. Nat. Commun. 2022, 13, 1163. [Google Scholar] [CrossRef]
- Ritter, J.; Schmitz, T.; Chew, L.-J.; Bührer, C.; Möbius, W.; Zonouzi, M.; Gallo, V. Neonatal Hyperoxia Exposure Disrupts Axon–Oligodendrocyte Integrity in the Subcortical White Matter. J. Neurosci. 2013, 33, 8990–9002. [Google Scholar] [CrossRef]
- Chang, J.L.; Bashir, M.; Santiago, C.; Farrow, K.; Fung, C.; Brown, A.S.; Dettman, R.W.; Dizon, M.L. Intrauterine Growth Restriction and Hyperoxia as a Cause of White Matter Injury. Dev. Neurosci. 2018, 40, 344–357. [Google Scholar] [CrossRef]
- Johnson, S.; Marlow, N. Preterm birth and childhood psychiatric disorders. Pediatr. Res. 2011, 69 Pt 2, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Khanbabaei, M.; Hughes, E.; Ellegood, J.; Qiu, L.R.; Yip, R.; Dobry, J.; Murari, K.; Lerch, J.P.; Rho, J.M.; Cheng, N. Precocious myelination in a mouse model of autism. Transl. Psychiatry 2019, 9, 251. [Google Scholar] [CrossRef] [PubMed]
- Bs, J.R.B.; Maire, J.; Riddle, A.; Gong, X.; Nguyen, T.; Nelson, K.; Luo, N.L.; Ren, J.; Struve, J.; Sherman, L.S.; et al. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann. Neurol. 2012, 71, 93–109. [Google Scholar] [CrossRef]
- Allin, M.; Nosarti, C.; Narberhaus, A.; Walshe, M.; Frearson, S.; Kalpakidou, A.; Wyatt, J.; Rifkin, L.; Murray, R. Growth of the Corpus Callosum in Adolescents Born Preterm. Arch. Pediatr. Adolesc. Med. 2007, 161, 1183–1189. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morken, T.S.; Nyman, A.K.G.; Sandvig, I.; Torp, S.H.; Skranes, J.; Goa, P.E.; Brubakk, A.-M.; Widerøe, M. Brain Development after Neonatal Intermittent Hyperoxia-Hypoxia in the Rat Studied by Longitudinal MRI and Immunohistochemistry. PLoS ONE 2013, 8, e84109. [Google Scholar] [CrossRef] [PubMed]
- Kıray, H.; Lindsay, S.L.; Hosseinzadeh, S.; Barnett, S.C. The multifaceted role of astrocytes in regulating myelination. Exp. Neurol. 2016, 283 Pt B, 541–549. [Google Scholar] [CrossRef]
- Li, M.; Li, Z.; Yao, Y.; Jin, W.-N.; Wood, K.; Liu, Q.; Shi, F.-D.; Hao, J. Astrocyte-derived interleukin-15 exacerbates ischemic brain injury via propagation of cellular immunity. Proc. Natl. Acad. Sci. USA 2017, 114, E396–E405. [Google Scholar] [CrossRef]
- Hagberg, H.; Mallard, C.; Ferriero, D.M.; Vannucci, S.J.; Levison, S.W.; Vexler, Z.S.; Gressens, P. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 2015, 11, 192–208. [Google Scholar] [CrossRef]
- Graf, A.E.; Haines, K.M.; Pierson, C.R.; Bolon, B.N.; Houston, R.H.; Velten, M.; Heyob, K.M.; Rogers, L.K. Perinatal inflammation results in decreased oligodendrocyte numbers in adulthood. Life Sci. 2014, 94, 164–171. [Google Scholar] [CrossRef]
- Barnett, S.C.; Linington, C. Myelination. Neurosci. 2013, 19, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Jurga, A.M.; Paleczna, M.; Kadluczka, J.; Kuter, K.Z. Beyond the GFAP-Astrocyte Protein Markers in the Brain. Biomolecules 2021, 11, 1361. [Google Scholar] [CrossRef] [PubMed]
- Ratner, V.; Slinko, S.; Utkina-Sosunova, I.; Starkov, A.; Polin, R.A.; Ten, V.S. Hypoxic Stress Exacerbates Hyperoxia-Induced Lung Injury in a Neonatal Mouse Model of Bronchopulmonary Dysplasia. Neonatology 2009, 95, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Faes, C.; Debevec, T.; Rytz, C.; Millet, G.; Pialoux, V. Preterm birth and oxidative stress: Effects of acute physical exercise and hypoxia physiological responses. Redox Biol. 2018, 17, 315–322. [Google Scholar] [CrossRef]
- Segovia, K.N.; McClure, M.; Moravec, M.; Luo, N.L.; Wan, Y.; Gong, X.; Riddle, A.; Craig, A.; Struve, J.; Sherman, L.S.; et al. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann. Neurol. 2008, 63, 520–530. [Google Scholar] [CrossRef]
- Ratner, V.; Kishkurno, S.V.; Slinko, S.K.; Sosunov, S.A.; Sosunov, A.A.; Polin, R.A.; Ten, V.S. The Contribution of Intermittent Hypoxemia to Late Neurological Handicap in Mice with Hyperoxia-Induced Lung Injury. Neonatology 2007, 92, 50–58. [Google Scholar] [CrossRef]
- Davidson, L.M.; Berkelhamer, S.K. Bronchopulmonary Dysplasia: Chronic Lung Disease of Infancy and Long-Term Pulmonary Outcomes. J. Clin. Med. 2017, 6, 4. [Google Scholar] [CrossRef]
- Muehlbacher, T.; Bassler, D.; Bryant, M.B. Evidence for the Management of Bronchopulmonary Dysplasia in Very Preterm Infants. Children 2021, 8, 298. [Google Scholar] [CrossRef]
- Tana, M.; Tirone, C.; Aurilia, C.; Lio, A.; Paladini, A.; Fattore, S.; Esposito, A.; De Tomaso, D.; Vento, G. Respiratory Management of the Preterm Infant: Supporting Evidence-Based Practice at the Bedside. Children 2023, 10, 535. [Google Scholar] [CrossRef]
- Giusto, K.; Wanczyk, H.; Jensen, T.; Finck, C. Hyperoxia-induced bronchopulmonary dysplasia: Better models for better therapies. Dis. Model. Mech. 2021, 14, dmm047753. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Wang, R.; Chen, C. Cerebral Myelination in a Bronchopulmonary Dysplasia Murine Model. Children 2023, 10, 1321. https://doi.org/10.3390/children10081321
Chen W, Wang R, Chen C. Cerebral Myelination in a Bronchopulmonary Dysplasia Murine Model. Children. 2023; 10(8):1321. https://doi.org/10.3390/children10081321
Chicago/Turabian StyleChen, Wenwen, Ran Wang, and Chao Chen. 2023. "Cerebral Myelination in a Bronchopulmonary Dysplasia Murine Model" Children 10, no. 8: 1321. https://doi.org/10.3390/children10081321
APA StyleChen, W., Wang, R., & Chen, C. (2023). Cerebral Myelination in a Bronchopulmonary Dysplasia Murine Model. Children, 10(8), 1321. https://doi.org/10.3390/children10081321