
Ovarian Masses in Children and Adolescents: A Review of the Literature with Emphasis on the Diagnostic Approach

 ,
,  ,
, 
Abstract
1. Introduction
2. Non-Neoplastic Lesions
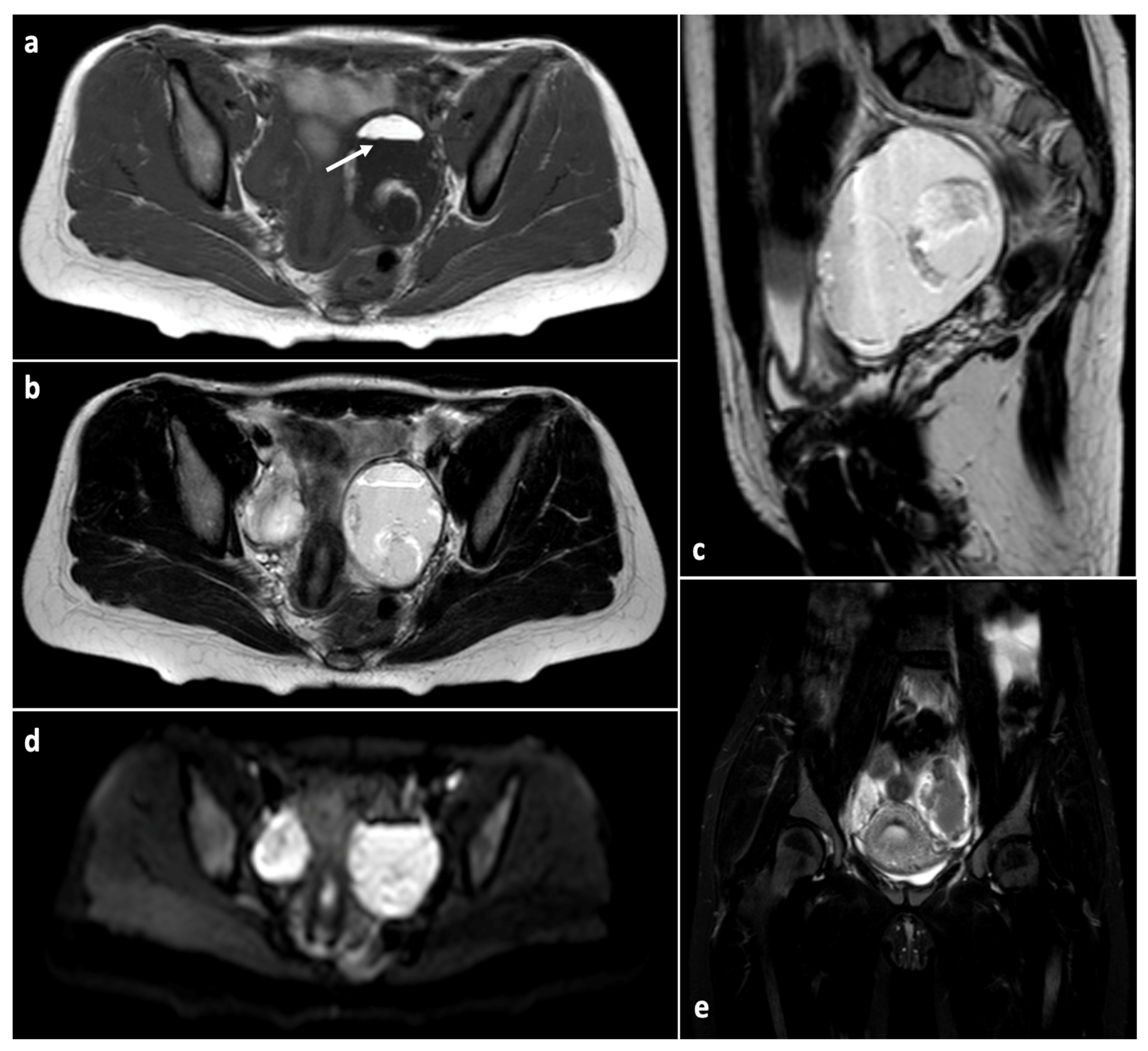
2.1. Functional Cysts
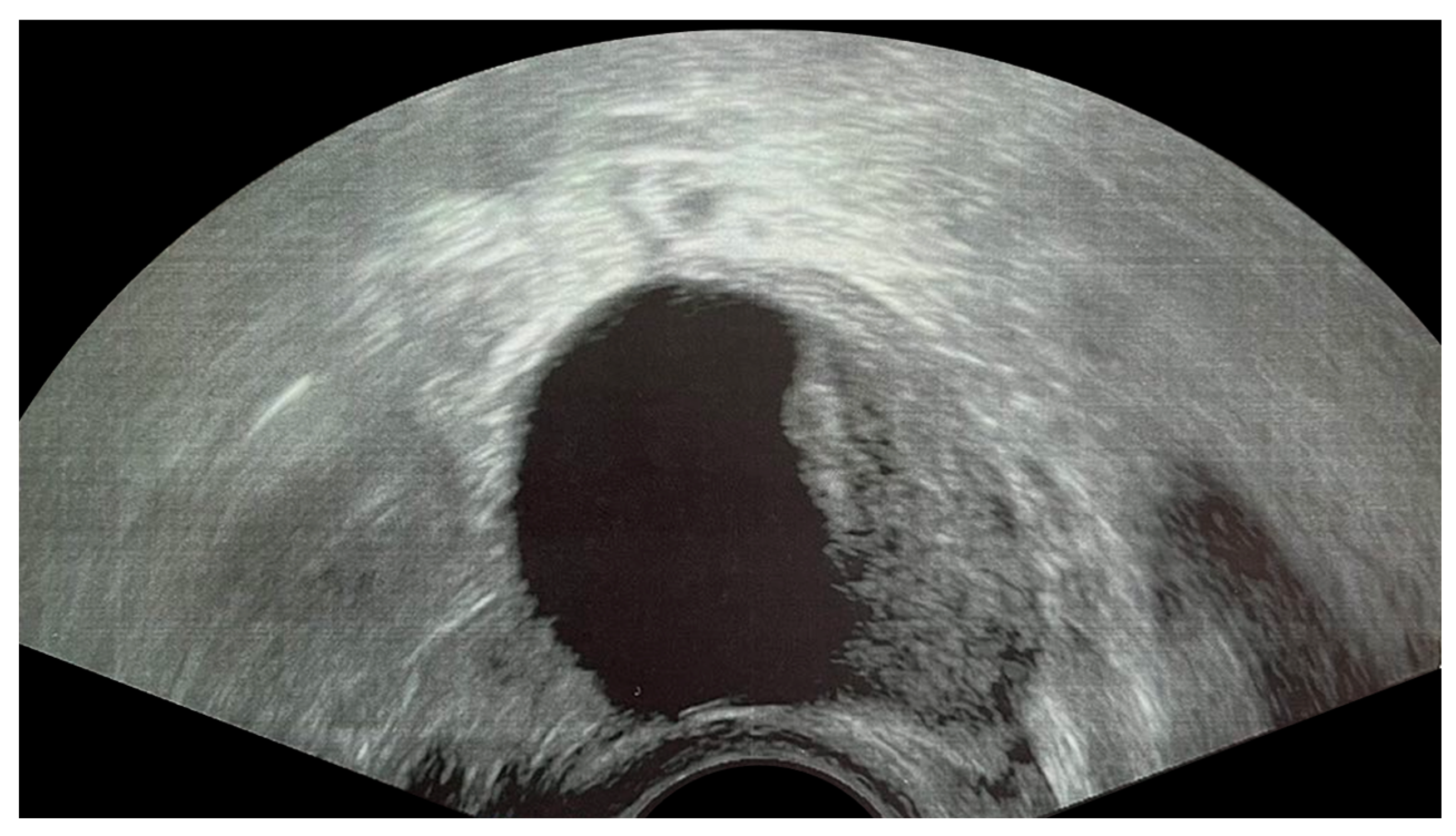
2.2. Endometrioma
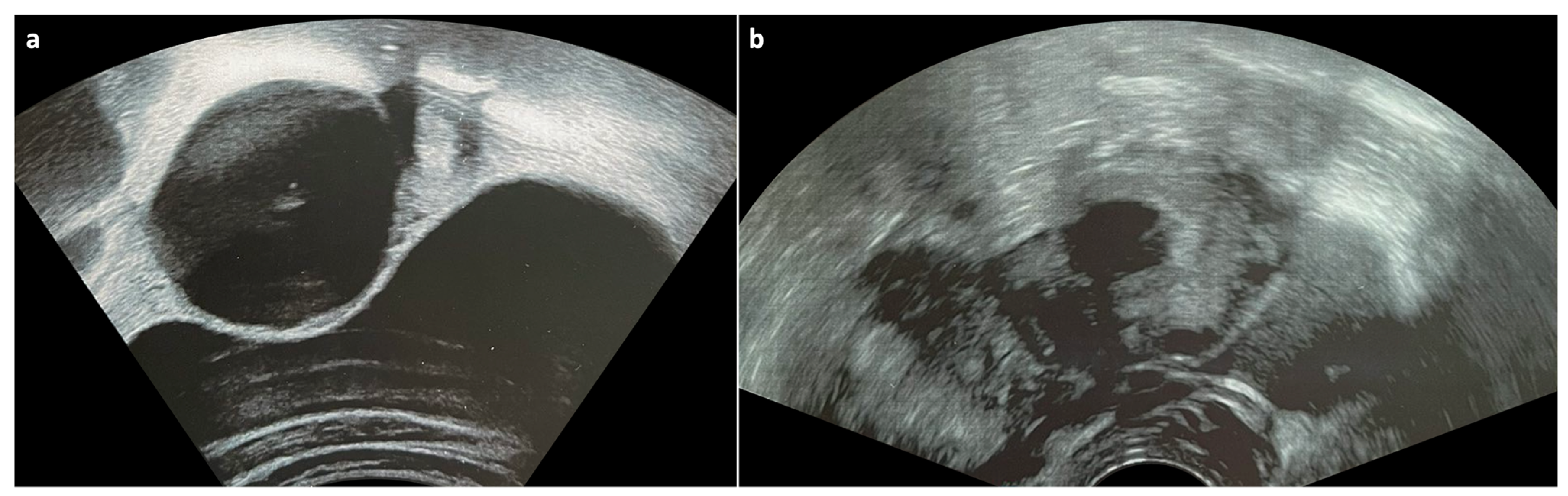
2.3. Ovarian Torsion
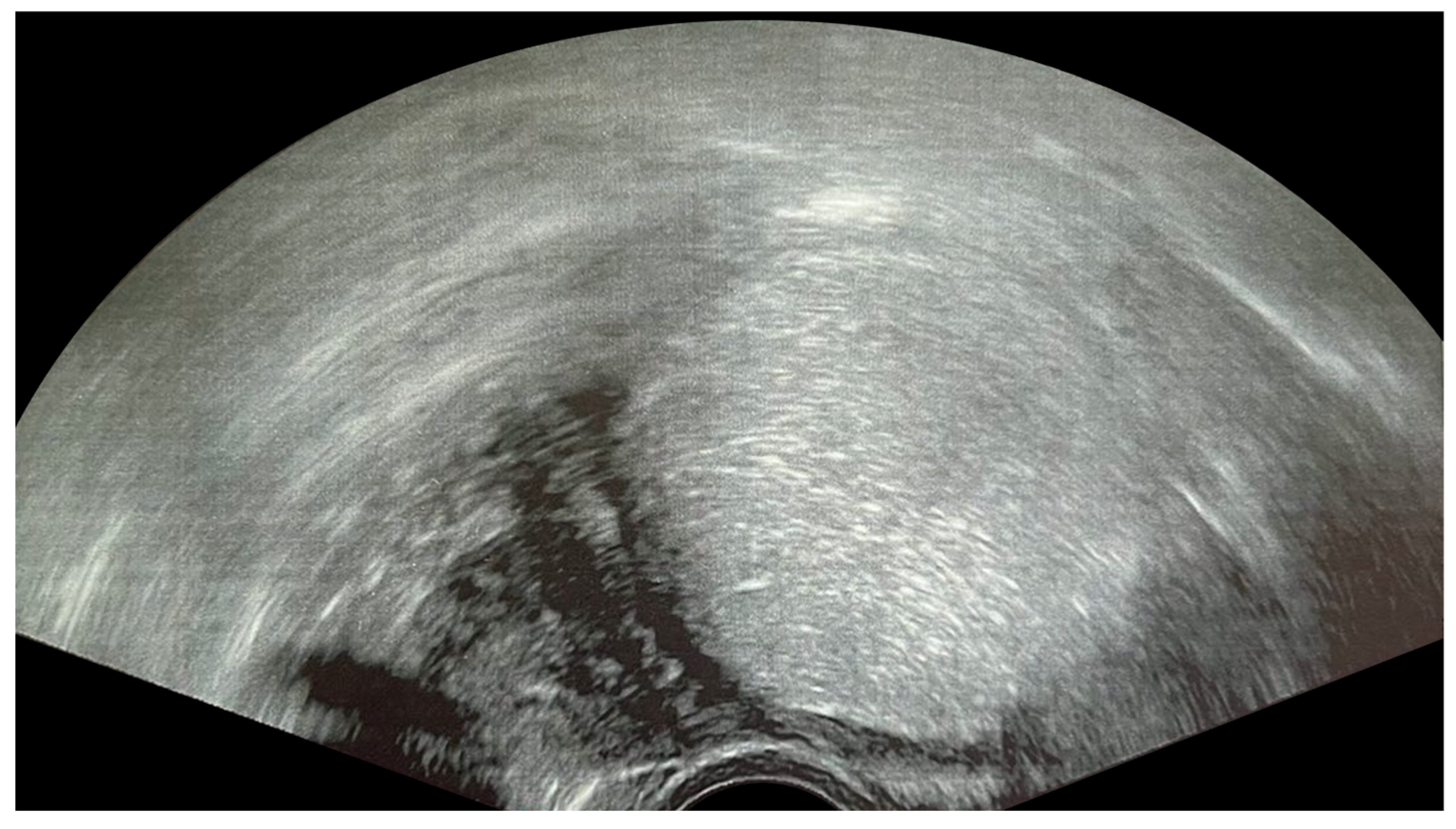
2.4. Tubo-Ovarian Abscess
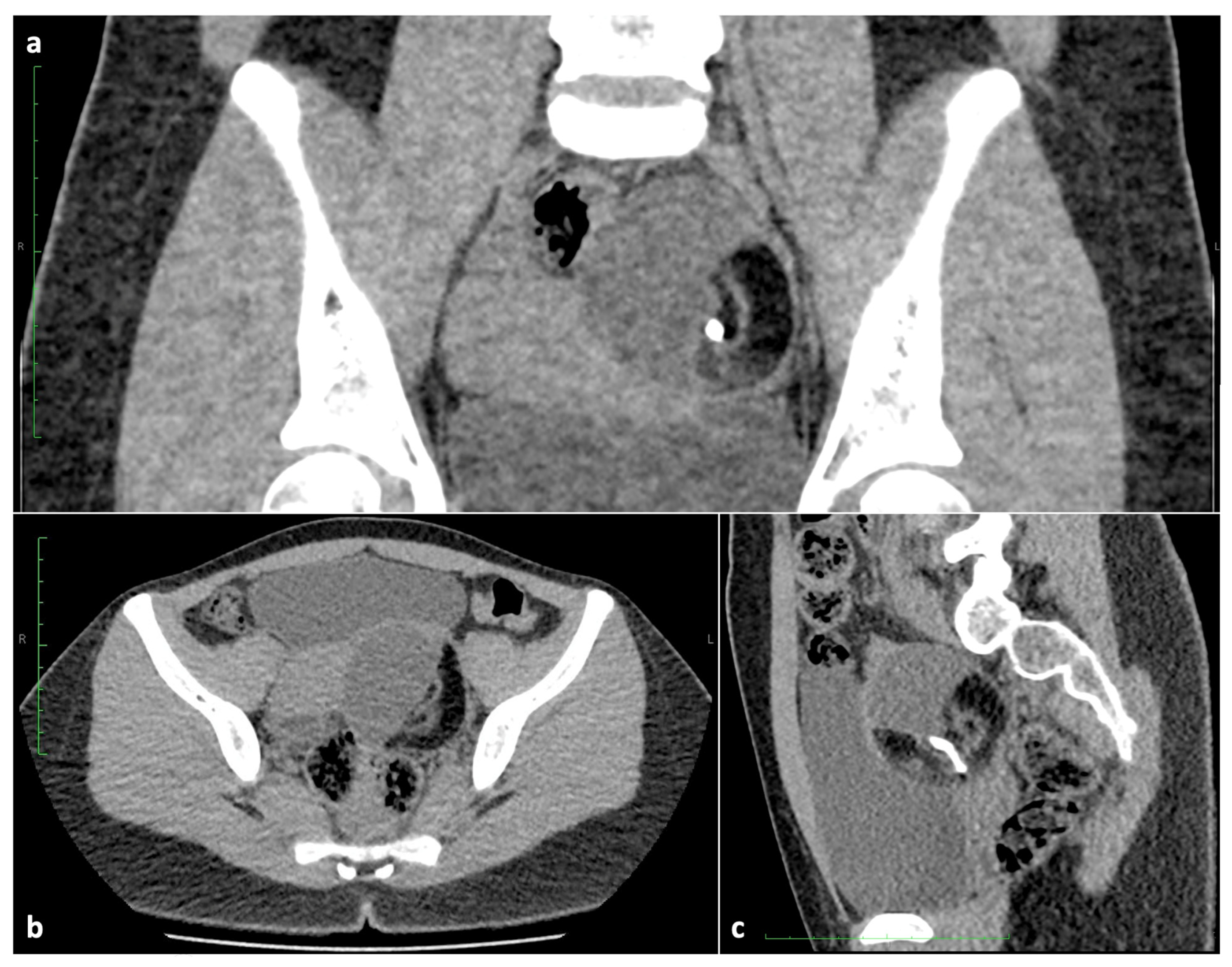
2.5. Lymphangioma
3. Neoplastic Tumors
3.1. Germ Cell Tumors
3.1.1. Mature Teratoma
3.1.2. Immature Teratoma
3.1.3. Dysgerminoma
3.1.4. Yolk Sac Tumor
3.1.5. Embryonal Carcinoma
3.1.6. Non-Gestational Choriocarcinoma
3.1.7. Mixed Germ Cell Tumor
3.1.8. Gonadoblastoma
3.2. Epithelial Tumors
3.3. Sex-Cord Stromal Tumors
3.3.1. Juvenile Granulosa Cell Tumor
3.3.2. Sertoli–Leydig Cell Tumor
3.4. Miscellaneous Tumors
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ciro, E.; Vincenzo, C.; Mariapina, C.; Fulvia, D.C.; Vincenzo, B.; Giorgia, E.; Roberto, C.; Lepore, B.; Castagnetti, M.; Califano, G.; et al. Review of a 25-Year Experience in the Management of Ovarian Masses in Neonates, Children and Adolescents: From Laparoscopy to Robotics and Indocyanine Green Fluorescence Technology. Children 2022, 9, 1219. [Google Scholar] [CrossRef]
- Tarca, E.; Trandafir, L.M.; Cojocaru, E.; Costea, C.F.; Rosu, S.T.; Butnariu, L.I.; Iordache, A.C.; Munteanu, V.; Luca, A.C. Diagnosis Difficulties and Minimally Invasive Treatment for Ovarian Masses in Adolescents. Int. J. Womens Health 2022, 14, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.Z.; Chavhan, G.B. Magnetic Resonance Imaging of Pediatric Adnexal Masses and Mimics. Pediatr. Radiol. 2018, 48, 1291–1306. [Google Scholar] [CrossRef]
- Heo, S.H.; Kim, J.W.; Shin, S.S.; Jeong, S.I.; Lim, H.S.; Choi, Y.D.; Lee, K.H.; Kang, W.D.; Jeong, Y.Y.; Kang, H.K. Review of Ovarian Tumors in Children and Adolescents: Radiologic-Pathologic Correlation. RadioGraphics 2014, 34, 2039–2055. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Lara-Torre, E. Surgical Considerations and Challenges in the Pediatric and Adolescent Gynecologic Patient. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 48, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Sintim-Damoa, A.; Majmudar, A.S.; Cohen, H.L.; Parvey, L.S. Pediatric Ovarian Torsion: Spectrum of Imaging Findings. RadioGraphics 2017, 37, 1892–1908. [Google Scholar] [CrossRef] [PubMed]
- Ngo, A.-V.; Otjen, J.P.; Parisi, M.T.; Ferguson, M.R.; Otto, R.K.; Stanescu, A.L. Pediatric Ovarian Torsion: A Pictorial Review. Pediatr. Radiol. 2015, 45, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Grigore, M.; Murarasu, M.; Himiniuc, L.M.; Toma, B.F.; Duma, O.; Popovici, R. Large Ovarian Tumors in Adolescents, a Systematic Review of Reported Cases, Diagnostic Findings and Surgical Management. Taiwan. J. Obstet. Gynecol. 2021, 60, 602–608. [Google Scholar] [CrossRef] [PubMed]
- AlDakhil, L.; Aljuhaimi, A.; AlKhattabi, M.; Alobaid, S.; Mattar, R.E.; Alobaid, A. Ovarian Neoplasia in Adolescence: A Retrospective Chart Review of Girls with Neoplastic Ovarian Tumors in Saudi Arabia. J. Ovarian Res. 2022, 15, 105. [Google Scholar] [CrossRef]
- Terzic, M.; Rapisarda, A.M.C.; Della Corte, L.; Manchanda, R.; Aimagambetova, G.; Norton, M.; Garzon, S.; Riemma, G.; King, C.R.; Chiofalo, B.; et al. Diagnostic Work-up in Paediatric and Adolescent Patients with Adnexal Masses: An Evidence-Based Approach. J. Obstet. Gynaecol. 2021, 41, 503–515. [Google Scholar] [CrossRef]
- Banlı-Cesur, İ.; Tanrıdan-Okcu, N.; Özçelik, Z. Ovarian Masses in Children and Adolescents: Analysis on 146 Patients. J. Gynecol. Obstet. Hum. Reprod. 2021, 50, 101901. [Google Scholar] [CrossRef] [PubMed]
- Kraikhong, C.; Laorwong, S.; Tongsin, A. Ovarian Tumors in Children: An 11-Year Review. Thai J. Surg. 2019, 40, 1–8. [Google Scholar]
- van Heerden, J.; Tjalma, W.A. The Multidisciplinary Approach to Ovarian Tumours in Children and Adolescents. Eur. J. Obstet. Gynecol. Reprod. Biol. 2019, 243, 103–110. [Google Scholar] [CrossRef]
- User, İ.R.; Karakuş, S.C.; Özokutan, B.H.; Akçaer, V.; Burulday, B.; Ceylana, H. Can Preoperative Findings Help to Interpret Neoplastic and Non-Neoplastic Lesions of Ovary and Affect Surgical Decisions in Children and Adolescents? Arch Argent Pediatr. 2019, 117, 294–400. [Google Scholar] [CrossRef] [PubMed]
- Pommert, L.; Bradley, W. Pediatric Gynecologic Cancers. Curr. Oncol. Rep. 2017, 19, 44. [Google Scholar] [CrossRef]
- Lala, S.V.; Strubel, N. Ovarian Neoplasms of Childhood. Pediatr. Radiol. 2019, 49, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.; Guleria, K.; Suneja, A.; Vaid, N.B.; Rajaram, S.; Wadhwa, N. Adolescent Ovarian Masses: A Retrospective Analysis. J. Obstet. Gynaecol. 2016, 36, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, S.; Fagerholm, R.; Lohi, J.; Taskinen, M. Pediatric Ovarian Neoplastic Tumors: Incidence, Age at Presentation, Tumor Markers and Outcome. Acta Obstet. Gynecol. Scand. 2015, 94, 425–429. [Google Scholar] [CrossRef]
- Tanksale, S.; Bendre, K.; Niyogi, G. Adolescent Ovarian Tumours: A Gynecologist’s Dilemma. Int. J. Reprod. Contracept. Obstet. Gynecol. 2015, 4, 833–836. [Google Scholar] [CrossRef]
- Goudie, C.; Witkowski, L.; Vairy, S.; McCluggage, W.G.; Foulkes, W.D. Paediatric Ovarian Tumours and Their Associated Cancer Susceptibility Syndromes. J. Med. Genet. 2018, 55, 1–10. [Google Scholar] [CrossRef]
- de Kock, L.; Terzic, T.; McCluggage, W.G.; Stewart, C.J.R.; Shaw, P.; Foulkes, W.D.; Clarke, B.A. DICER1 Mutations Are Consistently Present in Moderately and Poorly Differentiated Sertoli-Leydig Cell Tumors. Am. J. Surg. Pathol. 2017, 41, 1178–1187. [Google Scholar] [CrossRef]
- D’Oria, O.; Golia D’Auge, T.; Baiocco, E.; Vincenzoni, C.; Mancini, E.; Bruno, V.; Chiofalo, B.; Mancari, R.; Vizza, R.; Cutillo, G.; et al. The Role of Preoperative Frailty Assessment in Patients Affected by Gynecological Cancer: A Narrative Review. Ital. J. Gynaecol. Obstet. 2022, 34, 76. [Google Scholar] [CrossRef]
- Buzzaccarini, G.; Török, P.; Vitagliano, A.; Petousis, S.; Noventa, M.; Hortu, I.; Giannini, A.; Laganà, A.S. Predictors of Pain Development after Laparoscopic Adnexectomy: A Still Open Challenge. J. Investig. Surg. 2022, 35, 1392–1393. [Google Scholar] [CrossRef]
- Gonzalez, D.O.; Cooper, J.N.; Aldrink, J.H.; Hewitt, G.D.; Fallat, M.E.; Minneci, P.C.; Deans, K.J. Variability in Surgical Management of Benign Ovarian Neoplasms in Children. J. Pediatr. Surg. 2017, 52, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Hermans, A.J.; Kluivers, K.B.; Wijnen, M.H.; Bulten, J.; Massuger, L.F.; Coppus, S.F. Diagnosis and Treatment of Adnexal Masses in Children and Adolescents. Obstet. Gynecol. 2015, 125, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Mobeen, S.; Apostol, R. Ovarian Cyst. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Sayasneh, A.; Ekechi, C.; Ferrara, L.; Kaijser, J.; Stalder, C.; Sur, S.; Timmerman, D.; Bourne, T. The Characteristic Ultrasound Features of Specific Types of Ovarian Pathology (Review). Int. J. Oncol. 2015, 46, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Parasar, P.; Ozcan, P.; Terry, K.L. Endometriosis: Epidemiology, Diagnosis and Clinical Management. Curr. Obs. Gynecol. Rep. 2017, 6, 34–41. [Google Scholar] [CrossRef]
- Guo, S.-W.; Ding, D.; Shen, M.; Liu, X. Dating Endometriotic Ovarian Cysts Based on the Content of Cyst Fluid and Its Potential Clinical Implications. Reprod. Sci. 2015, 22, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Bortoletto, P.; Pollie, M. Management of Ovarian Endometrioma in Asymptomatic Reproductive Age Women. Curr. Obstet. Gynecol. Rep. 2021, 10, 53–60. [Google Scholar] [CrossRef]
- Hoyle, A.T.; Puckett, Y. Endometrioma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Van Holsbeke, C.; Van Calster, B.; Guerriero, S.; Savelli, L.; Paladini, D.; Lissoni, A.A.; Czekierdowski, A.; Fischerova, D.; Zhang, J.; Mestdagh, G.; et al. Endometriomas: Their Ultrasound Characteristics. Ultrasound Obs. Gynecol. 2010, 35, 730–740. [Google Scholar] [CrossRef]
- Thalluri, A.L.; Knox, S.; Nguyen, T. MRI Findings in Deep Infiltrating Endometriosis: A Pictorial Essay. J. Med. Imaging Radiat. Oncol 2017, 61, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Bhatt, S.; Dogra, V.S. Pearls and Pitfalls in Diagnosis of Ovarian Torsion. RadioGraphics 2008, 28, 1355–1368. [Google Scholar] [CrossRef]
- Gross, M.; Blumstein, S.L.; Chow, L.C. Isolated Fallopian Tube Torsion: A Rare Twist on a Common Theme. Am. J. Roentgenol. 2005, 185, 1590–1592. [Google Scholar] [CrossRef]
- Oltmann, S.C.; Fischer, A.; Barber, R.; Huang, R.; Hicks, B.; Garcia, N. Cannot Exclude Torsion—A 15-Year Review. J. Pediatr. Surg. 2009, 44, 1212–1217. [Google Scholar] [CrossRef]
- Cass, D.L. Ovarian Torsion. Semin. Pediatr. Surg. 2005, 14, 86–92. [Google Scholar] [CrossRef]
- Servaes, S.; Zurakowski, D.; Laufer, M.R.; Feins, N.; Chow, J.S. Sonographic Findings of Ovarian Torsion in Children. Pediatr. Radiol. 2007, 37, 446–451. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Tseng, J.-Y.; Yang, C.-Y. Tubo-Ovarian Abscess with Sepsis in a Nonagenarian Woman: A Case Report and Literature Review. BMC Women’s Health 2019, 19, 81. [Google Scholar] [CrossRef]
- Kairys, N.; Roepke, C. Tubo-Ovarian Abscess. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Ha, H.K.; Lim, G.Y.; Cha, E.S.; Lee, H.G.; Ro, H.J.; Kim, H.S.; Kim, H.H.; Joo, S.W.; Jee, M.K. MR Imaging of Tubo-Ovarian Abscess. Acta Radiol. 1995, 36, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Miceli, A.; Stewart, K.M. Lymphangioma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Pani, E.; Martin, A.; Buccoliero, A.; Ghionzoli, M.; Messineo, A. Giant Ovarian Lymphangioma: Case Report and Review of the Literature. Fetal Pediatr. Pathol. 2018, 37, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Akyildiz, E.U.; Peker, D.; Ilvan, S.; Calay, Z.; Cetinaslan, I.; Oruc, N. Lymphangioma of the Ovary: A Case Report and Review of the Literature. J. BUON 2006, 11, 91–93. [Google Scholar]
- Iwasa, T.; Tani, A.; Miyatani, Y.; Bekku, S.; Yamashita, M.; Nakanishi, K.; Fujii, Y.; Ino, H. Lymphangioma of the Ovary Accompanied by Chylous Ascites. J. Obstet. Gynaecol. Res. 2009, 35, 812–815. [Google Scholar] [CrossRef] [PubMed]
- Singer, T.; Filmar, G.; Jormark, S.; Seckin, T.; Divon, M. Rare Case of Ovarian Cystic Lymphangioma. J. Minim. Invasive Gynecol. 2010, 17, 97–99. [Google Scholar] [CrossRef]
- Ferrari, W.; De Angelis, V. Lymphangioma of the ovary. Rev. Bras. Cir. 1953, 25, 329–334. [Google Scholar] [PubMed]
- Bieniasz, A.; Sierant, E. A Case of Lymphangioma Cavernosum of the Ovary. Ginekol. Pol. 1961, 32, 667–669. [Google Scholar] [PubMed]
- Palliez, R.; Delecour, M.; Dupont, A.; Monnier, J.; Begueri, F.; Houcke, M. Ovarian Lymphangioma. A Case. Bull. Fed. Des Soc. Gynecol. Dobstetrique Lang. Fr. 1970, 22, 51–53. [Google Scholar]
- Aristizabal, S.A.; Galindo, J.H.; Davis, J.R.; Boone, M.L. Lymphangiomas Involving the Ovary. Report of a Case and Review of the Literature. Lymphology 1977, 10, 219–223. [Google Scholar]
- Khanna, S.; Mehrotra, M.L.; Basumallick, M.K. Lymphangioma Cavernosum of the Ovary. Int. Surg. 1978, 63, 104–105. [Google Scholar]
- Logani, K.B.; Agarwal, K. Lymphangioma of the Ovary. J. Indian Med. Assoc. 1997, 95, 146–152. [Google Scholar]
- Evans, A.; Lytwyn, A.; Urbach, G.; Chapman, W. Bilateral Lymphangiomas of the Ovary: An Immunohistochemical Characterization and Review of The Literature. Int. J. Gynecol. Pathol. 1999, 18, 87–90. [Google Scholar] [CrossRef]
- Ahluwalia, J.; Girish, V.; Saha, S.; Dey, P. Lymphangioma of the Ovary. Acta Obs. Gynecol. Scand. 2000, 79, 894–895. [Google Scholar] [CrossRef]
- Kearney, C.E.; Hall, G.H.; Purdie, D.W.; Turnbull, L.W. Ovarian Lymphangioma: MRI Appearances. Clin. Radiol. 2001, 56, 685–687. [Google Scholar] [CrossRef] [PubMed]
- Heinig, J.; Beckmann, V.; Bialas, T.; Diallo, R. Lymphangioma of the Ovary after Radiation Due to Wilms’ Tumor in the Childhood. Eur. J. Obstet. Gynecol. Reprod. Biol. 2002, 103, 191–194. [Google Scholar] [CrossRef]
- Park, C.; Lee, J.W.; Kim, S.J.; Kim, J. Sonographic Findings of Prenatal Torsion of Ovarian Lymphangioma. J. Clin. Ultrasound 2005, 33, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Saroha, V.; Singh, M. Lymphangioma of the Ovary. J. Obstet. Gynaecol. 2009, 29, 260–261. [Google Scholar] [CrossRef]
- Jallouli, M.; Trigui, L.; Gouiaa, N.; Gargouri, A.; Mhiri, R. Neonatal Ovarian Lymphangioma. J. Pediatr. Adolesc. Gynecol. 2011, 24, e9–e10. [Google Scholar] [CrossRef] [PubMed]
- Naik, S. Rare Case of Ovarian Cystic Lymphangioma Managed at Laparoscopy. J Gynec Endosc Surg 2011, 2, 97. [Google Scholar] [CrossRef]
- Pillai, S.; O’Brien, D.; Stewart, C.J.R. Bilateral Ovarian Lymphangioma (Lymphangioleiomyoma). Int. J. Gynecol. Pathol. 2013, 32, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Goyal, S.; Sharma, S.; Kotru, M.; Sharma, A. Ovarian Lymphangioma Masquerading as Ectopic Pregnancy: A Clinical Dilemma. J. Obstet. Gynaecol. 2015, 35, 535–536. [Google Scholar] [CrossRef] [PubMed]
- Sinhasan, S.; Nagesha, K. Intra-Abdominal Cystic Lymphangioma in an Adult Female Masquerading Ovarian Tumor. Indian J. Cancer 2015, 52, 380. [Google Scholar] [CrossRef] [PubMed]
- Radhouane, A.; Mayada, S.; Khaled, N. Lymphangioma of the Ovary: Etiology and Management. Eur. J. Obstet. Gynecol. Reprod. Biol. 2016, 203, 342–343. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, R.A.; Vora, P.H.; Deodhar, K.K.; Pisat, S.V.; Ganla, M.K.; Ganla, K.N. Rare Case of Bilateral Ovarian Lymphangioma with Chylous Ascites in Pregnancy with Review of Literature. J. Obs. Gynecol. India 2021, 71, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, K.M.; Shrestha, B. Ovarian Lymphangioma with Mature Cystic Teratoma. J. Nepal. Health Res. Counc. 2019, 17, 128–130. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumors Editorial Board. Female Genital Tumors, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2020; ISBN 978-92-832-4504-9.
- Kanneganti, A.; Bhadiraju, P.; Tong, P.S.Y. Extragonadal Teratomas in Women and Adolescent Girls: A Systematic Review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2021, 262, 134–141. [Google Scholar] [CrossRef]
- O’Neill, K.E.; Cooper, A.R. The Approach to Ovarian Dermoids in Adolescents and Young Women. J. Pediatr. Adolesc. Gynecol. 2011, 24, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Gkrozou, F.; Tsonis, O.; Vatopoulou, A.; Galaziou, G.; Paschopoulos, M. Ovarian Teratomas in Children and Adolescents: Our Own Experience and Review of Literature. Children 2022, 9, 1571. [Google Scholar] [CrossRef] [PubMed]
- Peterson, W.F.; Prevost, E.C.; Edmunds, F.T.; Hundley, J.M.; Morris, F.K. Benign Cystic Teratomas of the Ovary; a Clinico-Statistical Study of 1,007 Cases with a Review of the Literature. Am. J. Obs. Gynecol. 1955, 70, 368–382. [Google Scholar] [CrossRef]
- Comerci, J.T.; Licciardi, F.; Bergh, P.A.; Gregori, C.; Breen, J.L. Mature Cystic Teratoma: A Clinicopathologic Evaluation of 517 Cases and Review of the Literature. Obs. Gynecol. 1994, 84, 22–28. [Google Scholar]
- Ayhan, A.; Aksu, T.; Develioglu, O.; Tuncer, Z.S.; Ayhan, A. Complications and Bilaterality of Mature Ovarian Teratomas (Clinicopathological Evaluation of 286 Cases). Aust. N Z J. Obs. Gynaecol. 1991, 31, 83–85. [Google Scholar] [CrossRef]
- Stern, J.L.; Buscema, J.; Rosenshein, N.B.; Woodruff, J.D. Spontaneous Rupture of Benign Cystic Teratomas. Obs. Gynecol. 1981, 57, 363–366. [Google Scholar]
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rossi, J.E.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-Receptor Encephalitis: Case Series and Analysis of the Effects of Antibodies. Lancet Neurol. 2008, 7, 1091–1098. [Google Scholar] [CrossRef]
- Nii, M.; Kondo, E.; Maki, S.; Kubo, M.; Yoshida, K.; Zhang, L.; Kobayashi, Y.; Tabata, T.; Ikeda, T. Safety and Efficacy of Laparoscopic Oophorocystectomy for Ovarian Dermoid Cyst Associated With Autoimmune Hemolytic Anemia. Gynecol. Minim. Invasive 2018, 7, 27–30. [Google Scholar] [CrossRef]
- Glorieux, I.; Chabbert, V.; Rubie, H.; Baunin, C.; Gaspard, M.; Guitard, J.; Duga, I.; Suc, A.; Puget, C.; Robert, A. Autoimmune Hemolytic Anemia Associated with a Mature Ovarian Teratoma. Arch. Pediatr. Organe Off. Soc. Fr. Pediatr. 1998, 5, 41–44. [Google Scholar] [CrossRef]
- Nokura, K.; Yamamoto, H.; Okawara, Y.; Koga, H.; Osawa, H.; Sakai, K. Reversible Limbic Encephalitis Caused by Ovarian Teratoma. Acta Neurol. Scand. 1997, 95, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Terai, Y.; Terada, S.; Maruoka, H.; Kogata, Y.; Ashihara, K.; Tanaka, Y.; Tanaka, T.; Sasaki, H.; Tsunetoh, S.; et al. A Case of Ovarian Clear Cell Carcinoma Arising from Ovarian Mature Cystic Teratoma. J. Ovarian. Res. 2018, 11, 74. [Google Scholar] [CrossRef]
- Chen, V.W.; Ruiz, B.; Killeen, J.L.; Coté, T.R.; Wu, X.C.; Correa, C.N.; Howe, H.L. Pathology and Classification of Ovarian Tumors. Cancer 2003, 97, 2631–2642. [Google Scholar] [CrossRef] [PubMed]
- Sachs, J.R.; Dyer, R.B. The “Tip of the Iceberg” Sign. Abdom. Imaging 2015, 40, 934–935. [Google Scholar] [CrossRef]
- Kite, L.; Uppal, T. Ultrasound of Ovarian Dermoids—Sonographic Findings of a Dermoid Cyst in a 41-Year-Old Woman with an Elevated Serum HCG. Australas. J. Ultrasound Med. 2011, 14, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.D.; Feldstein, V.A.; Lipson, S.D.; Chen, D.C.; Filly, R.A. Cystic Teratomas of the Ovary: Diagnostic Value of Sonography. Am. J. Roentgenol. 1998, 171, 1061–1065. [Google Scholar] [CrossRef]
- Sahin, H.; Abdullazade, S.; Sanci, M. Mature Cystic Teratoma of the Ovary: A Cutting Edge Overview on Imaging Features. Insights Imaging 2017, 8, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Buy, J.N.; Ghossain, M.A.; Moss, A.A.; Bazot, M.; Doucet, M.; Hugol, D.; Truc, J.B.; Poitout, P.; Ecoiffier, J. Cystic Teratoma of the Ovary: CT Detection. Radiology 1989, 171, 697–701. [Google Scholar] [CrossRef]
- Şahin, H.; Akdoğan, A.I.; Ayaz, D.; Karadeniz, T.; Sancı, M. Utility of the “Floating Ball Sign” in Diagnosis of Ovarian Cystic Teratoma. Turk. J. Obstet. Gynecol. 2019, 16, 118–123. [Google Scholar] [CrossRef]
- Szymon, O.; Kiszka-Wiłkojć, A.; Fryczek, M.; Taczanowska-Niemczuk, A.; Wyrobek, Ł.; Górecki, W. “Floating Ball Sign” in the Diagnostic Imaging of Mature Ovarian Teratomas in Children. Pediatr. Surg. Int. 2023, 39, 215. [Google Scholar] [CrossRef]
- Nakayama, T.; Yoshimitsu, K.; Irie, H.; Aibe, H.; Tajima, T.; Nishie, A.; Asayama, Y.; Matake, K.; Kakihara, D.; Matsuura, S.; et al. Diffusion-Weighted Echo-Planar MR Imaging and ADC Mapping in the Differential Diagnosis of Ovarian Cystic Masses: Usefulness of Detecting Keratinoid Substances in Mature Cystic Teratomas. J. Magn. Reson. Imaging 2005, 22, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Iltar, E.; Ureyen, I.; Toptas, T.; Savas, M.; Çekiç, S.; Uysal, A. A Rare Case: Struma Ovarii in a 14-Year-Old Girl. J. Adolesc. Young Adult Oncol. 2018, 7, 134–136. [Google Scholar] [CrossRef]
- Ezon, I.; Zilbert, N.; Pinkney, L.; Wei, J.-J.; Malik, R.; Nadler, E.P. A Large Struma Ovarii Tumor Removed via Laparoscopy in a 16-Year-Old Adolescent. J. Pediatr. Surg. 2007, 42, e19–e22. [Google Scholar] [CrossRef] [PubMed]
- Savelli, L.; Testa, A.C.; Timmerman, D.; Paladini, D.; Ljungberg, O.; Valentin, L. Imaging of Gynecological Disease (4): Clinical and Ultrasound Characteristics of Struma Ovarii. Ultrasound Obs. Gynecol. 2008, 32, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, M.I.; Sekhri, P.; Turnbull, L.W. Struma Ovarii: Role of Imaging? Insights Imaging 2014, 5, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Park, S.B.; Kim, J.K.; Kim, K.-R.; Cho, K.-S. Imaging Findings of Complications and Unusual Manifestations of Ovarian Teratomas. RadioGraphics 2008, 28, 969–983. [Google Scholar] [CrossRef]
- Pashankar, F.; Hale, J.P.; Dang, H.; Krailo, M.; Brady, W.E.; Rodriguez-Galindo, C.; Nicholson, J.C.; Murray, M.J.; Bilmire, D.F.; Stoneham, S.; et al. Is Adjuvant Chemotherapy Indicated in Ovarian Immature Teratomas? A Combined Data Analysis from the Malignant Germ Cell Tumor International Collaborative: Adjuvant Chemotherapy in Ovarian ITs. Cancer 2016, 122, 230–237. [Google Scholar] [CrossRef]
- Yamaoka, T.; Togashi, K.; Koyama, T.; Fujiwara, T.; Higuchi, T.; Iwasa, Y.; Fujii, S.; Konishi, J. Immature Teratoma of the Ovary: Correlation of MR Imaging and Pathologic Findings. Eur. Radiol. 2003, 13, 313–319. [Google Scholar] [CrossRef]
- Yanai-Inbar, H.; Scully, R.E. Relation of Ovarian Dermoid Cysts and Immature Teratomas: An Analysis of 350 Cases of Immature Teratoma and 10 Cases of Dermoid Cyst with Microscopic Foci of Immature Tissue. Int. J. Gynecol. Pathol. 1987, 6, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Outwater, E.K.; Siegelman, E.S.; Hunt, J.L. Ovarian Teratomas: Tumor Types and Imaging Characteristics. RadioGraphics 2001, 21, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, M.O.S.; Navarro, O.M. Imaging of Ovarian Teratomas in Children: A 9-Year Review. Can. Assoc. Radiol. J. 2010, 61, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Merino, M.J.; Jaffe, G. Age Contrast in Ovarian Pathology. Cancer 2010, 71, 537–544. [Google Scholar] [CrossRef]
- Adhikari, S.; Joti, S.; Chhetri, P.K. Paediatric Ovarian Dysgerminoma: A Case Report. J. Nepal. Med. Assoc. 2022, 60, 985–988. [Google Scholar] [CrossRef]
- Hyseni, N.; Llullaku, S.; Jashari, H.; Zahiti, K.; Hyseni, F.; Kurshumliu, F.; Luci, L.; Muqolli, F.; Hasani, A. Advanced Ovarian Dysgerminoma Infiltrating Both Ovaries and Uterus in a 7-Year-Old Girl. Case Rep. Oncol. Med. 2014, 2014, 1–4. [Google Scholar] [CrossRef]
- Ueland, F. A Perspective on Ovarian Cancer Biomarkers: Past, Present and Yet-To-Come. Diagnostics 2017, 7, 14. [Google Scholar] [CrossRef]
- Kawai, M.; Kano, T.; Kikkawa, F.; Morikawa, Y.; Oguchi, H.; Nakashima, N.; Ishizuka, T.; Kuzuya, K.; Ohta, M.; Arii, Y.; et al. Seven Tumor Markers in Benign and Malignant Germ Cell Tumors of the Ovary. Gynecol. Oncol. 1992, 45, 248–253. [Google Scholar] [CrossRef]
- Shaaban, A.M.; Rezvani, M.; Elsayes, K.M.; Baskin, H.; Mourad, A.; Foster, B.R.; Jarboe, E.A.; Menias, C.O. Ovarian Malignant Germ Cell Tumors: Cellular Classification and Clinical and Imaging Features. RadioGraphics 2014, 34, 777–801. [Google Scholar] [CrossRef]
- Tanaka, Y.O.; Kurosaki, Y.; Nishida, M.; Michishita, N.; Kuramoto, K.; Itai, Y.; Kubo, T. Ovarian Dysgerminoma: MR and CT Appearance. J. Comput. Assist. Tomogr. 1994, 18, 443–448. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, S.B. Ovarian Dysgerminoma: Color Doppler Ultrasonographic Findings and Comparison with CT and MR Imaging Findings. J. Ultrasound Med. 1995, 14, 843–848. [Google Scholar] [CrossRef]
- Duhil de Bénazé, G.; Pacquement, H.; Faure-Conter, C.; Patte, C.; Orbach, D.; Corradini, N.; Berger, C.; Sudour-Bonnange, H.; Vérité, C.; Martelli, H.; et al. Paediatric Dysgerminoma: Results of Three Consecutive French Germ Cell Tumours Clinical Studies (TGM-85/90/95) with Late Effects Study. Eur. J. Cancer 2018, 91, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, M.; Al-Othman, Y.; Hijazi, E.; McCluggage, W.G. A Report of Ovarian Sertoli-Leydig Cell Tumors With Heterologous Intestinal-Type Glands and Alpha Fetoprotein Elevation and Review of the Literature. Int. J. Gynecol. Pathol. 2018, 37, 275–283. [Google Scholar] [CrossRef]
- Li, Y.-K.; Zheng, Y.; Lin, J.-B.; Xu, G.-X.; Cai, A.-Q.; Zhou, X.-G.; Zhang, G.-J. CT Imaging of Ovarian Yolk Sac Tumor with Emphasis on Differential Diagnosis. Sci. Rep. 2015, 5, 11000. [Google Scholar] [CrossRef]
- Choi, H.J.; Moon, M.H.; Kim, S.H.; Cho, J.Y.; Jung, D.C.; Hong, S.R. Yolk Sac Tumor of the Ovary: CT Findings. Abdom. Imaging 2008, 33, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Togashi, K.; Koyama, T.; Ueda, H.; Nakai, A.; Fujii, S.; Yamabe, H.; Konishi, J. Yolk Sac Tumor of the Ovary: Radiologic-Pathologic Correlation in Four Cases. J. Comput. Assist. Tomogr. 2000, 24, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Levitin, A.; Haller, K.D.; Cohen, H.L.; Zinn, D.L.; O’Connor, M.T. Endodermal Sinus Tumor of the Ovary: Imaging Evaluation. Am. J. Roentgenol. 1996, 167, 791–793. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Norris, H.J. Embryonal Carcinoma of the Ovary.A Clinicopathologic Entity Distinct from Endodermal Sinus Tumor Resembling Embryonal Carcinoma of the Adult Testis. Cancer 1976, 38, 2420–2433. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.U.; Lawrence, C.; Fickenscher, K.A.; Shao, L.; Lowe, L.H. Imaging of Pediatric Pelvic Neoplasms. Radiol. Clin. North Am. 2011, 49, 729–748. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guo, C.; Zou, L.; Wang, Y.; Song, X.; Ma, Y.; Liu, A. Clinicopathological Analysis of Non-Gestational Ovarian Choriocarcinoma: Report of Two Cases and Review of the Literature. Oncol. Lett. 2016, 11, 2599–2604. [Google Scholar] [CrossRef] [PubMed]
- Oladipo, A.; Mathew, J.; Oriolowo, A.; Lindsay, I.; Fisher, R.; Seckl, M.; Yiannakis, D. Nongestational Choriocarcinoma Arising from a Primary Ovarian Tumour. BJOG Int. J. Obstet. Gynaecol. 2007, 114, 1298–1300. [Google Scholar] [CrossRef]
- Park, S.H.; Park, A.; Kim, J.Y.; Kwon, J.H.; Koh, S.B. A Case of Non-Gestational Choriocarcinoma Arising in the Ovary of a Postmenopausal Woman. J. Gynecol. Oncol. 2009, 20, 192. [Google Scholar] [CrossRef]
- Scully, R.E. Gonadoblastoma. A Review of 74 Cases. Cancer 1970, 25, 1340–1356. [Google Scholar] [CrossRef]
- Papaioannou, G.; Sebire, N.J.; McHugh, K. Imaging of the Unusual Pediatric “Blastomas”. Cancer Imaging 2009, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Seymour, E.Q.; Hood, J.B.; Underwood, P.B.; Williamson, H.O. Gonadoblastoma: An Ovarian Tumor with Characteristic Pelvic Calcifications. AJR Am. J. Roentgenol. 1976, 127, 1001–1002. [Google Scholar] [CrossRef] [PubMed]
- Roth, L.M.; Cheng, L. Classical Gonadoblastoma: Its Relationship to the “dissecting” Variant and Undifferentiated Gonadal Tissue. Histopathology 2018, 72, 545–555. [Google Scholar] [CrossRef]
- Tsai, J.Y.; Saigo, P.E.; Brown, C.; La Quaglia, M.P. Diagnosis, Pathology, Staging, Treatment, and Outcome of Epithelial Ovarian Neoplasia in Patients Age < 21 Years. Cancer 2001, 91, 2065–2070. [Google Scholar]
- Morowitz, M.; Huff, D.; von Allmen, D. Epithelial Ovarian Tumors in Children: A Retrospective Analysis. J. Pediatr. Surg. 2003, 38, 331–335; discussion 331–335. [Google Scholar] [CrossRef] [PubMed]
- Hazard, F.K.; Longacre, T.A. Ovarian Surface Epithelial Neoplasms in the Pediatric Population: Incidence, Histologic Subtype, and Natural History. Am. J. Surg. Pathol. 2013, 37, 548–553. [Google Scholar] [CrossRef]
- Ghossain, M.A.; Buy, J.N.; Lignères, C.; Bazot, M.; Hassen, K.; Malbec, L.; Hugol, D.; Truc, J.B.; Decroix, Y.; Poitout, P. Epithelial Tumors of the Ovary: Comparison of MR and CT Findings. Radiology 1991, 181, 863–870. [Google Scholar] [CrossRef]
- Jung, S.E.; Lee, J.M.; Rha, S.E.; Byun, J.Y.; Jung, J.I.; Hahn, S.T. CT and MR Imaging of Ovarian Tumors with Emphasis on Differential Diagnosis. Radiographics 2002, 22, 1305–1325. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.O.; Nishida, M.; Kurosaki, Y.; Itai, Y.; Tsunoda, H.; Kubo, T. Differential Diagnosis of Gynaecological “Stained Glass” Tumours on MRI. BJR 1999, 72, 414–420. [Google Scholar] [CrossRef]
- Outwater, E.K.; Huang, A.B.; Dunton, C.J.; Talerman, A.; Capuzzi, D.M. Papillary Projections in Ovarian Neoplasms: Appearance on MRI. J. Magn. Reson. Imaging 1997, 7, 689–695. [Google Scholar] [CrossRef]
- Ma, F.H.; Zhao, S.H.; Qiang, J.W.; Zhang, G.F.; Wang, X.Z.; Wang, L. MRI Appearances of Mucinous Borderline Ovarian Tumors: Pathological Correlation. J. Magn. Reson. Imaging 2014, 40, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Outwater, E.K.; Wagner, B.J.; Mannion, C.; McLarney, J.K.; Kim, B. Sex Cord-Stromal and Steroid Cell Tumors of the Ovary. RadioGraphics 1998, 18, 1523–1546. [Google Scholar] [CrossRef] [PubMed]
- Fresneau, B.; Orbach, D.; Faure-Conter, C.; Verité, C.; Castex, M.P.; Kalfa, N.; Martelli, H.; Patte, C. Sex-Cord Stromal Tumors in Children and Teenagers: Results of the TGM-95 Study: Sex-Cord Stromal Tumors in Children. Pediatr. Blood Cancer 2015, 62, 2114–2119. [Google Scholar] [CrossRef]
- Schneider, D.T.; Calaminus, G.; Wessalowksi, R.; Pathmanathan, R.; Selle, B.; Sternschulte, W.; Harms, D.; Göbel, U. Ovarian Sex Cord–Stromal Tumors in Children and Adolescents. JCO 2003, 21, 2357–2363. [Google Scholar] [CrossRef] [PubMed]
- Roth, L.M. Recent Advances in the Pathology and Classification of Ovarian Sex Cord-Stromal Tumors. Int. J. Gynecol. Pathol. 2006, 25, 199–215. [Google Scholar] [CrossRef]
- Schneider, D.T.; Jänig, U.; Calaminus, G.; Göbel, U.; Harms, D. Ovarian Sex Cord–Stromal Tumors—A Clinicopathological Study of 72 Cases from the Kiel Pediatric Tumor Registry. Virchows Arch. 2003, 443, 549–560. [Google Scholar] [CrossRef]
- Young, R.H.; Dickersin, G.R.; Scully, R.E. Juvenile Granulosa Cell Tumor of the Ovary: A Clinicopathological Analysis of 125 Cases. Am. J. Surg. Pathol. 1984, 8, 575–596. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sasaki, Y.; Nishihira, H.; Izawa, T.; Nishi, T. Ovarian Juvenile Granulosa Cell Tumor Associated with Maffucci’s Syndrome. Am. J. Clin. Pathol. 1992, 97, 523–527. [Google Scholar] [CrossRef]
- Sampagar, A.A.; Jahagirdar, R.R.; Bafna, V.S.; Bartakke, S.P. Juvenile Granulosa Cell Tumor Associated with Ollier Disease. Indian J. Med. Paediatr. Oncol. 2016, 37, 293–295. [Google Scholar] [CrossRef] [PubMed]
- Vaz, R.M.; Turner, C. Ollier Disease (Enchondromatosis) Associated with Ovarian Juvenile Granulosa Cell Tumor and Precocious Pseudopuberty. J. Pediatr. 1986, 108, 945–947. [Google Scholar] [CrossRef]
- Tamimi, H.K.; Bolen, J.W. Enchondromatosis (Ollier’s Disease) and Ovarian Juvenile Granulosa Cell Tumor. A Case Report and Review of the Literature. Cancer 1984, 53, 1605–1608. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, C.; Bouvier, R.; Chappuis, J.P.; Hermier, M. Ollier’s disease and juvenile ovarian granulosa tumor. Arch. Fr. Pediatr. 1991, 48, 115–118. [Google Scholar] [PubMed]
- Hachi, H.; Othmany, A.; Douayri, A.; Bouchikhi, C.; Tijami, F.; Laâlou, L.; Chami, M.; Boughtab, A.; Jalil, A.; Benjelloun, S.; et al. Association d’une tumeur ovarienne de la granulosa juvénile à un syndrome de Maffucci. Gynécologie Obs. Fertil. 2002, 30, 692–695. [Google Scholar] [CrossRef]
- Yuan, J.; Lin, X.; Xu, J.; Zhu, J.; Zheng, W. Ovarian Juvenile Granulosa Cell Tumor Associated with Maffucci’s Syndrome: Case Report. Chin. Med. J. 2004, 117, 1592–1594. [Google Scholar]
- Pansuriya, T.C.; Kroon, H.M.; Bovée, J.V.M.G. Enchondromatosis: Insights on the Different Subtypes. Int. J. Clin. Exp. Pathol. 2010, 3, 557–569. [Google Scholar]
- Gell, J.S.; Stannard, M.W.; Ramnani, D.M.; Bradshaw, K.D. Juvenile Granulosa Cell Tumor in a 13-Year-Old Girl With Enchondromatosis (Oilier’s Disease): A Case Report. J. Pediatr. Adolesc. Gynecol. 1998, 11, 147–150. [Google Scholar] [CrossRef]
- Silve, C.; Jüppner, H. Ollier Disease. Orphanet J. Rare Dis. 2006, 1, 37. [Google Scholar] [CrossRef]
- Zaloudek, C.; Norris, H.J. Granulosa Tumors of the Ovary in Children: A Clinical and Pathologic Study of 32 Cases. Am. J. Surg. Pathol. 1982, 6, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.E.; Rha, S.E.; Lee, J.M.; Park, S.Y.; Oh, S.N.; Cho, K.S.; Lee, E.J.; Byun, J.Y.; Hahn, S.T. CT and MRI Findings of Sex Cord–Stromal Tumor of the Ovary. Am. J. Roentgenol. 2005, 185, 207–215. [Google Scholar] [CrossRef]
- Morikawa, K.; Hatabu, H.; Togashi, K.; Kataoka, M.L.; Mori, T.; Konishi, J. Granulosa Cell Tumor of the Ovary: MR Findings. J. Comput. Assist. Tomogr. 1997, 21, 1001–1004. [Google Scholar] [CrossRef]
- Kitamura, Y.; Kanegawa, K.; Muraji, T.; Sugimura, K. MR Imaging of Juvenile Granulosa Cell Tumour of the Ovary: A Case Report. Pediatr. Radiol. 2000, 30, 360. [Google Scholar] [CrossRef]
- Rusterholz, K.R.; MacDonald, W. An Unusual Case of Juvenile Granulosa Cell Tumor of the Ovary. Radiol. Case Rep. 2009, 4, 178. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, S.H. Granulosa Cell Tumor of the Ovary: Common Findings and Unusual Appearances on CT and MR. J. Comput. Assist. Tomogr. 2002, 26, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Auguste, A.; Bessière, L.; Todeschini, A.-L.; Caburet, S.; Sarnacki, S.; Prat, J.; D’angelo, E.; De La Grange, P.; Ariste, O.; Lemoine, F.; et al. Molecular Analyses of Juvenile Granulosa Cell Tumors Bearing AKT1 Mutations Provide Insights into Tumor Biology and Therapeutic Leads. Hum. Mol. Genet. 2015, 24, 6687–6698. [Google Scholar] [CrossRef]
- Young, R.H.; Scully, R.E. Ovarian Sertoli—Leydig Cell Tumors: A Clinicopathological Analysis of 207 Cases. Am. J. Surg. Pathol. 1985, 9, 543–569. [Google Scholar] [CrossRef]
- Young, R.H.; Perez-Atayde, A.R.; Scully, R.E. Ovarian Sertoli-Leydig Cell Tumor with Retiform and Heterologous Components: Report of a Case with Hepatocytic Differentiation and Elevated Serum Alpha-Fetoprotein. Am. J. Surg. Pathol. 1984, 8, 709–718. [Google Scholar] [CrossRef]
- Cai, S.-Q.; Zhao, S.-H.; Qiang, J.-W.; Zhang, G.-F.; Wang, X.-Z.; Wang, L. Ovarian Sertoli–Leydig Cell Tumors: MRI Findings and Pathological Correlation. J. Ovarian Res. 2013, 6, 73. [Google Scholar] [CrossRef]
- Schneider, D.T.; Orbach, D.; Cecchetto, G.; Stachowicz-Stencel, T.; Brummel, B.; Brecht, I.B.; Bisogno, G.; Ferrari, A.; Reguerre, Y.; Godzinski, J.; et al. Ovarian Sertoli Leydig Cell Tumours in Children and Adolescents: An Analysis of the European Cooperative Study Group on Pediatric Rare Tumors (EXPeRT). Eur. J. Cancer 2015, 51, 543–550. [Google Scholar] [CrossRef]
- McCarville, M.B.; Hill, D.A.; Miller, B.E.; Pratt, C.B. Secondary Ovarian Neoplasms in Children: Imaging Features with Histopathologic Correlation. Pediatr. Radiol. 2001, 31, 358–364. [Google Scholar] [CrossRef]
- Young, R.H.; Oliva, E.; Scully, R.E. Small Cell Carcinoma of the Ovary, Hypercalcemic Type. A Clinicopathological Analysis of 150 Cases. Am. J. Surg. Pathol. 1994, 18, 1102–1116. [Google Scholar] [CrossRef]
- Witkowski, L.; Carrot-Zhang, J.; Albrecht, S.; Fahiminiya, S.; Hamel, N.; Tomiak, E.; Grynspan, D.; Saloustros, E.; Nadaf, J.; Rivera, B.; et al. Germline and Somatic SMARCA4 Mutations Characterize Small Cell Carcinoma of the Ovary, Hypercalcemic Type. Nat. Genet. 2014, 46, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, L.; Goudie, C.; Ramos, P.; Boshari, T.; Brunet, J.-S.; Karnezis, A.N.; Longy, M.; Knost, J.A.; Saloustros, E.; McCluggage, W.G.; et al. The Influence of Clinical and Genetic Factors on Patient Outcome in Small Cell Carcinoma of the Ovary, Hypercalcemic Type. Gynecol. Oncol. 2016, 141, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Hasselblatt, M.; Gesk, S.; Oyen, F.; Rossi, S.; Viscardi, E.; Giangaspero, F.; Giannini, C.; Judkins, A.R.; Frühwald, M.C.; Obser, T.; et al. Nonsense Mutation and Inactivation of SMARCA4 (BRG1) in an Atypical Teratoid/Rhabdoid Tumor Showing Retained SMARCB1 (INI1) Expression. Am. J. Surg. Pathol. 2011, 35, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Schneppenheim, R.; Frühwald, M.C.; Gesk, S.; Hasselblatt, M.; Jeibmann, A.; Kordes, U.; Kreuz, M.; Leuschner, I.; Martin Subero, J.I.; Obser, T.; et al. Germline Nonsense Mutation and Somatic Inactivation of SMARCA4/BRG1 in a Family with Rhabdoid Tumor Predisposition Syndrome. Am. J. Hum. Genet. 2010, 86, 279–284. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Malignant | Benign |
|---|---|---|
| Consistency | Solid or presence of solid components | Cystic |
| Size | Large (≥8–10 cm) | Small (<8–10 cm) |
| Composition | Heterogenous | Homogenous |
| Ovarian torsion | Less likely | More likely |
| Papillary projections | More likely | Less likely |
| Tumor Marker | Ovarian Neoplasm |
|---|---|
| AFP 1 | Immature teratoma Yolk sac tumor Embryonal carcinoma Sertoli–Leydig cell tumor (rare) |
| LDH 2 | Dysgerminoma |
| β-hCG 3 | Dysgerminoma (rare) Embryonal carcinoma Non-gestational choriocarcinoma |
| CA-125 4 | Malignant epithelial tumors |
| Inhibin | Juvenile granulosa cell tumor |
| Ovarian Neoplasm | Cancer Predisposition Syndrome |
|---|---|
| Gonadoblastoma | Frasier syndrome 1 Denys–Drash syndrome 1 WAGR syndrome 1 45, X/46, XY mosaicism |
| Juvenile granulosa cell tumor | Ollier disease 2 Maffucci syndrome 2 |
| Sertoli–Leydig cell tumor | DICER-1 syndrome |
| Small cell carcinoma of the ovary, hypercalcemic type | Rhabdoid tumor predisposition syndrome type 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birbas, E.; Kanavos, T.; Gkrozou, F.; Skentou, C.; Daniilidis, A.; Vatopoulou, A. Ovarian Masses in Children and Adolescents: A Review of the Literature with Emphasis on the Diagnostic Approach. Children 2023, 10, 1114. https://doi.org/10.3390/children10071114
Birbas E, Kanavos T, Gkrozou F, Skentou C, Daniilidis A, Vatopoulou A. Ovarian Masses in Children and Adolescents: A Review of the Literature with Emphasis on the Diagnostic Approach. Children. 2023; 10(7):1114. https://doi.org/10.3390/children10071114
Chicago/Turabian StyleBirbas, Effrosyni, Theofilos Kanavos, Fani Gkrozou, Chara Skentou, Angelos Daniilidis, and Anastasia Vatopoulou. 2023. "Ovarian Masses in Children and Adolescents: A Review of the Literature with Emphasis on the Diagnostic Approach" Children 10, no. 7: 1114. https://doi.org/10.3390/children10071114
APA StyleBirbas, E., Kanavos, T., Gkrozou, F., Skentou, C., Daniilidis, A., & Vatopoulou, A. (2023). Ovarian Masses in Children and Adolescents: A Review of the Literature with Emphasis on the Diagnostic Approach. Children, 10(7), 1114. https://doi.org/10.3390/children10071114