Nutritional Status and Circulating Levels of Fat-Soluble Vitamins in Cystic Fibrosis Patients: A Cohort Study and Evaluation of the Effect of CFTR Modulators

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
- -
- To cross-sectionally evaluate CF patients’ nutritional status and serum levels of fat-soluble vitamins.
- -
- To retrospectively evaluate the efficacy of treatment with CFTR protein modulators in terms of changes in nutritional status and circulating fat-soluble vitamin levels.
2. Materials and Methods
- -
- Subjects who had undergone organ transplantation;
- -
- Subjects diagnosed with CF Related Disorders;
- -
- Subjects categorized as screening positive with inconclusive diagnosis (CFSPID).
- ≥0 SDS;
- <0 SDS, ≥−1 SDS;
- <−1 SDS, ≥−2SDS;
- <−2 SDS.
- -
- 25(OH)D: normal values ≥ 30 ng/mL; vitamin insufficiency 10–29 ng/mL; vitamin deficiency < 10 ng/mL; toxicity > 100 ng/mL [22];
- -
- Vitamin A: normal values: 30–70 mcg/dL;
- -
- Vitamin E: normal values: 500–2000 mcg/dL.
Statistical Analysis
3. Results
3.1. Patients’ Demography
- <2 years: 3 patients (1 male and 2 females), mean age ± SD 1.67 ± 0.58 years;
- 2–18 years: 135 patients (69 males and 66 females) mean age ± SD 10.70 ± 3.90 years;
- >18 years: 180 patients (109 males and 71 females) mean age ± SD 34.85 years; ±11.80.
3.2. Nutritional Status
3.3. Fat-Soluble Vitamin Levels
3.4. Impact of CFTR Modulators
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 25(OH)D | 25-hydroxyvitamin D |
| BMI | body mass index |
| CDC | Centers for Disease Control and Prevention |
| CF | cystic fibrosis |
| CFSPID | cystic fibrosis screening positive with inconclusive diagnosis |
| CFTR | CF transmembrane conductance regulator |
| ETI | Elexacaftor/Tezacaftor/Ivacaftor |
| F | female(s) |
| Iva | Ivacaftor |
| LumIva | Lumacaftor/Ivacaftor |
| M | male(s) |
| WHO | World Health Organization |
References
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef] [PubMed]
- Emerson, J.; Rosenfeld, M.; McNamara, S.; Ramsey, B.; Gibson, R.L. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr. Pulmonol. 2002, 34, 91–100. [Google Scholar] [CrossRef]
- Ashkenazi, M.; Nathan, N.; Sarouk, I.; Bar Aluma, B.E.; Dagan, A.; Bezalel, Y.; Keler, S.; Vilozni, D.; Efrati, O. Nutritional status in childhood as a prognostic factor in patients with cystic fibrosis. Lung 2019, 197, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Orenti, A.; Zolin, A.; van Rens, J.; Fox, A.; Krasnyk, M.; Daneau, G.; Hatziagorou, E.; Mei-Zahav, M.; Naerlich, L.; Storms, V.; et al. ECFSPR Annual Report 2020; European Cystic Fibrosis Society: Karup, Denmark, 2022. [Google Scholar]
- UK Cystic Fibrosis Trust Registry 2021 Annual Data Report; Cystic Fibrosis Trust: London, UK, 2022.
- Cystic Fibrosis Foundation Patient Registry 2021 Annual Data Report; Cystic Fibrosis Foundation: Bethesda, MA, USA, 2022.
- Castellani, C.; Duff, A.J.; Bell, S.C.; Heijerman, H.G.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS best practice guidelines: The 2018 revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of cystic fibrosis: Consensus guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181S, S4–S15.e1. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.-R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef]
- Farrell, P.M.; Kosorok, M.R.; Rock, M.J.; Laxova, A.; Zeng, L.; Lai, H.-C.; Hoffman, G.; Laessig, R.H.; Splaingard, M.L.; the Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Pediatrics 2001, 107, 1–3. [Google Scholar] [CrossRef]
- Mason, K.A.; Rogol, A.D. Trends in growth and maturation in children with cystic fibrosis throughout nine decades. Front. Endocrinol. 2022, 13, 935354. [Google Scholar] [CrossRef]
- Slae, M.; Wilschanski, M. Nutrition in Cystic Fibrosis. World Rev. Nutr. Diet. 2022, 124, 374–381. [Google Scholar]
- Marks, M.P.; Heltshe, S.L.; Baines, A.; Ramsey, B.W.; Hoffman, L.R.; Stalvey, M.S. Most short children with cystic fibrosis do not catch up by adulthood. Nutrients 2021, 13, 4414. [Google Scholar] [CrossRef]
- Bell, S.C.; Bowerman, A.R.; Davies, C.A.; Campbell, I.A.; Shale, D.J.; Elborn, J.S. Nutrition in adults with cystic fibrosis. Clin. Nutr. 1998, 17, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Steinkamp, G.; Wiedemann, B. Relationship between nutritional status and lung function in cystic fibrosis: Cross sectional and longitudinal analyses from the German CF quality assurance (CFQA) project. Thorax 2002, 57, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Brownell, J.N.; Bashaw, H.; Stallings, V.A. Growth and nutrition in cystic fibrosis. Semin. Respir. Crit Care Med. 2019, 40, 775–791. [Google Scholar] [CrossRef] [PubMed]
- Slae, M.; Wilschanski, M. Prevention of malnutrition in cystic fibrosis. Curr. Opin. Pulm. Med. 2019, 25, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Schönenberger, K.A.; Reber, E.; Bally, L.; Geiser, T.; Lin, D.; Stanga, Z. Nutritional assessment in adults with cystic fibrosis. Nutrition 2019, 67–68, 110518. [Google Scholar] [CrossRef] [PubMed]
- Sommerburg, O.; Hämmerling, S.; Schneider, S.; Okun, J.; Langhans, C.-D.; Leutz-Schmidt, P.; Wielpütz, M.; Siems, W.; Gräber, S.; Mall, M.; et al. CFTR Modulator Therapy with Lumacaftor/Ivacaftor alters plasma concentrations of lipid-soluble vitamins A and E in patients with cystic fibrosis. Antioxidants 2021, 10, 483. [Google Scholar] [CrossRef]
- Lai, H.J.; Chin, L.H.; Murali, S.; Bach, T.; Sander, D.; Farrell, P.M. Vitamins A, D, E status as related to supplementation and lung disease markers in young children with cystic fibrosis. Pediatr. Pulmonol. 2022, 57, 935–944. [Google Scholar] [CrossRef]
- Timmers, N.K.L.M.; Stellato, R.K.; van der Ent, C.K.; Houwen, R.H.J.; Woestenenk, J.W. Vitamin D intake, serum 25-hydroxy vitamin D and pulmonary function in paediatric patients with cystic fibrosis: A longitudinal approach. Br. J. Nutr. 2019, 121, 195–201. [Google Scholar] [CrossRef]
- Tangpricha, V.; Kelly, A.; Stephenso, A.; Maguiness, K.; Enders, J.; Robinson, K.A.; Marshall, B.C.; Borowitz, D. Cystic Fibrosis Foundation Vitamin D Evidence-Based Review Committee. An update on the screening, diagnosis, management, and treatment of vitamin D deficiency in individuals with cystic fibrosis: Evidence-based recommendations from the Cystic Fibrosis Foundation. J. Clin. Endocrinol. Metab. 2012, 97, 1082–1093. [Google Scholar]
- Karpińska, J.; Mikołuć, B.; Motkowski, R.; Piotrowska-Jastrzebska, J. HPLC method for simultaneous determination of retinol, alpha-tocopherol and coenzyme Q10 in human plasma. J. Pharm. Biomed. Anal. 2006, 42, 232–236. [Google Scholar] [CrossRef]
- Mangas-Sánchez, C.; Garriga-García, M.; Serrano-Nieto, M.J.; García-Romero, R.; Álvarez-Beltrán, M.; Crehuá-Gaudiza, E.; Muñoz-Codoceo, R.; Suárez-Cortina, L.; Vicente-Santamaría, S.; Martínez-Costa, C.; et al. Vitamin D status in pediatric and young adult cystic fibrosis patients. are the new recommendations effective? Nutrients 2021, 13, 4413. [Google Scholar] [CrossRef] [PubMed]
- Vogeser, M.; Kyriatsoulis, A.; Huber, E.; Kobold, U. Candidate reference method for the quantification of circulating 25-hydroxyvitamin D3 by liquid chromatography-tandem mass spectrometry. Clin. Chem. 2004, 50, 1415–1417. [Google Scholar] [CrossRef] [PubMed]
- Botti, M.; Terlizzi, V.; Francalanci, M.; Dolce, D.; Cavicchi, M.C.; Neri, A.S.; Galici, V.; Mergni, G.; Zavataro, L.; Centrone, C.; et al. Cystic fibrosis in Tuscany: Evolution of newborn screening strategies over time to the present. Ital. J. Pediatr. 2021, 47, 2. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.; Krick, S.; Fontaine, K.R. The changing landscape of nutrition in cystic fibrosis: The emergence of overweight and obesity. Nutrients 2022, 14, 1216. [Google Scholar] [CrossRef]
- Szentpetery, S.; Fernandez, G.S.; Schechter, M.S.; Jain, R.; Flume, P.A.; Fink, A.K. Obesity in Cystic fibrosis: Prevalence, trends and associated factors data from the US Cystic Fibrosis Foundation Patient Registry. J. Cyst. Fibros. 2022, 21, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Schwarzenberg, S.J. Pancreatic insufficiency in cystic fibrosis. J. Cyst. Fibros. 2017, 16, S70–S78. [Google Scholar] [CrossRef]
- Gibson-Corley, K.N.; Meyerholz, D.K.; Engelhardt, J.F. Pancreatic patophysiology in cystic fibrosis. J. Pathol. 2016, 238, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Ooi, C.Y.; Durie, P.R. Cystic fibrosis from the gastroenterologist’s perspective. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 175–185. [Google Scholar] [CrossRef]
- Bass, R.; Brownell, J.N.; Stallings, V.A. The impact of highly effective CFTR modulators on growth and nutrition status. Nutrients 2021, 13, 2907. [Google Scholar] [CrossRef]
- Bailey, J.; Rozga, M.; McDonald, C.M.; Bowser, E.K.; Farnham, K.; Mangus, M.; Padula, L.; Porco, K.; Alvarez, J.A. Effect of CFTR modulators on anthropometric parameters in individuals with cystic fibrosis: An evidence analysis Center systematic review. J. Acad. Nutr. Diet. 2021, 121, 1364–1378.e2. [Google Scholar] [CrossRef]
- Regard, L.; Martin, C.; Burnet, E.; Da Silva, J.; Burgel, P.R. CFTR modulators in people with cystic fibrosis: Real-world evidence in France. Cells 2022, 11, 1769. [Google Scholar] [CrossRef] [PubMed]
- Gentzsch, M.; Mall, M.A. Ion channel modulators in cystic fibrosis. Chest 2018, 154, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Holguin, F. Triple CFTR Modulator Therapy for Cystic Fibrosis. N. Engl. J. Med. 2018, 379, 1671–1672. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M. CFTR Modulators: The changing face of cystic fibrosis in the era of precision medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef] [PubMed]
- Accurso, F.J.; Rowe, S.M.; Clancy, J.P.; Boyle, M.P.; Dunitz, J.M.; Durie, P.R.; Sagel, S.D.; Hornick, D.B.; Konstan, M.W.; Donaldson, S.H.; et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 2010, 363, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- Borowitz, D.; Lubarsky, B.; Wilschanski, M.; Munck, A.; Gelfond, D.; Bodewes, F.; Schwarzenberg, S.J. Nutritional status improved in cystic fibrosis patients with the G551D mutation after treatment with Ivacaftor. Dig. Dis. Sci. 2016, 61, 198–207. [Google Scholar] [CrossRef] [PubMed]
- King, S.J.; Tierney, A.C.; Edgeworth, D.; Keating, D.; Williams, E.; Kotsimbos, T.; Button, B.M.; Wilson, J.W. Body composition and weight changes after ivacaftor treatment in adults with cystic fibrosis carrying the G551 D cystic fibrosis transmembrane conductance regulator mutation: A double-blind, placebo-controlled, randomized, crossover study with open-label. Nutrition 2021, 85, 111124. [Google Scholar] [CrossRef]
- Hubert, D.; Dehillotte, C.; Munck, A.; David, V.; Baek, J.; Mely, L.; Dominique, S.; Ramel, S.; Boucher, I.D.; Lefeuvre, S.; et al. Retrospective observational study of French patients with cystic fibrosis and a Gly551Asp-CFTR mutation after 1 and 2 years of treatment with ivacaftor in a real-world setting. J. Cyst. Fibros. 2018, 17, 89–95. [Google Scholar] [CrossRef]
- McKone, E.F.; Borowitz, D.; Drevinek, P.; Griese, M.; Konstan, M.W.; Wainwright, C.; Ratjen, F.; Sermet-Gaudelus, I.; Plant, B.; Munck, A.; et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: A phase 3, open-label extension study. Lancet Respir. Med. 2014, 2, 902–910. [Google Scholar] [CrossRef]
- Davies, J.C.; Wainwright, C.E.; Canny, G.J.; Chilvers, M.A.; Howenstine, M.S.; Munck, A.; Mainz, J.G.; Rodriguez, S.; Li, H.; Yen, K.; et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013, 187, 1219–1225. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Hug, C.; Marigowda, G.; Tian, S.; Huang, X.; Stanojevic, S.; Milla, C.E.; Robinson, P.D.; Waltz, D.; Davies, J.C.; et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for F508del-CFTR: A randomised, placebo-controlled phase 3 trial. Lancet Respir. Med. 2017, 5, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.R.; Munck, A.; Durieu, I.; Chiron, R.; Mely, L.; Prevotat, A.; Murris-Espin, M.; Porzio, M.; Abely, M.; Reix, P.; et al. French Cystic Fibrosis Reference Network Study Group. Real-life safety and effectiveness of Lumacaftor-Ivacaftor in patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 188–197. [Google Scholar] [CrossRef] [PubMed]
- McColley, S.A.; Konstan, M.W.; Ramsey, B.W.; Elborn, J.S.; Boyle, M.P.; Wainwright, C.E.; Waltz, D.; Vera-Llonch, M.; Marigowda, G.; Jiang, J.G.; et al. Lumacaftor/Ivacaftor reduces pulmonary exacerbations in patients irrespective of initial changes in FEV1. J. Cyst. Fibros. 2019, 18, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Konstan, M.W.; McKone, E.F.; Moss, R.B.; Marigowda, G.; Tian, S.; Waltz, D.; Huang, X.; Lubarsky, B.; Rubin, J.; Millar, S.J.; et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation: A phase 3, extension study. Lancet Respir. Med. 2017, 5, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; Van Der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Pilewski, J.M.; Griese, M.; Cooke, J.; Viswanathan, L.; Tullis, E.; Davies, J.C.; Lekstrom-Himes, J.A.; Wang, L.T. Tezacaftor/ivacaftor in subjects with cystic fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit. Care Med. 2018, 197, 214–224. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for cystic fibrosis with a single Phe508del allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Heijerman, H.G.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and Safety of the elexacaftor/tezacaftor/ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double blind, randomised, phase 3 trial. Lancet 2020, 394, 1940–1948. [Google Scholar] [CrossRef]
- Ridley, K.; Condren, M. Elexacaftor-tezacaftor-ivacaftor: The first triple combination cystic fibrosis transmembrane conductance regulator modulating therapy. J. Pediatr. Pharmac. Ther. 2020, 25, 192–197. [Google Scholar] [CrossRef]
- Petersen, M.C.; Begnel, L.; Wallendorf, M.L. Effect of elexacaftor-tezacaftor-ivacaftor on body weight and metabolic parameters in adults with cystic fibrosis. J. Cyst. Fibros. 2021, 21, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Ramos, K.J.; Pilewski, J.M.; Taylor-Cousar, J.L. Challenges in the use of highly effective modulator treatment for cystic fibrosis. J. Cyst. Fibros. 2021, 20, 381–387. [Google Scholar] [CrossRef] [PubMed]




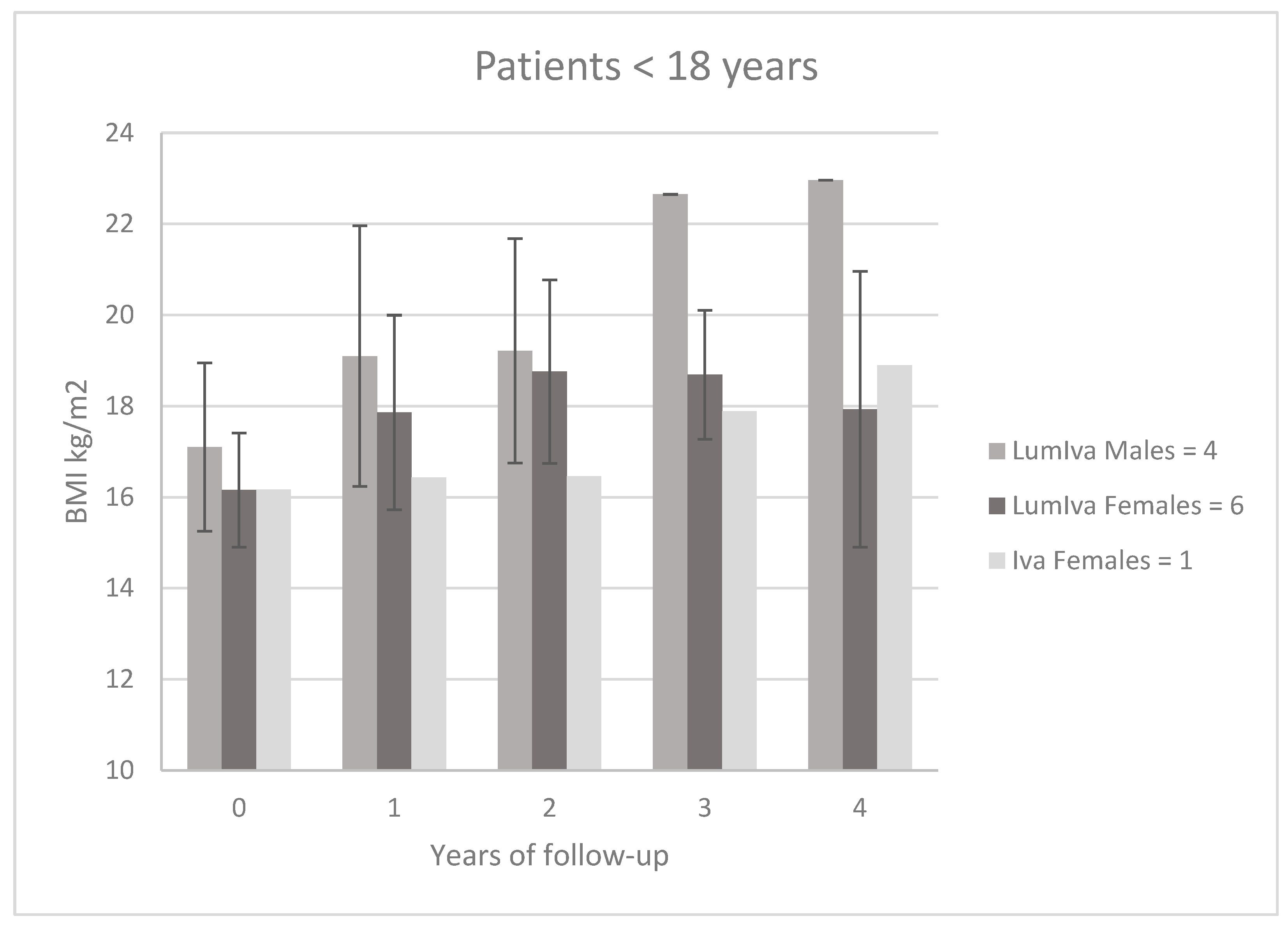{kind=link}
{kind=link}
{kind=link}
| M | F | |
|---|---|---|
| optimal | ≥23 | ≥22 |
| normal weight | 20–23 | 20–22 |
| underweight | 18–20 | 18–20 |
| malnutrition | <18 | <18 |
| BMI z-Score: | 69 Males N (%) | 66 Females N (%) |
|---|---|---|
| ≥0 SDS | 38 (55.1%) | 34 (51.5%) |
| <0 SDS ≥ −1 SDS | 21 (30.4%) | 21 (31.8%) |
| <−1 SDS ≥ −2 SDS | 7 (10.1%) | 9 (13.6%) |
| <−2 SDS | 3 (4.3%) | 2 (3.0%) |
| BMI Class | 109 Males N (%) | 71 Females N (%) |
|---|---|---|
| ≥23(M) o ≥22(F) optimal weight | 47 (43.1%) | 21 (29.6%) |
| 20–23(M) o 20–22(F) normal weight | 44 (40.4%) | 27 (38.0%) |
| 18–20 underweight | 15 (13.8%) | 18 (25.4%) |
| <18 malnutrition | 3 (2.7%) | 5 (7.0%) |
| 25(OH)D (ng/mL) | Vit A (mcg/dL) | Vit E (mcg/dL) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Basal | Post-Treatment | p Value | Basal | Post-Treatment | p Value | Basal | Post-Treatment | p Value | |
| Iva N = 9 | 28.4 ± 9.5 | 36.7 ± 11.6 | p = 0.12 | 44.7 ± 18.8 | 48.7 ± 7.6 | p = 0.56 | 1337.8 ± 378.9 | 1762 ± 408.5 | p = 0.036 |
| LumIva N = 42 | 25.9 ± 11.5 | 24.9 ± 14.7 | p = 0.73 | 36.4 ± 10.9 | 43.5 ± 13.1 | p = 0.008 | 967.1 ± 282.7 | 938.8 ± 273.6 | p = 0.64 |
| ETI N = 29 | 23.5 ± 7.6 | 29.2 ± 9.5 | p = 0.015 | 37.4 ± 13.0 | 47.4 ± 15.8 | p = 0.011 | 1054.3 ± 208.0 | 1213.4 ± 219.3 | p = 0.006 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francalanci, M.; Terlizzi, V.; Fevola, C.; Di Rosa, G.; Pierattini, V.; Roselli, E.; Bonomi, P.; Cavicchi, M.C.; Galici, V.; Neri, A.S.; et al. Nutritional Status and Circulating Levels of Fat-Soluble Vitamins in Cystic Fibrosis Patients: A Cohort Study and Evaluation of the Effect of CFTR Modulators. Children 2023, 10, 252. https://doi.org/10.3390/children10020252
Francalanci M, Terlizzi V, Fevola C, Di Rosa G, Pierattini V, Roselli E, Bonomi P, Cavicchi MC, Galici V, Neri AS, et al. Nutritional Status and Circulating Levels of Fat-Soluble Vitamins in Cystic Fibrosis Patients: A Cohort Study and Evaluation of the Effect of CFTR Modulators. Children. 2023; 10(2):252. https://doi.org/10.3390/children10020252
Chicago/Turabian StyleFrancalanci, Michela, Vito Terlizzi, Cristina Fevola, Giulia Di Rosa, Valentina Pierattini, Elena Roselli, Paolo Bonomi, Maria Chiara Cavicchi, Valeria Galici, Anna Silvia Neri, and et al. 2023. "Nutritional Status and Circulating Levels of Fat-Soluble Vitamins in Cystic Fibrosis Patients: A Cohort Study and Evaluation of the Effect of CFTR Modulators" Children 10, no. 2: 252. https://doi.org/10.3390/children10020252
APA StyleFrancalanci, M., Terlizzi, V., Fevola, C., Di Rosa, G., Pierattini, V., Roselli, E., Bonomi, P., Cavicchi, M. C., Galici, V., Neri, A. S., Bianchimani, C., Campana, S., Dolce, D., Ravenni, N., Camera, E., Orioli, T., & Taccetti, G. (2023). Nutritional Status and Circulating Levels of Fat-Soluble Vitamins in Cystic Fibrosis Patients: A Cohort Study and Evaluation of the Effect of CFTR Modulators. Children, 10(2), 252. https://doi.org/10.3390/children10020252