
Dendritic Spine in Autism Genetics: Whole-Exome Sequencing Identifying De Novo Variant of CTTNBP2 in a Quad Family Affected by Autism Spectrum Disorder
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Chromosome Microarray
2.3. Whole-Exome Sequencing
2.4. Bioinformatic Analysis
2.5. Sanger Sequencing
2.6. Genotyping
2.7. Tertiary Structure Prediction
2.8. Genetic Model Prediction
2.9. Genes Enrichment Analysis
3. Results
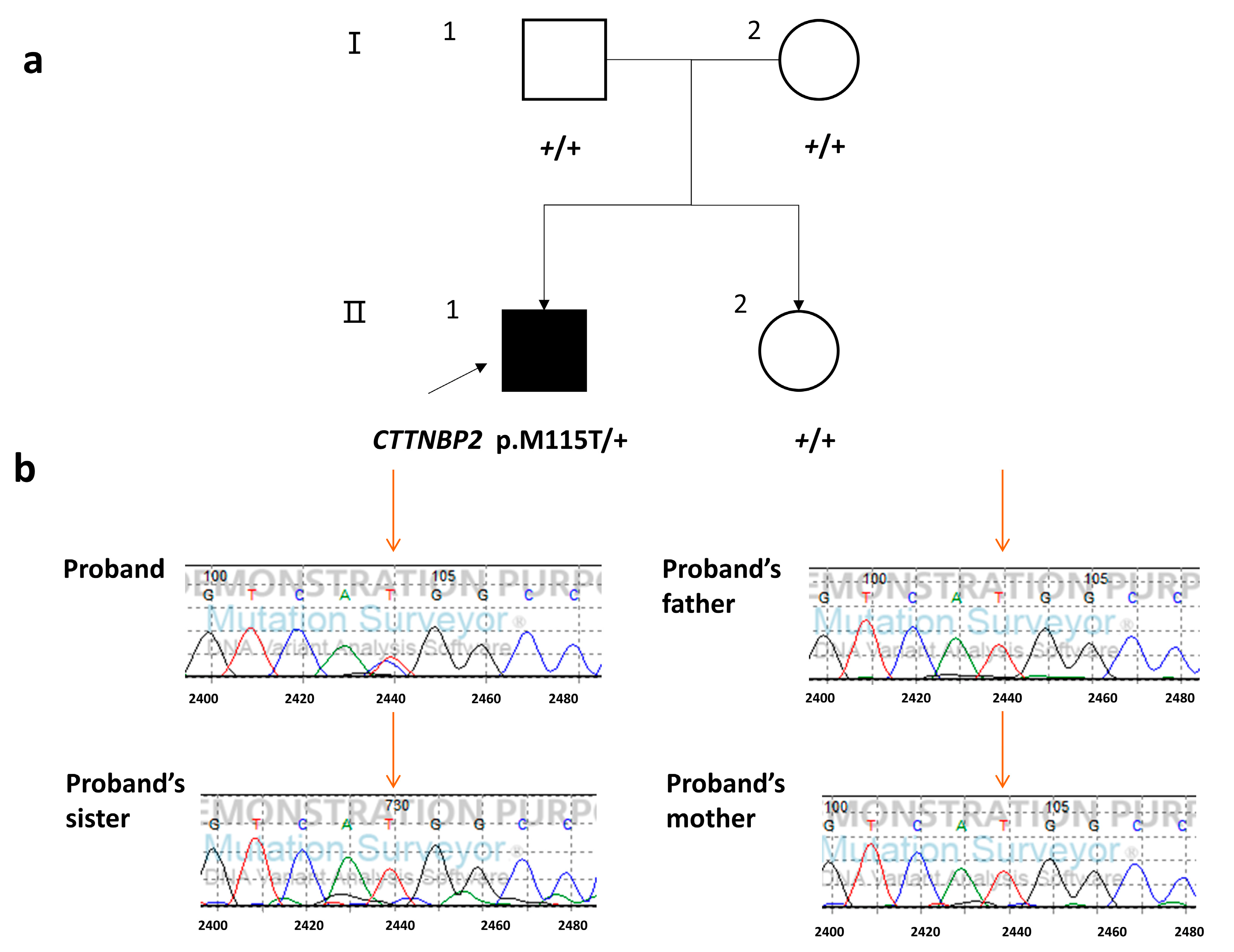
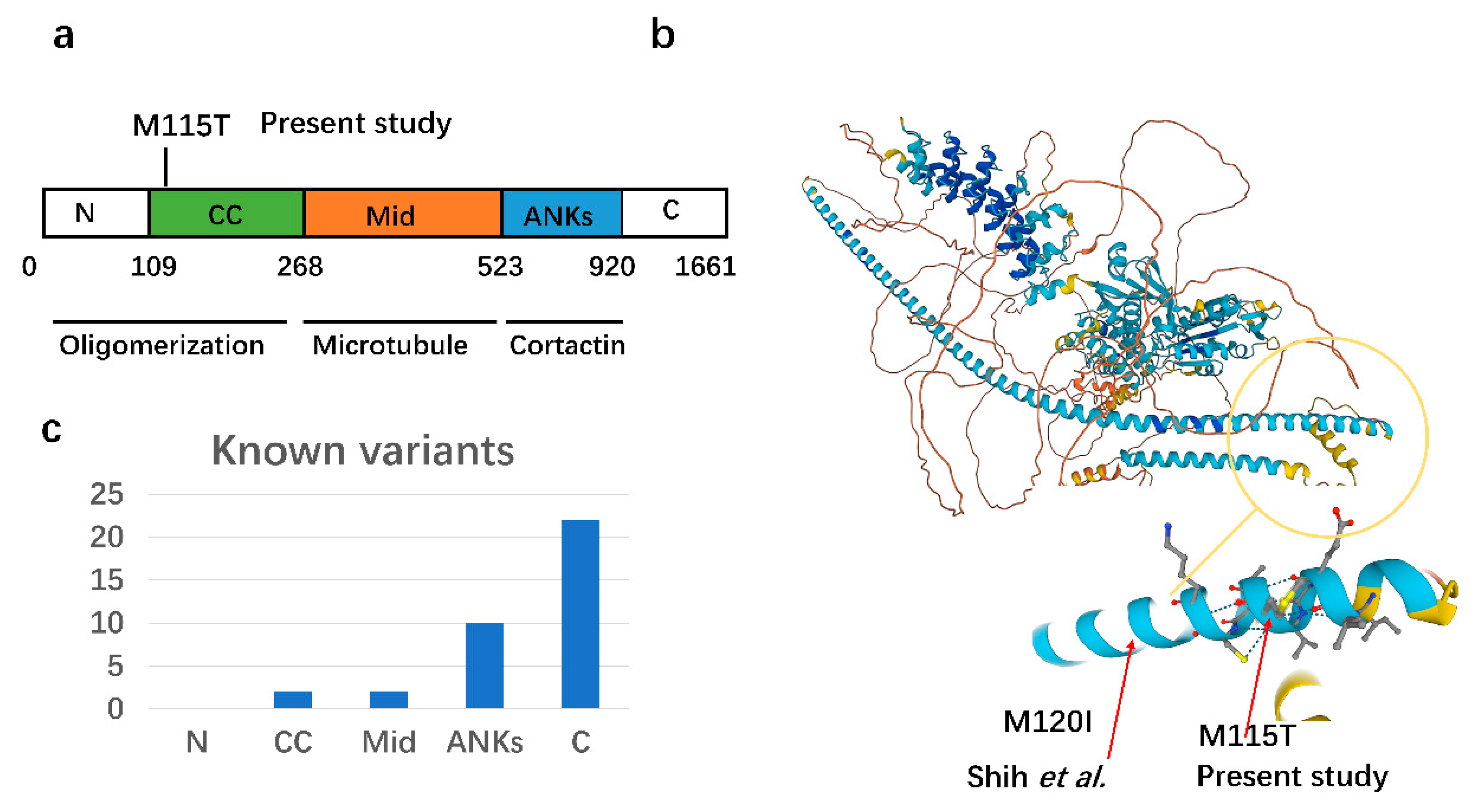
Variants Identified of the Proband
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lord, C.; Charman, T.; Havdahl, A.; Carbone, P.; Anagnostou, E.; Boyd, B.; Carr, T.; de Vries, P.J.; Dissanayake, C.; Divan, G.; et al. The Lancet Commission on the future of care and clinical research in autism. Lancet 2022, 399, 271–334. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, K.; He, X.; Zhou, J.; Jin, C.; Shen, L.; Gao, Y.; Tian, M.; Zhang, H. Structural, Functional, and Molecular Imaging of Autism Spectrum Disorder. Neurosci. Bull. 2021, 37, 1051–1071. [Google Scholar] [CrossRef] [PubMed]
- Colvert, E.; Tick, B.; McEwen, F.; Stewart, C.; Curran, S.R.; Woodhouse, E.; Gillan, N.; Hallett, V.; Lietz, S.; Garnett, T.; et al. Heritability of Autism Spectrum Disorder in a UK Population-Based Twin Sample. JAMA Psychiatry 2015, 72, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Constantino, J.N.; Zhang, Y.; Frazier, T.; Abbacchi, A.M.; Law, P. Sibling recurrence and the genetic epidemiology of autism. Am. J. Psychiatry 2010, 167, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef]
- Bai, D.; Yip, B.H.K.; Windham, G.C.; Sourander, A.; Francis, R.; Yoffe, R.; Glasson, E.; Mahjani, B.; Suominen, A.; Leonard, H.; et al. Association of Genetic and Environmental Factors With Autism in a 5-Country Cohort. JAMA Psychiatry 2019, 76, 1035–1043. [Google Scholar] [CrossRef]
- Maenner, M.J.; Shaw, K.A.; Bakian, A.V.; Bilder, D.A.; Durkin, M.S.; Esler, A.; Furnier, S.M.; Hallas, L.; Hall-Lande, J.; Hudson, A.; et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2018. MMWR Surveill. Summ. 2021, 70, 1–16. [Google Scholar] [CrossRef]
- Yasuda, Y.; Matsumoto, J.; Miura, K.; Hasegawa, N.; Hashimoto, R. Genetics of autism spectrum disorders and future direction. J. Hum. Genet. 2022. [Google Scholar] [CrossRef]
- Wang, T.; Zhao, P.A.; Eichler, E.E. Rare variants and the oligogenic architecture of autism. Trends Genet. 2022, 38, 895–903. [Google Scholar] [CrossRef]
- Wang, M.; Fan, X.; Wang, T.; Wu, J. High-throughput sequencing of autism spectrum disorders comes of age. Genet. Res. 2013, 95, 121–129. [Google Scholar] [CrossRef]
- Jiang, Y.H.; Yuen, R.K.; Jin, X.; Wang, M.; Chen, N.; Wu, X.; Ju, J.; Mei, J.; Shi, Y.; He, M.; et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 2013, 93, 249–263. [Google Scholar] [CrossRef]
- Yuen, R.K.; Merico, D.; Cao, H.; Pellecchia, G.; Alipanahi, B.; Thiruvahindrapuram, B.; Tong, X.; Sun, Y.; Cao, D.; Zhang, T.; et al. Genome-wide characteristics of de novo mutations in autism. NPJ Genom. Med. 2016, 1, 16027. [Google Scholar] [CrossRef]
- Wu, J.; Yu, P.; Jin, X.; Xu, X.; Li, J.; Li, Z.; Wang, M.; Wang, T.; Wu, X.; Jiang, Y.; et al. Genomic landscapes of Chinese sporadic autism spectrum disorders revealed by whole-genome sequencing. J. Genet. Genom. 2018, 45, 527–538. [Google Scholar] [CrossRef]
- Talli, I.; Dovrolis, N.; Oulas, A.; Stavrakaki, S.; Makedou, K.; Spyrou, G.M.; Maroulakou, I. Novel clinical, molecular and bioinformatics insights into the genetic background of autism. Hum. Genom. 2022, 16, 39. [Google Scholar] [CrossRef]
- Willsey, H.R.; Willsey, A.J.; Wang, B.; State, M.W. Genomics, convergent neuroscience and progress in understanding autism spectrum disorder. Nat. Rev. Neurosci. 2022, 23, 323–341. [Google Scholar] [CrossRef]
- Manoli, D.S.; State, M.W. Autism Spectrum Disorder Genetics and the Search for Pathological Mechanisms. Am. J. Psychiatry 2021, 178, 30–38. [Google Scholar] [CrossRef]
- Paulsen, B.; Velasco, S.; Kedaigle, A.J.; Pigoni, M.; Quadrato, G.; Deo, A.J.; Adiconis, X.; Uzquiano, A.; Sartore, R.; Yang, S.M.; et al. Autism genes converge on asynchronous development of shared neuron classes. Nature 2022, 602, 268–273. [Google Scholar] [CrossRef]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef]
- Quesnel-Vallieres, M.; Weatheritt, R.J.; Cordes, S.P.; Blencowe, B.J. Autism spectrum disorder: Insights into convergent mechanisms from transcriptomics. Nat. Rev. Genet. 2019, 20, 51–63. [Google Scholar] [CrossRef]
- Adhya, D.; Swarup, V.; Nagy, R.; Dutan, L.; Shum, C.; Valencia-Alarcon, E.P.; Jozwik, K.M.; Mendez, M.A.; Horder, J.; Loth, E.; et al. Atypical Neurogenesis in Induced Pluripotent Stem Cells From Autistic Individuals. Biol. Psychiatry 2021, 89, 486–496. [Google Scholar] [CrossRef]
- Sacai, H.; Sakoori, K.; Konno, K.; Nagahama, K.; Suzuki, H.; Watanabe, T.; Watanabe, M.; Uesaka, N.; Kano, M. Autism spectrum disorder-like behavior caused by reduced excitatory synaptic transmission in pyramidal neurons of mouse prefrontal cortex. Nat. Commun. 2020, 11, 5140. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [PubMed]
- Ruzzo, E.K.; Perez-Cano, L.; Jung, J.Y.; Wang, L.K.; Kashef-Haghighi, D.; Hartl, C.; Singh, C.; Xu, J.; Hoekstra, J.N.; Leventhal, O.; et al. Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell 2019, 178, 850–866.e26. [Google Scholar] [CrossRef]
- Shih, P.Y.; Hsieh, B.Y.; Tsai, C.Y.; Lo, C.A.; Chen, B.E.; Hsueh, Y.P. Autism-linked mutations of CTTNBP2 reduce social interaction and impair dendritic spine formation via diverse mechanisms. Acta Neuropathol. Commun. 2020, 8, 185. [Google Scholar] [CrossRef]
- Chen, C.; Wei, J.; Ma, X.; Xia, B.; Shakir, N.; Zhang, J.K.; Zhang, L.; Cui, Y.; Ferguson, D.; Qiu, S.; et al. Disrupted Maturation of Prefrontal Layer 5 Neuronal Circuits in an Alzheimer’s Mouse Model of Amyloid Deposition. Neurosci. Bull. 2022. [Google Scholar] [CrossRef]
- Song, X.L.; Liu, D.S.; Qiang, M.; Li, Q.; Liu, M.G.; Li, W.G.; Qi, X.; Xu, N.J.; Yang, G.; Zhu, M.X.; et al. Postsynaptic Targeting and Mobility of Membrane Surface-Localized hASIC1a. Neurosci. Bull. 2021, 37, 145–165. [Google Scholar] [CrossRef]
- Hu, H.T.; Shih, P.Y.; Shih, Y.T.; Hsueh, Y.P. The Involvement of Neuron-Specific Factors in Dendritic Spinogenesis: Molecular Regulation and Association with Neurological Disorders. Neural Plast. 2016, 2016, 5136286. [Google Scholar] [CrossRef]
- Nanou, E.; Catterall, W.A. Calcium Channels, Synaptic Plasticity, and Neuropsychiatric Disease. Neuron 2018, 98, 466–481. [Google Scholar] [CrossRef]
- Vorstman, J.A.S.; Parr, J.R.; Moreno-De-Luca, D.; Anney, R.J.L.; Nurnberger, J.I., Jr.; Hallmayer, J.F. Autism genetics: Opportunities and challenges for clinical translation. Nat. Rev. Genet. 2017, 18, 362–376. [Google Scholar] [CrossRef]
- McClellan, J.; King, M.C. Genetic heterogeneity in human disease. Cell 2010, 141, 210–217. [Google Scholar] [CrossRef]
- Xie, Y.; Yuan, H.; Wang, M.; Zhong, L.; Zhou, J.; Song, B.; Yin, Q.; Sun, X. Copy number variations independently induce autism spectrum disorder. Biosci. Rep. 2017, 37, BSR20160570. [Google Scholar]
- Zhou, J.; Yang, F.; Li, X.; Zhou, S.; He, F.; Liang, J.; Wang, Y.; Wang, M.; Xu, R. Whole Exome Sequencing Revealed Clinical Relevant Variants in A Family Affected with Autism Spectrum Disorder. J. Psychiatry Brain Sci. 2016, 1, 3. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Li, J.; Jiang, Y.; Wang, T.; Chen, H.; Xie, Q.; Shao, Q.; Ran, X.; Xia, K.; Sun, Z.S.; Wu, J. mirTrios: An integrated pipeline for detection of de novo and rare inherited mutations from trios-based next-generation sequencing. J. Med. Genet. 2015, 52, 275–281. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Abrahams, B.S.; Arking, D.E.; Campbell, D.B.; Mefford, H.C.; Morrow, E.M.; Weiss, L.A.; Menashe, I.; Wadkins, T.; Banerjee-Basu, S.; Packer, A. SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 2013, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Robinson, P.N.; Wang, K. Phenolyzer: Phenotype-based prioritization of candidate genes for human diseases. Nat. Methods 2015, 12, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.; Ziaugra, L.; Tabbaa, D. SNP genotyping using the Sequenom MassARRAY iPLEX platform. Curr. Protoc. Hum. Genet. 2009, 60, 2.12.1–2.12.18. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Quinodoz, M.; Royer-Bertrand, B.; Cisarova, K.; Di Gioia, S.A.; Superti-Furga, A.; Rivolta, C. DOMINO: Using Machine Learning to Predict Genes Associated with Dominant Disorders. Am. J. Hum. Genet. 2017, 101, 623–629. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Jeste, S.S.; Geschwind, D.H. Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat. Rev. Neurol. 2014, 10, 74–81. [Google Scholar] [CrossRef]
- Chen, J.A.; Penagarikano, O.; Belgard, T.G.; Swarup, V.; Geschwind, D.H. The emerging picture of autism spectrum disorder: Genetics and pathology. Annu. Rev. Pathol. 2015, 10, 111–144. [Google Scholar] [CrossRef]
- de la Torre-Ubieta, L.; Won, H.; Stein, J.L.; Geschwind, D.H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 2016, 22, 345–361. [Google Scholar] [CrossRef]
- Tammimies, K.; Marshall, C.R.; Walker, S.; Kaur, G.; Thiruvahindrapuram, B.; Lionel, A.C.; Yuen, R.K.; Uddin, M.; Roberts, W.; Weksberg, R.; et al. Molecular Diagnostic Yield of Chromosomal Microarray Analysis and Whole-Exome Sequencing in Children With Autism Spectrum Disorder. JAMA 2015, 314, 895–903. [Google Scholar] [CrossRef]
- Cheung, J.; Petek, E.; Nakabayashi, K.; Tsui, L.C.; Vincent, J.B.; Scherer, S.W. Identification of the human cortactin-binding protein-2 gene from the autism candidate region at 7q31. Genomics 2001, 78, 7–11. [Google Scholar] [CrossRef]
- Chen, Y.K.; Hsueh, Y.P. Cortactin-binding protein 2 modulates the mobility of cortactin and regulates dendritic spine formation and maintenance. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 1043–1055. [Google Scholar] [CrossRef]
- Chen, Y.K.; Chen, C.Y.; Hu, H.T.; Hsueh, Y.P. CTTNBP2, but not CTTNBP2NL, regulates dendritic spinogenesis and synaptic distribution of the striatin-PP2A complex. Mol. Biol. Cell 2012, 23, 4383–4392. [Google Scholar] [CrossRef]
- Shih, P.Y.; Hsieh, B.Y.; Lin, M.H.; Huang, T.N.; Tsai, C.Y.; Pong, W.L.; Lee, S.P.; Hsueh, Y.P. CTTNBP2 Controls Synaptic Expression of Zinc-Related Autism-Associated Proteins and Regulates Synapse Formation and Autism-like Behaviors. Cell Rep. 2020, 31, 107700. [Google Scholar] [CrossRef]
- Shih, P.Y.; Fang, Y.L.; Shankar, S.; Lee, S.P.; Hu, H.T.; Chen, H.; Wang, T.F.; Hsia, K.C.; Hsueh, Y.P. Phase separation and zinc-induced transition modulate synaptic distribution and association of autism-linked CTTNBP2 and SHANK3. Nat. Commun. 2022, 13, 2664. [Google Scholar] [CrossRef]
- Saldarriaga, W.; Payan-Gomez, C.; Gonzalez-Teshima, L.Y.; Rosa, L.; Tassone, F.; Hagerman, R.J. Double Genetic Hit: Fragile X Syndrome and Partial Deletion of Protein Patched Homolog 1 Antisense as Cause of Severe Autism Spectrum Disorder. J. Dev. Behav. Pediatr. 2020, 41, 724–728. [Google Scholar] [CrossRef]
- Guo, H.; Wang, T.; Wu, H.; Long, M.; Coe, B.P.; Li, H.; Xun, G.; Ou, J.; Chen, B.; Duan, G.; et al. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol. Autism 2018, 9, 64. [Google Scholar] [CrossRef]
- Tau, G.Z.; Peterson, B.S. Normal development of brain circuits. Neuropsychopharmacology 2010, 35, 147–168. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Wang, Y.; Shao, B.; Zhou, H.; Li, X.; Zhang, C.; Sun, N.; Shi, J. Multiple Mild Stimulations Reduce Membrane Distribution of CX3CR1 Promoted by Annexin a1 in Microglia to Attenuate Excessive Dendritic Spine Pruning and Cognitive Deficits Caused by a Transient Ischemic Attack in Mice. Neurosci. Bull. 2022, 38, 753–768. [Google Scholar] [CrossRef] [PubMed]
- MacGillavry, H.D.; Kerr, J.M.; Kassner, J.; Frost, N.A.; Blanpied, T.A. Shank-cortactin interactions control actin dynamics to maintain flexibility of neuronal spines and synapses. Eur. J. Neurosci. 2016, 43, 179–193. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Y.; Wang, H.; Hu, B.; Zhang, X.; Liu, A.; Cai, C.; Li, S.; Chen, C.; Wang, Z.; Yin, Z.; et al. Dendritic Spine in Autism Genetics: Whole-Exome Sequencing Identifying De Novo Variant of CTTNBP2 in a Quad Family Affected by Autism Spectrum Disorder. Children 2023, 10, 80. https://doi.org/10.3390/children10010080
Xie Y, Wang H, Hu B, Zhang X, Liu A, Cai C, Li S, Chen C, Wang Z, Yin Z, et al. Dendritic Spine in Autism Genetics: Whole-Exome Sequencing Identifying De Novo Variant of CTTNBP2 in a Quad Family Affected by Autism Spectrum Disorder. Children. 2023; 10(1):80. https://doi.org/10.3390/children10010080
Chicago/Turabian StyleXie, Yingmei, Hui Wang, Bing Hu, Xueli Zhang, Aiping Liu, Chunquan Cai, Shijun Li, Cheng Chen, Zhangxing Wang, Zhaoqing Yin, and et al. 2023. "Dendritic Spine in Autism Genetics: Whole-Exome Sequencing Identifying De Novo Variant of CTTNBP2 in a Quad Family Affected by Autism Spectrum Disorder" Children 10, no. 1: 80. https://doi.org/10.3390/children10010080
APA StyleXie, Y., Wang, H., Hu, B., Zhang, X., Liu, A., Cai, C., Li, S., Chen, C., Wang, Z., Yin, Z., & Wang, M. (2023). Dendritic Spine in Autism Genetics: Whole-Exome Sequencing Identifying De Novo Variant of CTTNBP2 in a Quad Family Affected by Autism Spectrum Disorder. Children, 10(1), 80. https://doi.org/10.3390/children10010080