
Involvement of Angiogenesis in the Pathogenesis of Coronary Aneurysms

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
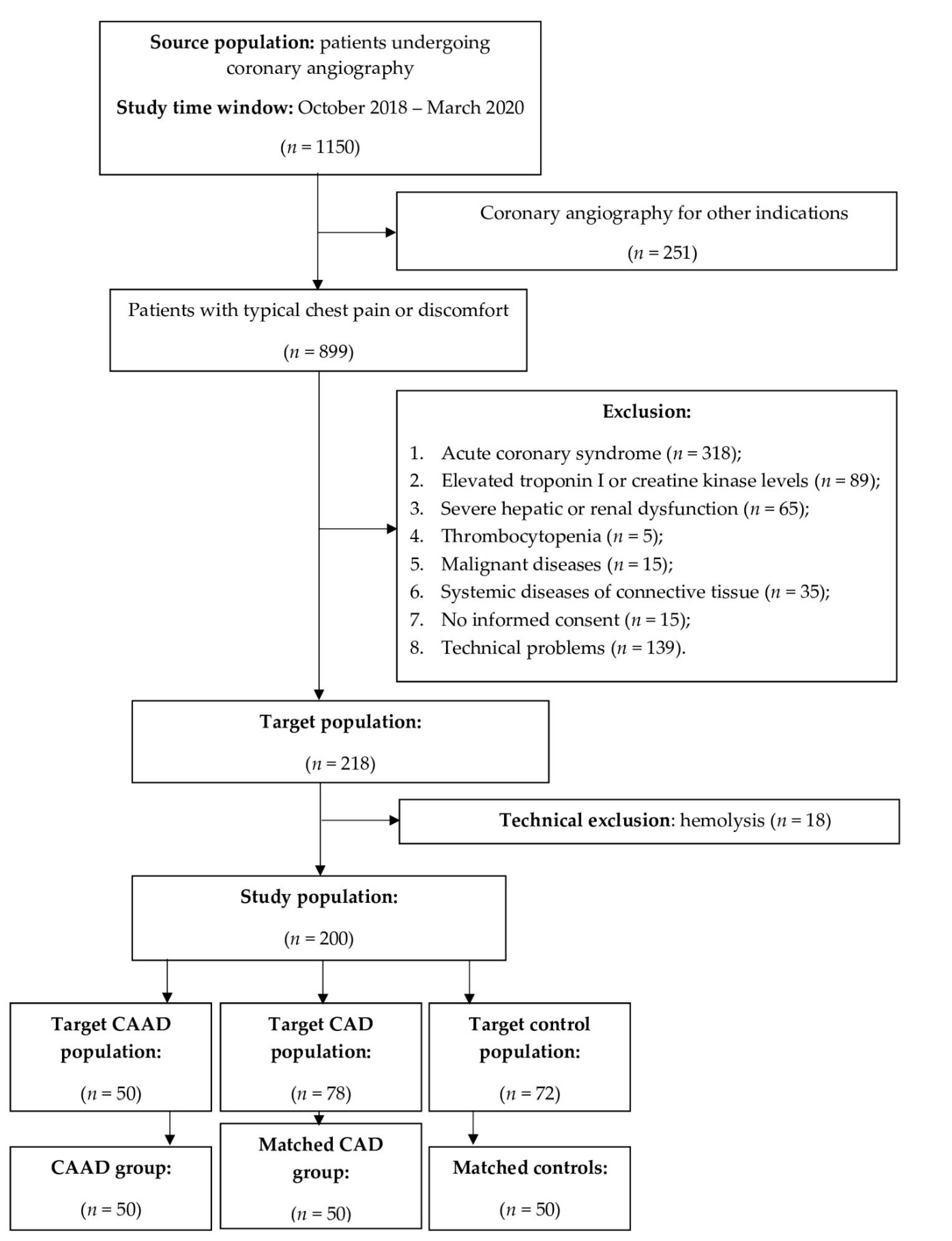
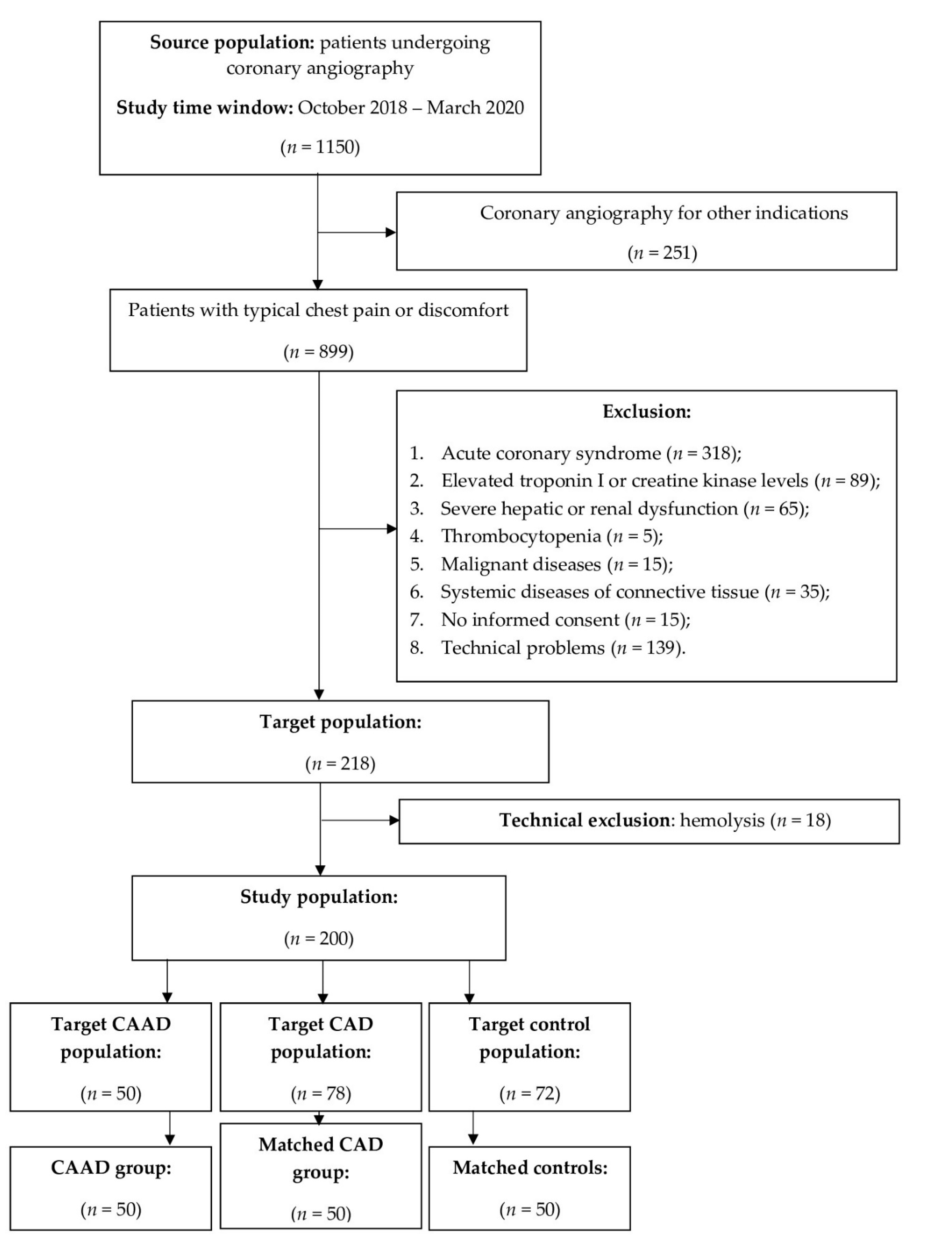
2.1. Study Design and Patient Selection
2.2. ELISA Analysis
2.3. Statistical Analysis
3. Results
3.1. Clinical Characteristics of the Study Population
3.2. Angiographic Characteristics of Group 1 (CAAD Group)
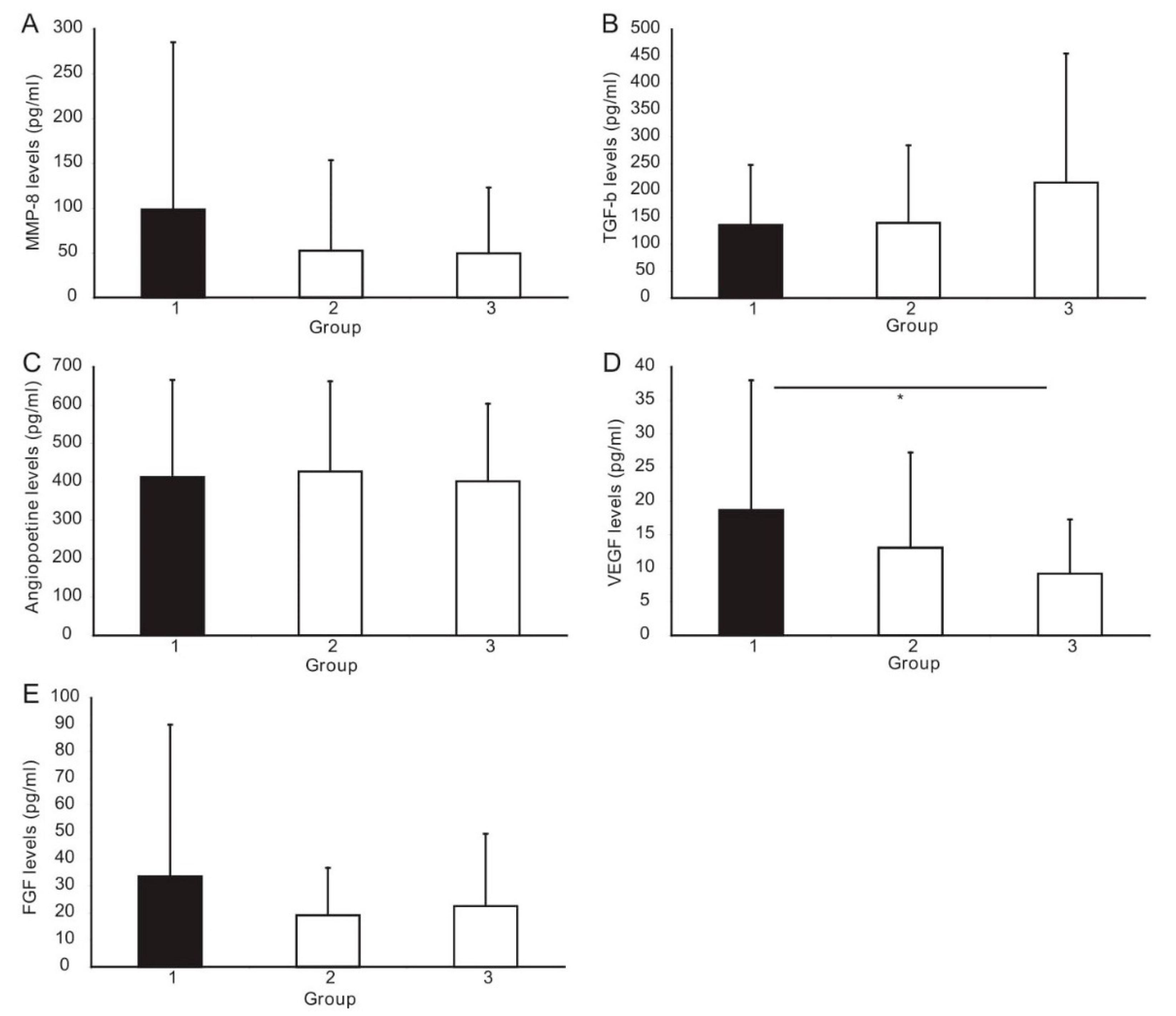
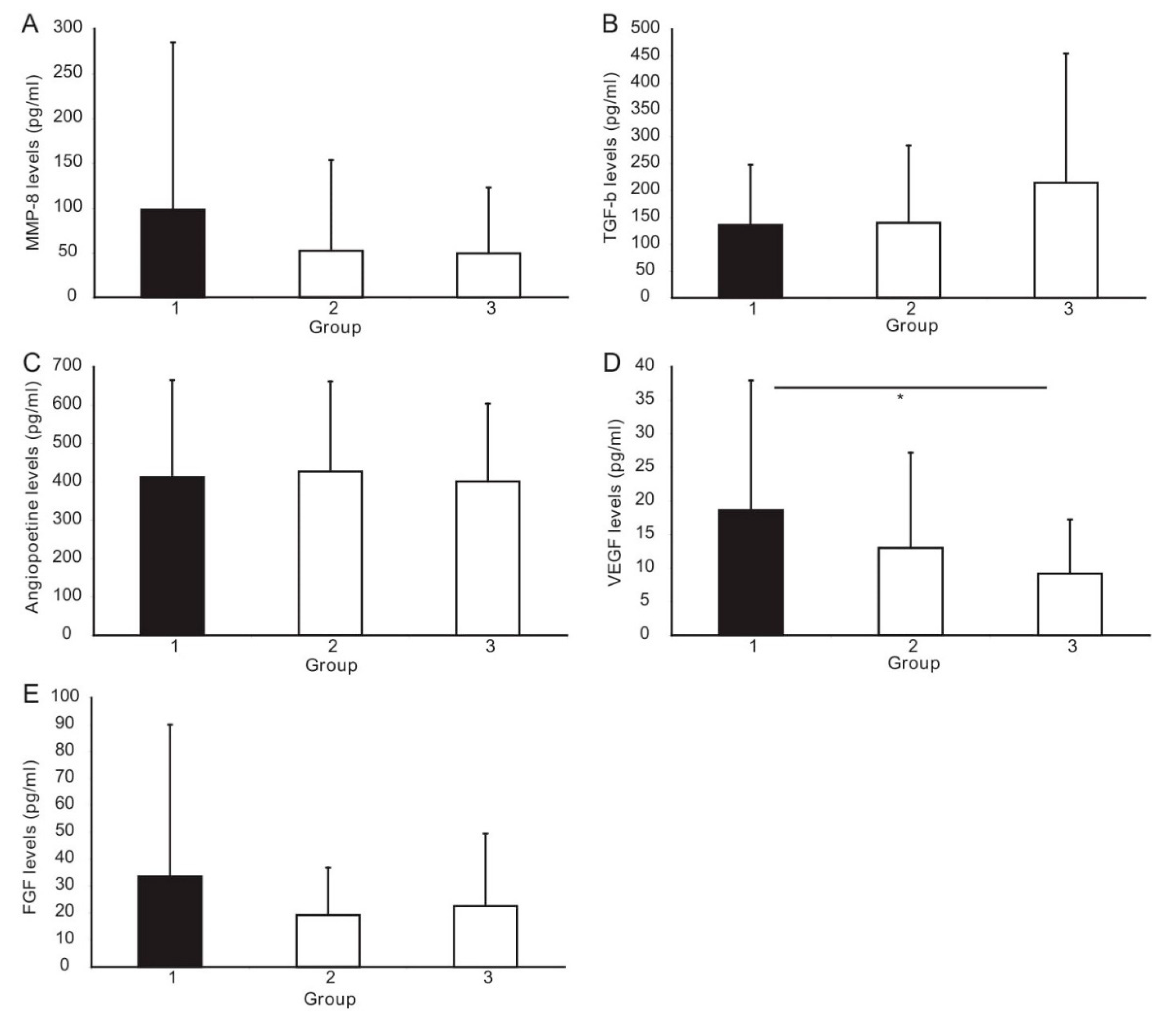
3.3. ELISA Analysis of Angiogenesis Factors
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Swaye, P.S.; Fisher, L.D.; Litwin, P.; Vignola, P.A.; Judkins, M.P.; Kemp, H.G.; Mudd, J.G.; Gosselin, A.J. Aneurysmal coronary artery disease. Circulation 1983, 67, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Baman, T.S.; Cole, J.H.; Devireddy, C.M.; Sperling, L.S. Risk factors and outcomes in patients with coronary artery aneurysms. Am. J. Cardiol. 2004, 93, 1549–1551. [Google Scholar] [CrossRef] [PubMed]
- Warisawa, T.; Naganuma, T.; Tomizawa, N.; Fujino, Y.; Ishiguro, H.; Tahara, S.; Kurita, N.; Nojo, T.; Nakamura, S.; Nakamura, S. High prevalence of coronary artery events and non-coronary events in patients with coronary artery aneurysm in the observational group. Int. J. Cardiol. Heart Vasc. 2015, 10, 29–31. [Google Scholar] [CrossRef] [Green Version]
- Markis, J.E.; Joffe, C.D.; Cohn, P.F.; Feen, D.J.; Herman, M.V.; Gorlin, R. Clinical significance of coronary arterial ectasia. Am. J. Cardiol. 1976, 37, 217–222. [Google Scholar] [CrossRef]
- Li, J.-J.; Nie, S.-P.; Qian, X.-W.; Zeng, H.-S.; Zhang, C.-Y. Chronic inflammatory status in patients with coronary artery ectasia. Cytokine 2009, 46, 61–64. [Google Scholar] [CrossRef]
- Moreno, P.R.; Purushothaman, K.R.; Zias, E.; Sanz, J.; Fuster, V. Neovascularization in Human Atherosclerosis. Curr. Mol. Med. 2006, 6, 457–477. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Schaper, W. Mechanisms of arteriogenesis. Acta Biochim. Biophys. Sin. 2008, 40, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Khatri, J.J. Matrix Metalloproteinases in Vascular Remodeling and Atherogenesis. Circ. Res. 2002, 90, 251–262. [Google Scholar] [CrossRef]
- Sedding, D.G.; Boyle, E.C.; Demandt, J.A.F.; Sluimer, J.C.; Dutzmann, J.; Haverich, A.; Bauersachs, J. Vasa Vasorum Angiogenesis: Key Player in the Initiation and Progression of Atherosclerosis and Potential Target for the Treatment of Cardiovascular Disease. Front Immunol. 2018, 9, 706. [Google Scholar] [CrossRef] [Green Version]
- El Guindy, M.S.; El Guindy, A.M. Aneurysmal coronary artery disease: An overview. Glob. Cardiol. Sci. Pract. 2017, 3, e201726. [Google Scholar]
- Iwańczyk, S.; Araszkiewicz, A.; Borger, M.; Kamiński, M.; Wrotyński, M.; Chmara, E.; Cieślewicz, A.; Radziemski, A.; Lesiak, M. Endocan expression correlated with total volume of coronary artery dilation in patients with coronary artery ectasia. Postepy Kardiol. Interwencyjnej 2020, 16, 294–299. [Google Scholar]
- Paulus, P.; Jennewein, C.; Zacharowski, K. Biomarkers of endothelial dysfunction: Can they help us deciphering systemic inflammation and sepsis? Biomarkers 2011, 16 (Suppl. 1), S11–S21. [Google Scholar] [CrossRef]
- Courtois, A.; Nusgens, B.; Garbacki, N.; Hustinx, R.; Gomez, P.; Defraigne, J.-O.; Colige, A.C.; Sakalihasan, N. Circulating microRNAs signature correlates with positive [18F]fluorodeoxyglucose-positron emission tomography in patients with abdominal aortic aneurysm. J. Vasc. Surg. 2018, 67, 585–595.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, T.; Yao, Y.; Zhang, B.; Hao, D.-C.; Sun, Q.-F.; Li, J.-B.; Yuan, C.; Jing, B.; Wang, Y.-P.; Wang, H.-Y. Role of MicroRNA-103a Targeting ADAM10 in Abdominal Aortic Aneurysm. Biomed Res. Int. 2017, 2017, 9645874. [Google Scholar] [CrossRef] [PubMed]
- Neumann, F.-J.; Sousa-Uva, M.; Ahlsson, A.; Alfonso, F.; Banning, A.P.; Benedetto, U.; Byrne, R.A.; Collet, J.P.; Falk, V.; Head, S.J.; et al. 2018 ESC/EACTS Guidelines on myocardial revascularization. Eur. Heart J. 2019, 40, 87–165. [Google Scholar] [CrossRef]
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar]
- Yamazaki, Y.; Morita, T. Molecular and functional diversity of vascular endothelial growth factors. Mol. Divers. 2006, 10, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef]
- Ucuzian, A.A.; Gassman, A.A.; East, A.T.; Greisler, H.P. Molecular mediators of angiogenesis. J. Burn Care Res. 2010, 31, 158–175. [Google Scholar] [CrossRef]
- Clauss, M.; Weich, H.; Breier, G.; Knies, U.; Röckl, W.; Waltenberger, J.; Risau, W. The vascular endothelial growth factor receptor Flt-1 mediates biological activities. Implications for a functional role of placenta growth factor in monocyte activation and chemotaxis. J. Biol. Chem. 1996, 271, 17629–17634. [Google Scholar] [CrossRef] [Green Version]
- Tchaikovski, V.; Fellbrich, G.; Waltenberger, J. The molecular basis of VEGFR-1 signal transduction pathways in primary human monocytes. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 322–328. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, S.K.; Giri, R.K.; Perelman, N.; Johnson, C.; Malik, P.; Kalra, V.K. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood 2003, 102, 1515–1524. [Google Scholar] [CrossRef] [Green Version]
- Ishida, A.; Murray, J.; Saito, Y.; Kanthou, C.; Benzakour, O.; Shibuya, M.; Wijelath, E.S. Expression of vascular endothelial growth factor receptors in smooth muscle cells. J. Cell Physiol. 2001, 188, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etulain, J.; Negrotto, S.; Tribulatti, M.V.; Croci, D.O.; Carabelli, J.; Campetella, O.; Rabinovich, G.A.; Schattner, M. Control of angiogenesis by galectins involves the release of platelet-derived proangiogenic factors. PLoS ONE 2014, 9, e96402. [Google Scholar]
- Raica, M.; Cimpean, A.M. Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, L.S. Clinical experience with angiogenesis signaling inhibitors: Focus on vascular endothelial growth factor (VEGF) blockers. Cancer Control. 2002, 9 (Suppl. 2), 36–44. [Google Scholar] [CrossRef]
- Van Hove, A.H.; Benoit, D.S.W. Depot-Based Delivery Systems for Pro-Angiogenic Peptides: A Review. Front. Bioeng. Biotechnol. 2015, 3, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parenti, A.; Brogelli, L.; Filippi, S.; Donnini, S.; Ledda, F. Effect of hypoxia and endothelial loss on vascular smooth muscle cell responsiveness to VEGF-A: Role of flt-1/VEGF-receptor-1. Cardiovasc. Res. 2002, 55, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Ley, C.D.; Olsen, M.W.B.; Lund, E.L.; Kristjansen, P.E.G. Angiogenic synergy of bFGF and VEGF is antagonized by Angiopoietin-2 in a modified in vivo Matrigel assay. Microvasc. Res. 2004, 68, 161–168. [Google Scholar] [CrossRef]
- Raza, A.; Franklin, M.J.; Dudek, A.Z. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am. J. Hematol. 2010, 85, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Miron, L.; Gafton, B.; Marinca, M. Angiogeneza Tumorală—Implicaţii în Terapia Cancerelor. J. Chir. 2010, 6, 8. [Google Scholar]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Banda, M.J.; Leppert, D. Matrix metalloproteinases in immunity. J. Immunol. 1996, 156, 1–4. [Google Scholar] [PubMed]
- Sangiorgi, G.; D’Averio, R.; Mauriello, A.; Bondio, M.; Pontillo, M.; Castelvecchio, S.; Trimarchi, S.; Tolva, V.; Nano, G.; Rampoldi, V.; et al. Plasma levels of metalloproteinases-3 and -9 as markers of successful abdominal aortic aneurysm exclusion after endovascular graft treatment. Circulation 2001, 18 (Suppl. 1), I288–I295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovsepian, D.M.; Ziporin, S.J.; Sakurai, M.K.; Lee, J.K.; Curci, J.A.; Thompson, R.W. Elevated plasma levels of matrix metalloproteinase-9 in patients with abdominal aortic aneurysms: A circulating marker of degenerative aneurysm disease. J. Vasc. Interv. Radiol. 2000, 11, 1345–1352. [Google Scholar] [CrossRef]
- McMillan, W.D.; Pearce, W.H. Increased plasma levels of metalloproteinase-9 are associated with abdominal aortic aneurysms. J. Vasc. Surg. 1999, 29, 122–127; discussion 127–129. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Matsubara, J.; Matsushita, M.; Nishikimi, N.; Sakurai, T.; Nimura, Y. Expression of angiogenesis and angiogenic factors in human aortic vascular disease. J. Surg. Res. 2002, 106, 239–245. [Google Scholar] [CrossRef]
- Saito, S.; Zempo, N.; Yamashita, A.; Takenaka, H.; Fujioka, K.; Esato, K. Matrix metalloproteinase expressions in arteriosclerotic aneurysmal disease. Vasc. Endovasc. Surg. 2002, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Longo, G.M.; Xiong, W.; Greiner, T.C.; Zhao, Y.; Fiotti, N.; Baxter, B.T. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J. Clin. Investig. 2002, 110, 625–632. [Google Scholar] [CrossRef]
- Chua, P.K.; Melish, M.E.; Yu, Q.; Yanagihara, R.; Yamamoto, K.S.; Nerurkar, V.R. Elevated levels of matrix metalloproteinase 9 and tissue inhibitor of metalloproteinase 1 during the acute phase of Kawasaki disease. Clin. Diagn. Lab. Immunol. 2003, 10, 308–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terai, M.; Yasukawa, K.; Narumoto, S.; Tateno, S.; Oana, S.; Kohno, Y. Vascular endothelial growth factor in acute Kawasaki disease. Am. J. Cardiol. 1999, 83, 337–339. [Google Scholar] [CrossRef]
- Ohno, T.; Igarashi, H.; Inoue, K.; Akazawa, K.; Joho, K.; Hara, T. Serum vascular endothelial growth factor: A new predictive indicator for the occurrence of coronary artery lesions in Kawasaki disease. Eur. J. Pediatr. 2000, 159, 424–429. [Google Scholar] [CrossRef]
- Savino, M.; Parisi, Q.; Biondi-Zoccai, G.G.L.; Pristipino, C.; Cianflone, D.; Crea, F. New insights into molecular mechanisms of diffuse coronary ectasiae: A possible role for VEGF. Int. J. Cardiol. 2006, 106, 307–312. [Google Scholar] [CrossRef]
- Seghezzi, G.; Patel, S.; Ren, C.J.; Gualandris, A.; Pintucci, G.; Robbins, E.S.; Shapiro, R.L.; Galloway, A.C.; Rifkin, D.B.; Mignatti, P. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: An autocrine mechanism contributing to angiogenesis. J. Cell Biol. 1998, 141, 1659–1673. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Baseline Data | Group 1 (n = 50) | Group 2 (n = 50) | Group 3 (n = 50) |
|---|---|---|---|
| Sex, male, n (%) | 40 (80.0) | 40 (80.0) | 40 (80.0) |
| Age (yrs), mean ± SD | 65.8 ± 8.6 | 66.4 ± 7.9 | 63.5 ± 10.1 |
| BMI (kg/m2), mean ± SD | 31.4 + 4.7 * | 28.6 ± 4.8 * | 29.1 ± 6.1 |
| Previous MI, n (%) | 16 (32.0) **,*** | 19 (38.0) **,*** | 0 **,*** |
| Previous PCI, n (%) | 19 (38.0) **,*** | 27 (54.0) **,*** | 0 **,*** |
| Previous CABG, n (%) | 3 (6.0) **,*** | 6 (12.0) **,*** | 0 **,*** |
| Hypertension, n (%) | 45 (90.0) **,*** | 46 (92.0) **,*** | 39 (78.0) **,*** |
| Heart failure, n (%) | 19 (38.0) | 19 (38.0) | 12 (24.0) |
| LVEF (%), mean ± s.d. | 53.7 ± 11.9 | 52.8 ± 10.6 | 55.6 ± 8.6 |
| Diabetes mellitus, n (%) | 17 (34.0) | 14 (28.0) | 13 (26.0) |
| Hyperlipidemia, n (%) | 38 (76.0) | 40 (80.0) | 34 (68.0) |
| Cigarette smoking, n (%) | 20 (40.0) | 18 (36.0) | 15 (30.0) |
| Aortic aneurysm, n (%) | 9 (18.0) | 9 (18.0) | 8 (16.0) |
| CKD class ≥ 2, n (%) | 9 (18.0) | 7 (14.0) | 4 (8.0) |
| Drug administration | |||
| Statin, n (%) | 46 (92.0) | 49 (98.0) *** | 41 (82.0) *** |
| CCB, n (%) | 22 (44.0) | 20 (40.0) | 20 (40.0) |
| Beta blocker, n (%) | 44 (88.0) **,*** | 46 (92.0) **,*** | 36 (72.0) **,*** |
| Aspirin, n (%) | 43 (86.0) **,*** | 46 (92.0) **,*** | 27 (54.0) **,*** |
| Clopidogrel, n (%) | 23 (46.0) **,*** | 35 (70.0) **,*** | 0 **,*** |
| ACEI/ARB, n (%) | 44 (88.0) | 45 (90.0) | 39 (78.0) |
| Laboratory tests | |||
| LDL cholesterol (mmol/L) | 2.3 ± 1.7 | 2.2 ± 0.8 | 2.4 ± 0.9 |
| CRP (mg/L) | 1.7 (1.25–2.35) | 2.9 (2.8–3.0) | 2.1 (1.8–2.9) |
| Baseline Data | Group 1 (n = 50), n (%) |
|---|---|
| CAE | 37 (74.0) |
| CAA | 10 (20.0) |
| Both | 3 (6.0) |
| Number of vessels involved | |
| 1 | 35 (70.0) |
| 2 | 12 (24.0) |
| 3 | 3 (6.0) |
| Vessel localization | |
| LM | 3 (6.0) |
| RCA | 25 (50.0) |
| LAD | 20 (40.0) |
| LCx | 20 (40.0) |
| Concomitant CAD | 28 (56.0) |
| Angiogenic Factors | Group 1 | Group 2 | Group 3 | 1 vs. 2 | 1 vs. 3 | 2 vs. 3 |
|---|---|---|---|---|---|---|
| MMP-8, pg/mL | 40.7 (23.1–87.4) | 15.6 (12.4–42.1) | 31.7 (11.3–56.8) | 0.06 | 0.58 | 0.58 |
| TGF-β1, pg/mL | 114.2 (42.2–184.0) | 77.1 (40.3–217.0) | 139.0 (82.9–253.6) | 0.48 | 0.06 | 0.03 |
| Angiopoietin-2, pg/mL | 325.4 (252.2 457.0) | 336.2 (283.8–492.3) | 361.8 (273.8–486.5) | 0.94 | 0.94 | 0.94 |
| VEGF, pg/mL | 18.7 ± 19.3 | 13.1 ± 14.2 | 9.2 ± 8.1 | 0.21 | 0.002 | 0.51 |
| FGF, pg/mL | 14.9 (8–30) | 14.9 (7.5–29) | 12.7 (4–33.5) | 0.97 | 0.99 | 0.98 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwańczyk, S.; Lehmann, T.; Cieślewicz, A.; Radziemski, A.; Malesza, K.; Wrotyński, M.; Jagodziński, P.P.; Grygier, M.; Lesiak, M.; Araszkiewicz, A. Involvement of Angiogenesis in the Pathogenesis of Coronary Aneurysms. Biomedicines 2021, 9, 1269. https://doi.org/10.3390/biomedicines9091269
Iwańczyk S, Lehmann T, Cieślewicz A, Radziemski A, Malesza K, Wrotyński M, Jagodziński PP, Grygier M, Lesiak M, Araszkiewicz A. Involvement of Angiogenesis in the Pathogenesis of Coronary Aneurysms. Biomedicines. 2021; 9(9):1269. https://doi.org/10.3390/biomedicines9091269
Chicago/Turabian StyleIwańczyk, Sylwia, Tomasz Lehmann, Artur Cieślewicz, Artur Radziemski, Katarzyna Malesza, Michał Wrotyński, Paweł Piotr Jagodziński, Marek Grygier, Maciej Lesiak, and Aleksander Araszkiewicz. 2021. "Involvement of Angiogenesis in the Pathogenesis of Coronary Aneurysms" Biomedicines 9, no. 9: 1269. https://doi.org/10.3390/biomedicines9091269
APA StyleIwańczyk, S., Lehmann, T., Cieślewicz, A., Radziemski, A., Malesza, K., Wrotyński, M., Jagodziński, P. P., Grygier, M., Lesiak, M., & Araszkiewicz, A. (2021). Involvement of Angiogenesis in the Pathogenesis of Coronary Aneurysms. Biomedicines, 9(9), 1269. https://doi.org/10.3390/biomedicines9091269