
Dietary Patterns Influence Target Gene Expression through Emerging Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease




Abstract
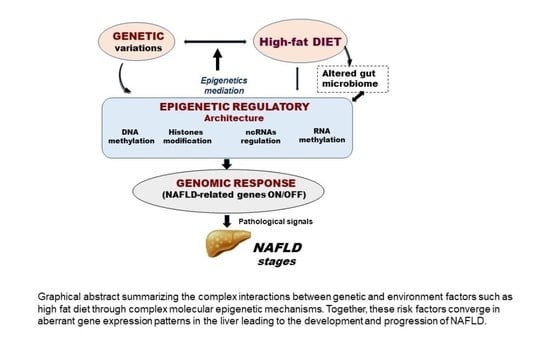
:
1. Introduction
2. Epigenetic Mechanisms Underlying the Link between Nutrition and Aberrant Gene Expression in NAFLD
2.1. DNA Methylation and NAFLD
2.2. Histone Post-Translational Modifications in NAFLD
3. Epigenetic Studies’ Limitations
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Arnlt | Aryl hydrocarbon receptor nuclear translocator-like |
| CpG | Cytosine-phospho-guanine |
| CXCL5 | C-X-C Motif Chemokine Ligand 5 |
| DNMT | DNA methyltransferase |
| HATs | Histone acetyltransferases |
| HCC | Hepatocellular carcinoma |
| HDACis | Histone deacetylase inhibitors |
| HDACs | Histone deacetylases |
| HDMs | Histone demethylases |
| HFD | High-fat diet |
| HMTs | Histone methyltransferases |
| HSC | Hepatic stellate cell |
| IR | Insulin resistance |
| MetS | Metabolic syndrome |
| mTOR | mammalian target of rapamycin |
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | Nonalcoholic steatohepatitis |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| PTEN | Phosphatase and tensin homolog |
| S6K1 | Ribosomal protein S6 kinase beta-1 |
| SAM | S-adenosyl methionine |
| Sirt1 | Sirtuin 1 |
| STAT5 | Signal transducer and activator of transcription 5 |
| T2DM Type 2 | diabetes mellitus |
| TG | Triglyceride |
| UPS10 | Ubiquitin-Specific Protease 10 |
| α-SMA | Alpha Smooth Muscle Actin |
References
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef]
- Van Kleef, L.; Ayada, I.; Alferink, L.; Pan, Q.; de Knegt, R. Metabolic dysfunction associated fatty liver disease improves detection of high liver stiffness: The rotterdam study. Hepatology 2021. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Eslam, M.; Kawaguchi, T.; Tsutsumi, T.; Nakano, D.; Yoshinaga, S.; Takahashi, H.; Anzai, K.; George, J.; Torimura, T. MAFLD identifies patients with significant hepatic fibrosis better than NAFLD. Liver Int. 2020, 40, 3018–3030. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Huang, J.; Wang, M.; Kumar, R.; Liu, Y.; Liu, S.; Wu, Y.; Wang, X.; Zhu, Y. Comparison of MAFLD and NAFLD diagnostic criteria in real world. Liver Int. 2020, 40, 2082–2089. [Google Scholar] [CrossRef]
- Niriella, M.A.; Ediriweera, D.S.; Kasturiratne, A.; de Silva, S.T.; Dassanayaka, A.S.; de Silva, A.P.; Kato, N.; Pathmeswaran, A.; Wickramasinghe, A.R.; de Silva, H.J. Outcomes of NAFLD and MAFLD: Results from a community-based, prospective cohort study. PLoS ONE 2021, 16, e0245762. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.; Kennedy, L.; Hargrove, L.; Demieville, J.; Thomson, J.; Alpini, G.; Francis, H. Updates on Dietary Models of Nonalcoholic Fatty Liver Disease: Current Studies and Insights. Gene Expr. 2018, 18, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E. Nonalcoholic Fatty Liver Disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef]
- Godoy-Matos, A.F.; Júnior, W.S.S.; Valerio, C.M. NAFLD as a continuum: From obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Mantovani, A.; Byrne, C.D.; Bonora, E.; Targher, G. Nonalcoholic Fatty Liver Disease and Risk of Incident Type 2 Diabetes: A Meta-analysis. Diabetes Care 2018, 41, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- Chiu, L.S.; Pedley, A.; Massaro, J.M.; Benjamin, E.J.; Mitchell, G.F.; McManus, D.D.; Aragam, J.; Vasan, R.S.; Cheng, S.; Long, M.T. The association of non-alcoholic fatty liver disease and cardiac structure and function—Framingham Heart Study. Liver Int. 2020, 40, 2445–2454. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G. What about non-alcoholic fatty liver disease as a new criterion to define metabolic syndrome? World J. Gastroenterol. 2013, 19, 3375–3384. [Google Scholar] [CrossRef]
- Lujan, P.V.; Esmel, E.V.; Meseguer, E.S. Overview of Non-Alcoholic Fatty Liver Disease (NAFLD) and the Role of Sugary Food Consumption and Other Dietary Components in Its Development. Nutrients 2021, 13, 1442. [Google Scholar] [CrossRef]
- Jonas, W.; Schürmann, A. Genetic and epigenetic factors determining NAFLD risk. Mol. Metab. 2020, 50, 101111. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- Di Costanzo, A.; Belardinilli, F.; Bailetti, D.; Sponziello, M.; D’Erasmo, L.; Polimeni, L.; Baratta, F.; Pastori, D.; Ceci, F.; Montali, A.; et al. Evaluation of Polygenic Determinants of Non-Alcoholic Fatty Liver Disease (NAFLD) By a Candidate Genes Resequencing Strategy. Sci. Rep. 2018, 8, 3702. [Google Scholar] [CrossRef] [PubMed]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e6. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, M.; Rau, M.; Schattenberg, J.M.; Bantel, H.; Pathil, A.; Demir, M.; Kluwe, J.; Boettler, T.; Lammert, F.; Geier, A. NAFLD Clinical Study Group. Combined effects of the PNPLA3 rs738409, TM6SF2 rs58542926, and MBOAT7 rs641738 variants on NAFLD severity: A multicenter biopsy-based study. J. Lipid Res. 2017, 58, 247–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Campo, J.A.; Gallego-Durán, R.; Gallego, P.; Grande, L. Genetic and Epigenetic Regulation in Nonalcoholic Fatty Liver Disease (NAFLD). Int. J. Mol. Sci. 2018, 19, 911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.-C.; Chen, Y.-Z.; Wang, C.-H.; Lin, F.-J. The nonalcoholic fatty liver disease-like phenotype and lowered serum VLDL are associated with decreased expression and DNA hypermethylation of hepatic ApoB in male offspring of ApoE deficient mothers fed a with Western diet. J. Nutr. Biochem. 2020, 77, 108319. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M.; Nonalcoholic Steatohepatitis Clinical Research Network. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [Green Version]
- Lustig, R.H.; Mulligan, K.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Erkin-Cakmak, A.; Gugliucci, A.; Schwarz, J.-M. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obesity 2016, 24, 453–460. [Google Scholar] [CrossRef]
- Nobili, V.; Mosca, A.; de Vito, R.; Raponi, M.; Scorletti, E.; Byrne, C.D. Liver zonation in children with non-alcoholic fatty liver disease: Associations with dietary fructose and uric acid concentrations. Liver Int. 2018, 38, 1102–1109. [Google Scholar] [CrossRef] [Green Version]
- Maersk, M.; Belza, A.; Stødkilde-Jørgensen, H.; Ringgaard, S.; Chabanova, E.; Thomsen, H.; Pedersen, S.B.; Astrup, A.; Richelsen, B. Sucrose-sweetened beverages increase fat storage in the liver, muscle, and visceral fat depot: A 6-mo randomized intervention study. Am. J. Clin. Nutr. 2011, 95, 283–289. [Google Scholar] [CrossRef]
- Baker, P.R.; Friedman, J.E. Mitochondrial role in the neonatal predisposition to developing nonalcoholic fatty liver disease. J. Clin. Investig. 2018, 128, 3692–3703. [Google Scholar] [CrossRef]
- Giannetto, A.; Esposito, E.; Lanza, M.; Oliva, S.; Riolo, K.; di Pietro, S.; Abbate, J.M.; Briguglio, G.; Cassata, G.; Cicero, L.; et al. Protein Hydrolysates from Anchovy (Engraulis encrasicolus) Waste: In Vitro and In Vivo Biological Activities. Mar. Drugs 2020, 18, 86. [Google Scholar] [CrossRef]
- Abbate, J.M.; Macrì, F.; Capparucci, F.; Iaria, C.; Briguglio, G.; Cicero, L.; Salvo, A.; Arfuso, F.; Ieni, A.; Piccione, G.; et al. Administration of Protein Hydrolysates from Anchovy (Engraulis encrasicolus) Waste for Twelve Weeks Decreases Metabolic Dysfunction-Associated Fatty Liver Disease Severity in ApoE–/–Mice. Animals 2020, 10, 2303. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Day, C.P. The genetics of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-W.; Friso, S. Epigenetics: A New Bridge between Nutrition and Health. Adv. Nutr. 2010, 1, 8–16. [Google Scholar] [CrossRef]
- Gautam, S. Epigenetic Mechanism and Current Advances in Epigenetic Programming in the Context of Non-Alcoholic Fatty Liver Disease (NAFLD). J. Clin. Epigenetics 2018, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Leung, A.; Trac, C.; Du, J.; Natarajan, R.; Schones, D.E. Persistent Chromatin Modifications Induced by High Fat Diet*. J. Biol. Chem. 2016, 291, 10446–10455. [Google Scholar] [CrossRef] [Green Version]
- Lyall, M.J.; Thomson, J.P.; Cartier, J.; Ottaviano, R.; Kendall, T.J.; Meehan, R.R.; Drake, A.J. Non-alcoholic fatty liver disease (NAFLD) is associated with dynamic changes in DNA hydroxymethylation. Epigenetics 2020, 15, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Hajri, T.; Zaiou, M.; Fungwe, T.; Ouguerram, K.; Besong, S. Epigenetic Regulation of Peroxisome Proliferator-Activated Receptor Gamma Mediates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 1355. [Google Scholar] [CrossRef]
- Eslam, M.; George, J. Genetic and epigenetic mechanisms of NASH. Hepatol. Int. 2016, 10, 394–406. [Google Scholar] [CrossRef]
- Kaelin, W.G.; McKnight, S.L. Influence of Metabolism on Epigenetics and Disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [Green Version]
- Gut, P.; Verdin, E. The nexus of chromatin regulation and intermediary metabolism. Nature 2013, 502, 489–498. [Google Scholar] [CrossRef]
- Zaiou, M. The Emerging Role and Promise of Circular RNAs in Obesity and Related Metabolic Disorders. Cells 2020, 9, 1473. [Google Scholar] [CrossRef] [PubMed]
- Zaiou, M. Circular RNAs as Potential Biomarkers and Therapeutic Targets for Metabolic Diseases. Adv. Exp. Med. Biol. 2019, 1134, 177–191. [Google Scholar] [CrossRef]
- Zhao, X.-Y.; Xiong, X.; Liu, T.; Mi, L.; Peng, X.; Rui, C.; Guo, L.; Li, S.; Li, X.; Lin, J.D. Long noncoding RNA licensing of obesity-linked hepatic lipogenesis and NAFLD pathogenesis. Nat. Commun. 2018, 9, 2986. [Google Scholar] [CrossRef] [Green Version]
- Zaiou, M.; Bakillah, A. Epigenetic Regulation of ATP-Binding Cassette Protein A1 (ABCA1) Gene Expression: A New Era to Alleviate Atherosclerotic Cardiovascular Disease. Diseases 2018, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, C.; Guo, X. The clinical potential of circulating microRNAs in obesity. Nat. Rev. Endocrinol. 2019, 15, 731–743. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, D.-H.; Park, S.-H.; Jeon, T.-I.; Jung, C.H. The interplay of microRNAs and transcription factors in autophagy regulation in nonalcoholic fatty liver disease. Exp. Mol. Med. 2021, 53, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-Y.; Yang, Y.-L.; Wang, P.-W.; Wang, F.-S.; Huang, Y.-H. The Emerging Role of MicroRNAs in NAFLD: Highlight of MicroRNA-29a in Modulating Oxidative Stress, Inflammation, and Beyond. Cells 2020, 9, 1041. [Google Scholar] [CrossRef]
- Liu, C.H.; Ampuero, J.; Gil-Gómez, A.; Montero-Vallejo, R.; Rojas, Á.; Muñoz-Hernández, R.; Gallego-Durán, R.; Romero-Gómez, M. miRNAs in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2018, 69, 1335–1348. [Google Scholar] [CrossRef]
- Gjorgjieva, M.; Sobolewski, C.; Dolicka, D.; de Sousa, M.C.; Foti, M. miRNAs and NAFLD: From pathophysiology to therapy. Gut 2019, 68, 2065–2079. [Google Scholar] [CrossRef]
- Newman, L.A.; Sorich, M.J.; Rowland, A. Role of Extracellular Vesicles in the Pathophysiology, Diagnosis and Tracking of Non-Alcoholic Fatty Liver Disease. J. Clin. Med. 2020, 9, 2032. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Lee, Y.; Lee, Y.-S.; Gim, J.-A.; Ko, E.; Yim, S.Y.; Jung, Y.K.; Kang, S.; Kim, M.Y.; Kim, H.; et al. Circulating miRNA is a useful diagnostic biomarker for nonalcoholic steatohepatitis in nonalcoholic fatty liver disease. Sci. Rep. 2021, 11, 14639. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Dou, G.; Wang, L. MicroRNAs in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Int. J. Biol. Sci. 2021, 17, 1851–1863. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, O.; Errafii, K.; Al-Akl, N.S.; Arredouani, A. Noncoding RNAs in Nonalcoholic Fatty Liver Disease: Potential Diagnosis and Prognosis Biomarkers. Dis. Markers 2020, 2020, 8822859. [Google Scholar] [CrossRef]
- Sulaiman, S.A.; Muhsin, N.I.A.; Jamal, R. Regulatory Non-coding RNAs Network in Non-alcoholic Fatty Liver Disease. Front. Physiol. 2019, 10, 279. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.; Kohlbacher, O.; de Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nat. Cell Biol. 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Aguilera, O.; Depreux, P.; Halby, L.; Arimondo, P.B.; Goossens, L. DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge. Biomolecules 2017, 7, 3. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’His, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Page, A.; Paoli, P.; Salvador, E.M.; White, S.; French, J.; Mann, J. Hepatic stellate cell transdifferentiation involves genome-wide remodeling of the DNA methylation landscape. J. Hepatol. 2016, 64, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Bestor, T.H.; Edwards, J.R.; Boulard, M. Notes on the role of dynamic DNA methylation in mammalian development. Proc. Natl. Acad. Sci. USA 2015, 112, 6796–6799. [Google Scholar] [CrossRef] [Green Version]
- Barres, R.; Osler, M.E.; Yan, J.; Rune, A.; Fritz, T.; Caidahl, K.; Krook, A.; Zierath, J.R. Non-CpG Methylation of the PGC-1α Promoter through DNMT3B Controls Mitochondrial Density. Cell Metab. 2009, 10, 189–198. [Google Scholar] [CrossRef] [Green Version]
- González-Becerra, K.; Ramos-Lopez, O.; Barrón-Cabrera, E.; Riezu-Boj, J.I.; Milagro, F.I.; Martinez-Lopez, E.; Martínez, J.A. Fatty acids, epigenetic mechanisms and chronic diseases: A systematic review. Lipids Health Dis. 2019, 18, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, H.; Zhou, D.; Moody, L.; Lezmi, S.; Chen, H.; Pan, Y.-X. High-fat diet caused widespread epigenomic differences on hepatic methylome in rat. Physiol. Genom. 2015, 47, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Zwamborn, R.A.J.; Slieker, R.C.; Mulder, P.C.A.; Zoetemelk, I.; Verschuren, L.; Suchiman, H.E.D.; Toet, K.H.; Droog, S.; Slagboom, P.E.; Kooistra, T.; et al. Prolonged high-fat diet induces gradual and fat depot-specific DNA methylation changes in adult mice. Sci. Rep. 2017, 7, 43261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Matinez, G.A.; Rodríguez-Ríos, D.; Alvarado-Caudillo, Y.; Vaquero, A.; Esteller, M.; Carmona, F.J.; Moran, S.; Nielsen, F.C.; Wickström-Lindholm, M.; Wrobel, K.; et al. Arachidonic and oleic acid exert distinct effects on the DNA methylome. Epigenetics 2016, 11, 321–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wankhade, U.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Thakali, K.M.; Shankar, K. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS ONE 2017, 12, e0175675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, K.J.; Sloboda, D.M.; Connor, K.L.; Beltrand, J.; Vickers, M.H. Offspring of Mothers Fed a High Fat Diet Display Hepatic Cell Cycle Inhibition and Associated Changes in Gene Expression and DNA Methylation. PLoS ONE 2011, 6, e21662. [Google Scholar] [CrossRef] [Green Version]
- Cordero, P.; Campion, J.; Milagro, F.; Martinez, J.A. Transcriptomic and epigenetic changes in early liver steatosis associated to obesity: Effect of dietary methyl donor supplementation. Mol. Genet. Metab. 2013, 110, 388–395. [Google Scholar] [CrossRef]
- Farkas, S.A.; Befekadu, R.; Hahn-Strömberg, V.; Nilsson, T.K. DNA methylation and expression of the folate transporter genes in colorectal cancer. Tumor Biol. 2015, 36, 5581–5590. [Google Scholar] [CrossRef]
- Pogribny, I.P.; Tryndyak, V.P.; Bagnyukova, T.V.; Melnyk, S.; Montgomery, B.; Ross, S.A.; Latendresse, J.R.; Rusyn, I.; Beland, F.A. Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J. Hepatol. 2009, 51, 176–186. [Google Scholar] [CrossRef] [Green Version]
- Mato, J.M.; Chantar, M.L.M.; Lu, S.C. S-adenosylmethionine metabolism and liver disease. Ann. Hepatol. 2013, 12, 183–189. [Google Scholar] [CrossRef]
- Chang, X.; Yan, H.; Fei, J.; Jiang, M.; Zhu, H.; Lu, D.; Gao, X. Berberine reduces methylation of the MTTP promoter and alleviates fatty liver induced by a high-fat diet in rats. J. Lipid Res. 2010, 51, 2504–2515. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Fan, J.-G.; Qiao, L. Potential Epigenetic Mechanism in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2015, 16, 5161–5179. [Google Scholar] [CrossRef]
- Da Silva, R.P.; Kelly, K.B.; Al Rajabi, A.; Jacobs, R.L. Novel insights on interactions between folate and lipid metabolism. BioFactors 2014, 40, 277–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.-J.; Zhang, H.-W.; Zhou, J.-Y.; Liu, Y.; Yang, Y.; Chen, X.-L.; Zhu, C.-H.; Zheng, R.-D.; Ling, W.-H.; Zhu, H.-L. Betaine attenuates hepatic steatosis by reducing methylation of the MTTP promoter and elevating genomic methylation in mice fed a high-fat diet. J. Nutr. Biochem. 2014, 25, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Shiraishi, S.; Kishimoto, K.; Miura, S.; Ezaki, O. An increase in liver PPARγ2 is an initial event to induce fatty liver in response to a diet high in butter: PPARγ2 knockdown improves fatty liver induced by high-saturated fat. J. Nutr. Biochem. 2011, 22, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Moreno, M.; Lucano-Landeros, S.; Monroy-Ramirez, H.C.; Silva-Gomez, J.; Gutierrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Roles of Nrf2 in Liver Diseases: Molecular, Pharmacological, and Epigenetic Aspects. Antioxidants 2020, 9, 980. [Google Scholar] [CrossRef]
- Hosseini, H.; Teimouri, M.; Shabani, M.; Koushki, M.; Khorzoughi, R.B.; Namvarjah, F.; Izadi, P.; Meshkani, R. Resveratrol alleviates non-alcoholic fatty liver disease through epigenetic modification of the Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2020, 119, 105667. [Google Scholar] [CrossRef]
- Fernández-Tussy, P.; Fernández-Ramos, D.; Lopitz-Otsoa, F.; Simón, J.; Barbier-Torres, L.; Gomez-Santos, B.; Nuñez-Garcia, M.; Azkargorta, M.; Juan, V.G.-D.; Serrano-Macia, M.; et al. miR-873-5p targets mitochondrial GNMT-Complex II interface contributing to non-alcoholic fatty liver disease. Mol. Metab. 2019, 29, 40–54. [Google Scholar] [CrossRef]
- Borowa-Mazgaj, B.; de Conti, A.; Tryndyak, V.; Steward, C.R.; Jimenez, L.; Melnyk, S.; Seneshaw, M.; Mirshahi, F.; Rusyn, I.; Beland, F.A.; et al. Gene Expression and DNA Methylation Alterations in the Glycine N-Methyltransferase Gene in Diet-Induced Nonalcoholic Fatty Liver Disease-Associated Carcinogenesis. Toxicol. Sci. 2019, 170, 273–282. [Google Scholar] [CrossRef]
- Barbier-Torres, L.; Fortner, K.A.; Iruzubieta, P.; Delgado, T.C.; Giddings, E.; Chen, Y.; Champagne, D.; Fernández-Ramos, D.; Mestre, D.; Gomez-Santos, B.; et al. Silencing hepatic MCJ attenuates non-alcoholic fatty liver disease (NAFLD) by increasing mitochondrial fatty acid oxidation. Nat. Commun. 2020, 11, 3360. [Google Scholar] [CrossRef]
- Ahrens, M.; Ammerpohl, O.; von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA Methylation Analysis in Nonalcoholic Fatty Liver Disease Suggests Distinct Disease-Specific and Remodeling Signatures after Bariatric Surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeybel, M.; Hardy, T.; Robinson, S.M.; Fox, C.; Anstee, Q.M.; Ness, T.; Masson, S.; Mathers, J.C.; French, J.; White, S.; et al. Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin. Epigenetics 2015, 7, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Z.; Chen, J.; Ding, C.; Wong, K.; Chen, X.; Pu, L.; Huang, Q.; Chen, X.; Cheng, Z.; Liu, Y.; et al. Association of Hepatic Global DNA Methylation and Serum One-Carbon Metabolites with Histological Severity in Patients with NAFLD. Obesity 2020, 28, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueño, A.L.; Gianotti, T.F.; Castaño, G.O.; Pirola, C.J. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator-activated receptor γ coactivator 1α promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Gianotti, T.F.; Burgueño, A.L.; Rey-Funes, M.; Loidl, C.F.; Mallardi, P.; Martino, J.S.; Castaño, G.O.; Sookoian, S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2012, 62, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Hyun, J.; Jung, Y. DNA Methylation in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 8138. [Google Scholar] [CrossRef]
- Tian, Y.; Wong, V.W.-S.; Chan, H.L.-Y.; Cheng, A.S.-L. Epigenetic regulation of hepatocellular carcinoma in non-alcoholic fatty liver disease. Semin. Cancer Biol. 2013, 23, 471–482. [Google Scholar] [CrossRef]
- Hardy, T.; Zeybel, M.; Day, C.P.; Dipper, C.; Masson, S.; McPherson, S.; Henderson, E.; Tiniakos, D.; White, S.; French, J.; et al. Plasma DNA methylation: A potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut 2017, 66, 1321–1328. [Google Scholar] [CrossRef]
- Zeybel, M.; Hardy, T.; Wong, Y.K.; Mathers, J.C.; Fox, C.R.; Gackowska, A.; Oakley, F.; Burt, A.D.; Wilson, C.L.; Anstee, Q.M.; et al. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat. Med. 2012, 18, 1369–1377. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhou, W.; Wu, Y.; Xu, L.; Wang, Y.; Xu, Z.; Yang, Y. Circulating IGFBP-2 levels are inversely associated with the incidence of nonalcoholic fatty liver disease: A cohort study. J. Int. Med Res. 2020, 48, 300060520935219. [Google Scholar] [CrossRef]
- Fahlbusch, P.; Knebel, B.; Hörbelt, T.; Barbosa, D.M.; Nikolic, A.; Jacob, S.; Al-Hasani, H.; van de Velde, F.; van Nieuwenhove, Y.; Müller-Wieland, D.; et al. Physiological Disturbance in Fatty Liver Energy Metabolism Converges on IGFBP2 Abundance and Regulation in Mice and Men. Int. J. Mol. Sci. 2020, 21, 4144. [Google Scholar] [CrossRef]
- Murphy, S.K.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.; Garrett, M.E.; Ashley-Koch, A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship Between Methylome and Transcriptome in Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2013, 145, 1076–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Li, L.; Li, M.; Zhang, X.; Hao, C.; Yu, L.; Zeng, S.; Xu, H.; Fang, M.; Shen, A.; et al. The histone methyltransferase Suv39h2 contributes to nonalcoholic steatohepatitis in mice. Hepatology 2017, 65, 1904–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, M.; Jellyman, J.K.; Ross, M.G. Epigenomics, gestational programming and risk of metabolic syndrome. Int. J. Obes. 2015, 39, 633–641. [Google Scholar] [CrossRef]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harr, J.; Sandoval, A.V.G.; Gasser, S.M. Histones and histone modifications in perinuclear chromatin anchoring: From yeast to man. EMBO Rep. 2016, 17, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Groop, L. Epigenetics: A Molecular Link Between Environmental Factors and Type 2 Diabetes. Diabetes 2009, 58, 2718–2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, A.; Lopaschuk, G.D. Acetylation control of cardiac fatty acid β-oxidation and energy metabolism in obesity, diabetes, and heart failure. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2016, 1862, 2211–2220. [Google Scholar] [CrossRef]
- Chung, M.-Y.; Song, J.-H.; Lee, J.; Shin, E.J.; Park, J.H.; Lee, S.-H.; Hwang, J.-T.; Choi, H.-K. Tannic acid, a novel histone acetyltransferase inhibitor, prevents non-alcoholic fatty liver disease both in vivo and in vitro model. Mol. Metab. 2019, 19, 34–48. [Google Scholar] [CrossRef]
- Morral, N.; Liu, S.; Conteh, A.M.; Chu, X.; Wang, Y.; Dong, X.C.; Liu, Y.; Linnemann, A.K.; Wan, J. Aberrant gene expression induced by a high fat diet is linked to H3K9 acetylation in the promoter-proximal region. Biochim. Biophys. Acta (BBA)—Bioenerg. 2021, 1864, 194691. [Google Scholar] [CrossRef]
- Jun, H.-J.; Kim, J.; Hoang, M.-H.; Lee, S.-J. Hepatic Lipid Accumulation Alters Global Histone H3 Lysine 9 and 4 Trimethylation in the Peroxisome Proliferator-Activated Receptor Alpha Network. PLoS ONE 2012, 7, e44345. [Google Scholar] [CrossRef] [PubMed]
- Bricambert, J.; Miranda, J.; Benhamed, F.; Girard, J.; Postic, C.; Dentin, R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J. Clin. Investig. 2010, 120, 4316–4331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-H.; Kim, J.; Kwon, J.-S.; Sandhu, J.; Tontonoz, P.; Lee, S.-K.; Lee, S.; Lee, J.W. Critical Roles of the Histone Methyltransferase MLL4/KMT2D in Murine Hepatic Steatosis Directed by ABL1 and PPARγ2. Cell Rep. 2016, 17, 1671–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-H.; Jung, D.Y.; Nagappan, A.; Jung, M.H. Histone H3K9 demethylase JMJD2B induces hepatic steatosis through upregulation of PPARγ2. Sci. Rep. 2018, 8, 13734. [Google Scholar] [CrossRef]
- Schwer, B.; Verdin, E. Conserved Metabolic Regulatory Functions of Sirtuins. Cell Metab. 2008, 7, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colak, Y.; Yesil, A.; Mutlu, H.H.; Caklili, O.T.; Ulasoglu, C.; Senates, E.; Takir, M.; Kostek, O.; Yilmaz, Y.; Enc, F.Y.; et al. A Potential Treatment of Non-Alcoholic Fatty Liver Disease with SIRT1 Activators. J. Gastrointest. Liver Dis. 2014, 23, 311–319. [Google Scholar] [CrossRef]
- Sathyanarayan, A.; Mashek, M.T.; Mashek, D. ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism. Cell Rep. 2017, 19, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Cao, Y.; Xue, Y.; Xue, L.; Jiang, X.; Wang, X.; Zhang, Z.; Yang, J.; Lu, J.; Zhang, C.; Wang, W.; et al. Hepatic menin recruits SIRT1 to control liver steatosis through histone deacetylation. J. Hepatol. 2013, 59, 1299–1306. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-Specific Deletion of SIRT1 Alters Fatty Acid Metabolism and Results in Hepatic Steatosis and Inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Vilà, L.; Elias, I.; Roca, C.; Ribera, A.; Ferre, T.; Casellas, A.; Lage, R.; Franckhauser, S.; Bosch, F. AAV8-mediated Sirt1 gene transfer to the liver prevents high carbohydrate diet-induced nonalcoholic fatty liver disease. Mol. Ther.—Methods Clin. Dev. 2014, 1, 14039. [Google Scholar] [CrossRef]
- Deng, X.-Q.; Chen, L.-L.; Li, N.-X. The expression of SIRT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver Int. 2007, 27, 708–715. [Google Scholar] [CrossRef]
- Mariani, S.; Fiore, D.; Basciani, S.; Persichetti, A.; Contini, S.; Lubrano, C.; Salvatori, L.; Lenzi, A.; Gnessi, L. Plasma levels of SIRT1 associate with non-alcoholic fatty liver disease in obese patients. Endocrine 2015, 49, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Herranz, D.; Muñoz-Martin, M.; Cañamero, M.; Mulero, F.; Martinez-Pastor, B.; Fernandez-Capetillo, O.; Serrano, M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat. Commun. 2010, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; He, Z.; Sun, X.; Gu, X.; Zhang, W.; Gao, J.; Li, X.; Jia, R.; Wei, J.; Yu, Y.; et al. DHA Protects Against Hepatic Steatosis by Activating Sirt1 in a High Fat Diet-Induced Nonalcoholic Fatty Liver Disease Mouse Model. Diabetes Metab. Syndr. Obesity Targets Ther. 2020, 13, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.-L.; Bonine-Summers, A.R.; et al. Hdac3 Is Essential for the Maintenance of Chromatin Structure and Genome Stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Miller, R.A.; Patel, R.T.; Chen, J.; Dhir, R.; Wang, H.; Zhang, D.; Graham, M.J.; Unterman, T.G.; Shulman, G.; et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat. Med. 2012, 18, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Liu, T.; Sun, Z.; Bugge, A.; Mullican, S.E.; Alenghat, T.; Liu, X.S.; Lazar, M.A. A Circadian Rhythm Orchestrated by Histone Deacetylase 3 Controls Hepatic Lipid Metabolism. Science 2011, 331, 1315–1319. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Wong, V.W.; Wong, G.L.-H.; Yang, W.; Sun, H.; Shen, J.; Tong, J.H.; Go, M.Y.; Cheung, Y.S.; Lai, P.B.; et al. Histone Deacetylase HDAC8 Promotes Insulin Resistance and β-Catenin Activation in NAFLD-Associated Hepatocellular Carcinoma. Cancer Res. 2015, 75, 4803–4816. [Google Scholar] [CrossRef] [Green Version]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Reinberg, D. Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails. Genes Dev. 2001, 15, 2343–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vella, S.; Gnani, D.; Crudele, A.; Ceccarelli, S.; de Stefanis, C.; Gaspari, S.; Nobili, V.; Locatelli, F.; Marquez, V.E.; Rota, R.; et al. EZH2 Down-Regulation Exacerbates Lipid Accumulation and Inflammation in In Vitro and In Vivo NAFLD. Int. J. Mol. Sci. 2013, 14, 24154–24168. [Google Scholar] [CrossRef] [Green Version]
- Mosammaparast, N.; Shi, Y. Reversal of Histone Methylation: Biochemical and Molecular Mechanisms of Histone Demethylases. Annu. Rev. Biochem. 2010, 79, 155–179. [Google Scholar] [CrossRef] [PubMed]
- Viscarra, J.A.; Wang, Y.; Nguyen, H.; Choi, Y.G.; Sul, H.S. Histone demethylase JMJD1C is phosphorylated by mTOR to activate de novo lipogenesis. Nat. Commun. 2020, 11, 796. [Google Scholar] [CrossRef]
- Kim, J.-H.; Jung, D.Y.; Kim, H.-R.; Jung, M.H. Histone H3K9 Demethylase JMJD2B Plays a Role in LXRα-Dependent Lipogenesis. Int. J. Mol. Sci. 2020, 21, 8313. [Google Scholar] [CrossRef] [PubMed]
- Bricambert, J.; Alves-Guerra, M.-C.; Esteves, P.; Prip-Buus, C.; Bertrand-Michel, J.; Guillou, H.; Chang, C.J.; Wal, M.N.V.; Canonne-Hergaux, F.; Mathurin, P.; et al. The histone demethylase Phf2 acts as a molecular checkpoint to prevent NAFLD progression during obesity. Nat. Commun. 2018, 9, 2092. [Google Scholar] [CrossRef] [Green Version]
- Hardy, T.; Mann, D.A. Epigenetics in liver disease: From biology to therapeutics. Gut 2016, 65, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Oussalah, A.; Rischer, S.; Bensenane, M.; Conroy, G.; Filhine-Tresarrieu, P.; Debard, R.; Forest-Tramoy, D.; Josse, T.; Reinicke, D.; Garcia, M.; et al. Plasma mSEPT9: A Novel Circulating Cell-free DNA-Based Epigenetic Biomarker to Diagnose Hepatocellular Carcinoma. EBioMedicine 2018, 30, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Huan, T.; Joehanes, R.; McKeown, N.M.; Horvath, S.; Levy, D.; Ma, J. Higher diet quality relates to decelerated epigenetic aging. Am. J. Clin. Nutr. 2021, 201, nqab201. [Google Scholar] [CrossRef]
- Ma, J.; Rebholz, C.M.; Braun, K.V.; Reynolds, L.M.; Aslibekyan, S.; Xia, R.; Biligowda, N.G.; Huan, T.; Liu, C.; Mendelson, M.M.; et al. Whole Blood DNA Methylation Signatures of Diet Are Associated with Cardiovascular Disease Risk Factors and All-Cause Mortality. Circ. Genom. Precis. Med. 2020, 13, 002766. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Meng, J.; Su, R.; Zhang, J.; Chen, J.; Ma, X.; Xia, Q. Epitranscriptomics in liver disease: Basic concepts and therapeutic potential. J. Hepatol. 2020, 73, 664–679. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Zhang, Z.; Tai, L.; Zhang, L.; Sun, Z.; Zhou, L. Comprehensive analysis of differences of N6-methyladenosine RNA methylomes between high-fat-fed and normal mouse livers. Epigenomics 2019, 11, 1267–1282. [Google Scholar] [CrossRef] [PubMed]
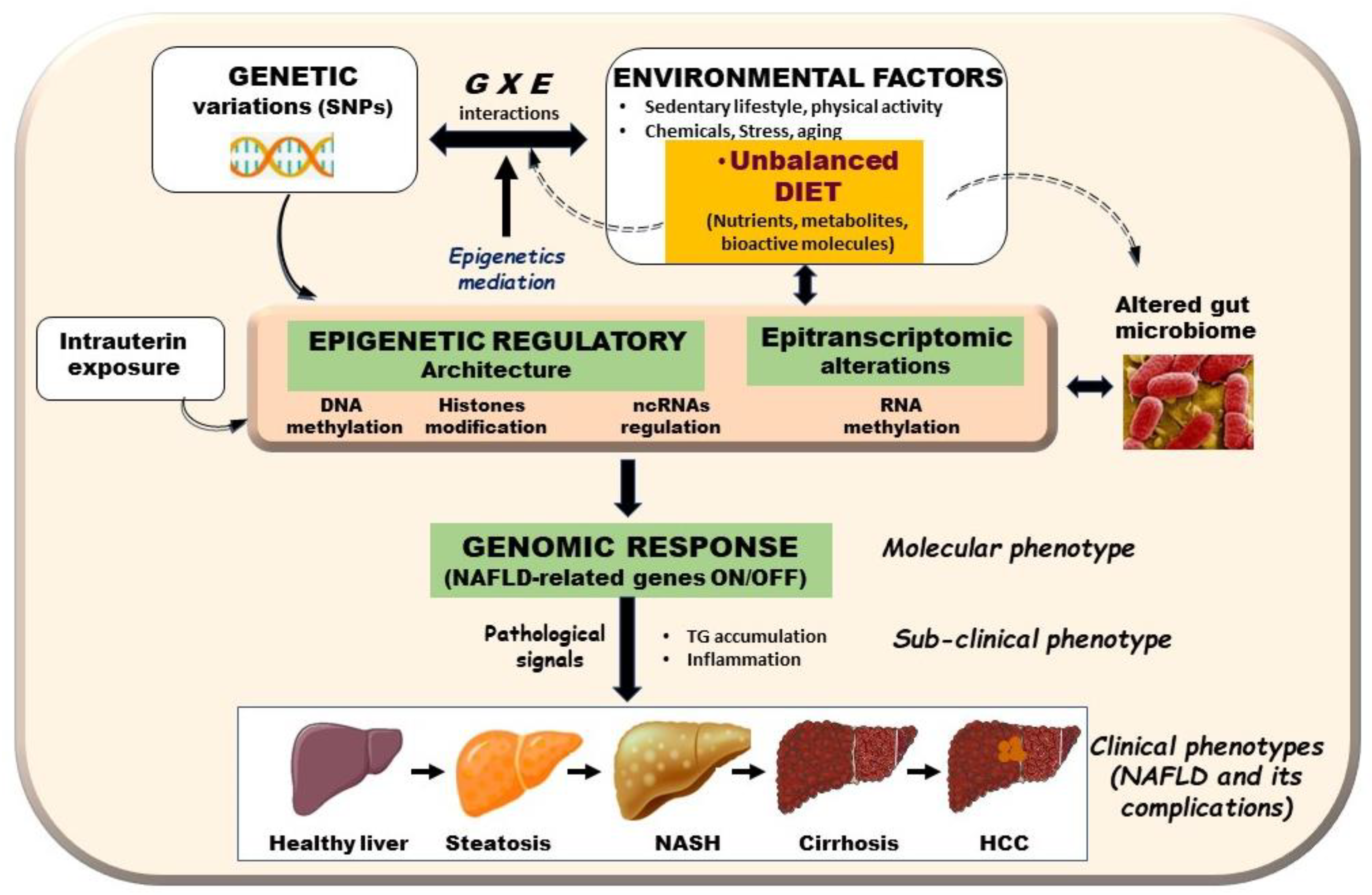

{kind=link}
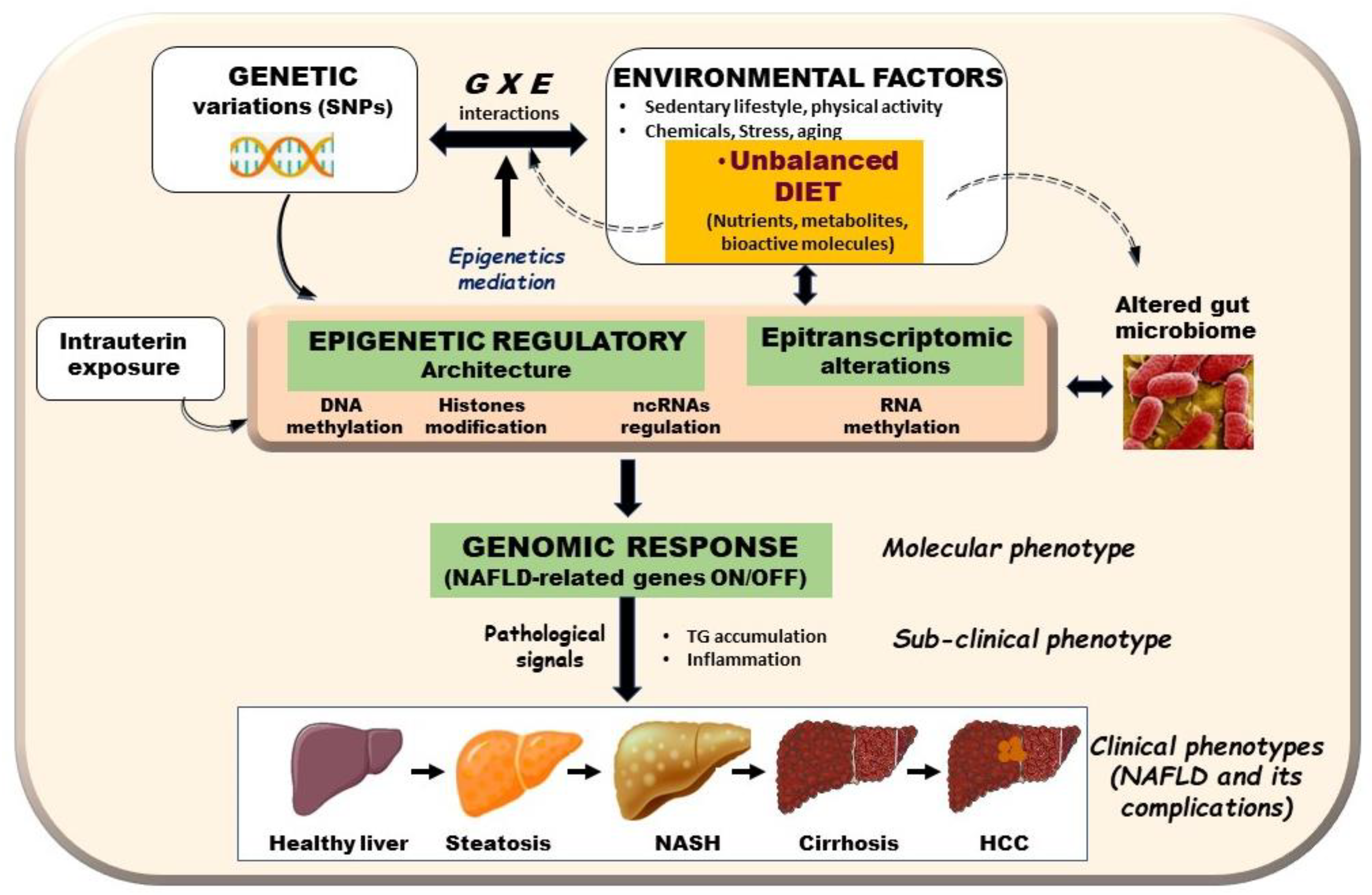{kind=link}
| Gene | Stage | Associated Disease Mechanisms | References |
|---|---|---|---|
| Srebf2 | Hepatic steatosis | Supplementation with methyl donors containing folic acid, choline, betaine, and Vitamin B12 improved liver steatosis by reversing the methylation status in the promoter region of sterol regulatory element binding transcription factor 2 (Srebf2) | [68,73] |
| Mttp | Hepatic steatosis | Betaine supplementation decreased DNA methylation of the microsomal triglyceride transfer protein (Mttp) gene promoter in mice and improved HFD-induced hepatic steatosis | [75] |
| Pparγ | NAFLD | HFD and palmitic acid alter Pparγ promoter DNA methylation leading to a significant induction of PPARγ expression and enhanced fat accumulation in mice liver, which may lead to NAFLD | [38] |
| Nrf2 | NAFLD | Treatment of HepG2 cells with high glucose enhanced methylation level of the Nrf2 promoter whereas Resveratrol reversed the effect, which led to a reduction in TG levels and the expression of lipogenesis-related genes | [78] |
| Gnmt | HCC | Reduced Gnmt expression caused by promoter cytosine DNA hypermethylation is one of the key molecular events in the development of NAFLD-derived HCC | [80] |
| Pparγ | NAFLD | Hypermethylation at the Pparγ promoter of plasma DNA correlated with with fibrosis severity in patients with NAFLD | [89] |
| PGC1-α | NAFLD | Hepatic DNA methylation of of PPARγ coactivator 1- α (PGC1-α) promoter significantly correlates with peripheral insulin resistance and is associated with decreased PGC1-α mRNA expression | [85] |
| Pparα, Pparγ, TGFβ1, Collagen 1A1,PDGFα | NAFLDfibrosis | DNA methylation at specific CpGs within PPARα, PPARγ, TGFβ1, Collagen 1A1, and PDGFα genes can distinguish mild from severe fibrosis in NAFLD patients | [83] |
| IGFBP2 | NASH | The IGFBP2 (insulin-like growth factor binding protein 2) locuswas hypermethylated and its mRNA downregulated in NASH | [82] |
| MT-ND6 | NAFLD | Hepatic methylation and transcriptional activity of the MT-ND6 gene are significantly associated with the histological severity of NAFLD | [86] |
| Sirt1, Pparγ | NAFLD | Suv39h2 is significantly elevated in diet-induced obese mice and NAFLD patients, and it increases the methylation levels at histone H3 lysine 9 of both Sirt1 and Pparγ to suppress the gene expression | [94] |
| Gene | Stage | Association between Epigenetic Determinant and Gene Expression | References |
|---|---|---|---|
| SREBP1c, FASN ACLYS, Pparγ | NAFLD | Blocking the hyperacetylation of lysine 9 and 36 at histone 3 (H3K9 and H3K36) in the promoter of lipogenesis-related genes (SREBP1c, FASN, ACLYS, Pparγ) prevented NAFLD | [100] |
| Pparα | NAFLDSteatosis | Hepatic lipid accumulation induced aberrant H3K9me3 and H3K4me3 status in Pparα gene and other hepatic lipid catabolism network genes, which may contribute to hepatic steatosis and the pathogenesis of NAFLD | [102] |
| ChREBP | Hepatic Steatosis | p300 associates and regulates carbohydrate-responsive element–binding protein (ChREBP) transcriptional activity by acetylation. Inhibition of hepatic p300 activity may be beneficial for treating hepatic steatosis | [103] |
| Pparγ2 | Hepatic steatosis | Histone H3 lysine 4 (H3K4) methyltransferase MLL4/KMT2D directs overnutrition-induced murine steatosis via its coactivator function for PPARγ2 | [104] |
| Pparγ2, CD36, FABP4, PLIN2, CIDEC, | Hepatic steatosis | Overexpressing JMJD2B upregulated Pparγ2 expression which lead to a concomitant increase in its steatosis target genes by removing repressive histone marks H3K9me2 and H3K9me3 near LXREs of lipogenic gene promoters leading to the development of NAFLD | [105] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaiou, M.; Amrani, R.; Rihn, B.; Hajri, T. Dietary Patterns Influence Target Gene Expression through Emerging Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease. Biomedicines 2021, 9, 1256. https://doi.org/10.3390/biomedicines9091256
Zaiou M, Amrani R, Rihn B, Hajri T. Dietary Patterns Influence Target Gene Expression through Emerging Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease. Biomedicines. 2021; 9(9):1256. https://doi.org/10.3390/biomedicines9091256
Chicago/Turabian StyleZaiou, Mohamed, Rim Amrani, Bertrand Rihn, and Tahar Hajri. 2021. "Dietary Patterns Influence Target Gene Expression through Emerging Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease" Biomedicines 9, no. 9: 1256. https://doi.org/10.3390/biomedicines9091256
APA StyleZaiou, M., Amrani, R., Rihn, B., & Hajri, T. (2021). Dietary Patterns Influence Target Gene Expression through Emerging Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease. Biomedicines, 9(9), 1256. https://doi.org/10.3390/biomedicines9091256