
The Cholinergic Anti-Inflammatory Pathway Attenuates the Development of Atherosclerosis in Apoe-/- Mice through Modulating Macrophage Functions

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
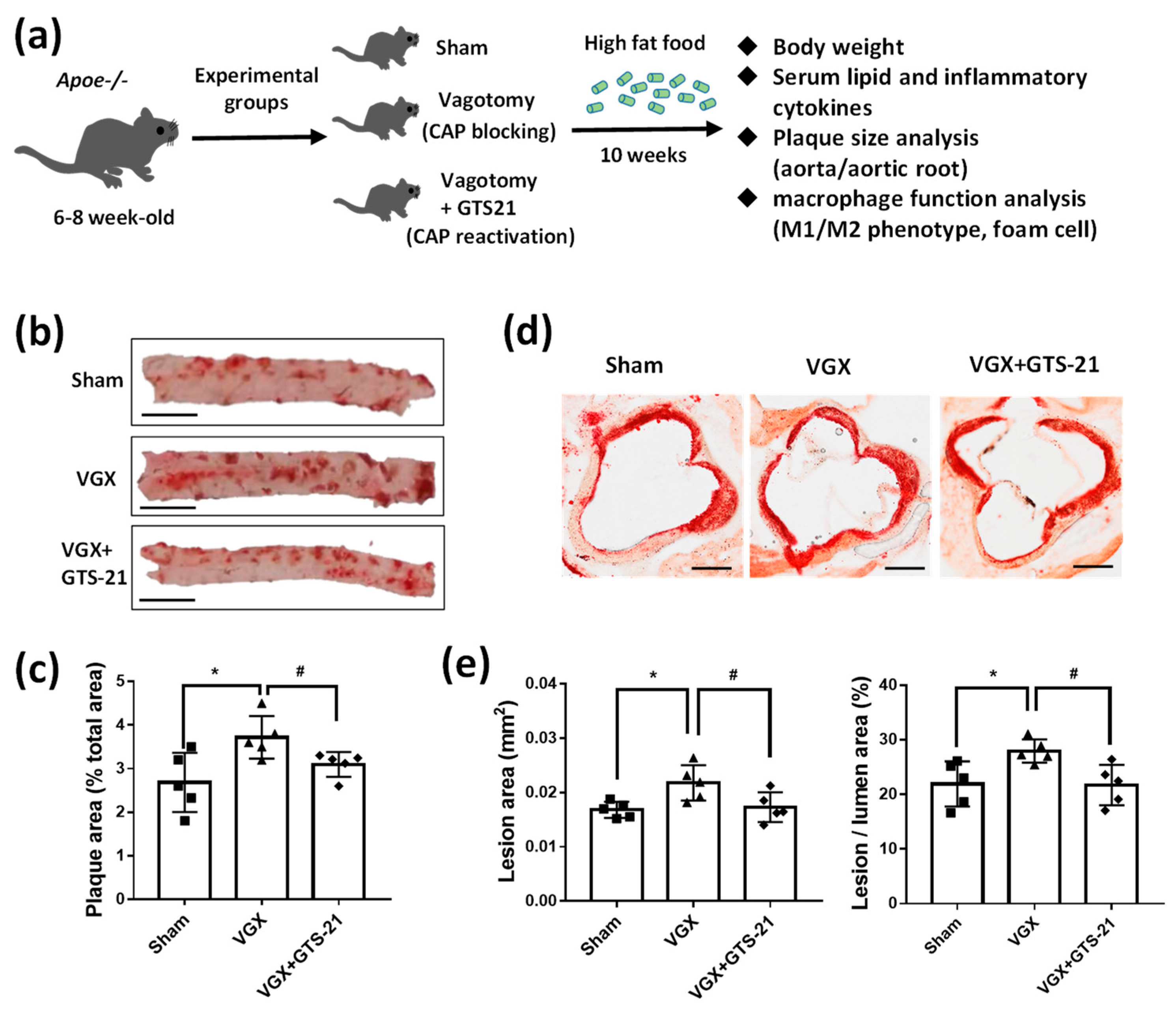
2.1. Animals and Treatments
2.2. Serum Lipid and Cytokines Analysis
2.3. Oil-Red-O Analysis of Atherosclerotic Lesion
2.4. Flow Cytometry
2.5. Cell Culture and Treatment
2.6. Evaluation of Foam Cell Formation
2.7. RNA Extraction and qRT-PCR
2.8. Western Blotting
2.9. Transfection of Small Interfering RNA (siRNA)
2.10. Cholesterol Efflux Assay
2.11. Statistical Analysis
3. Results
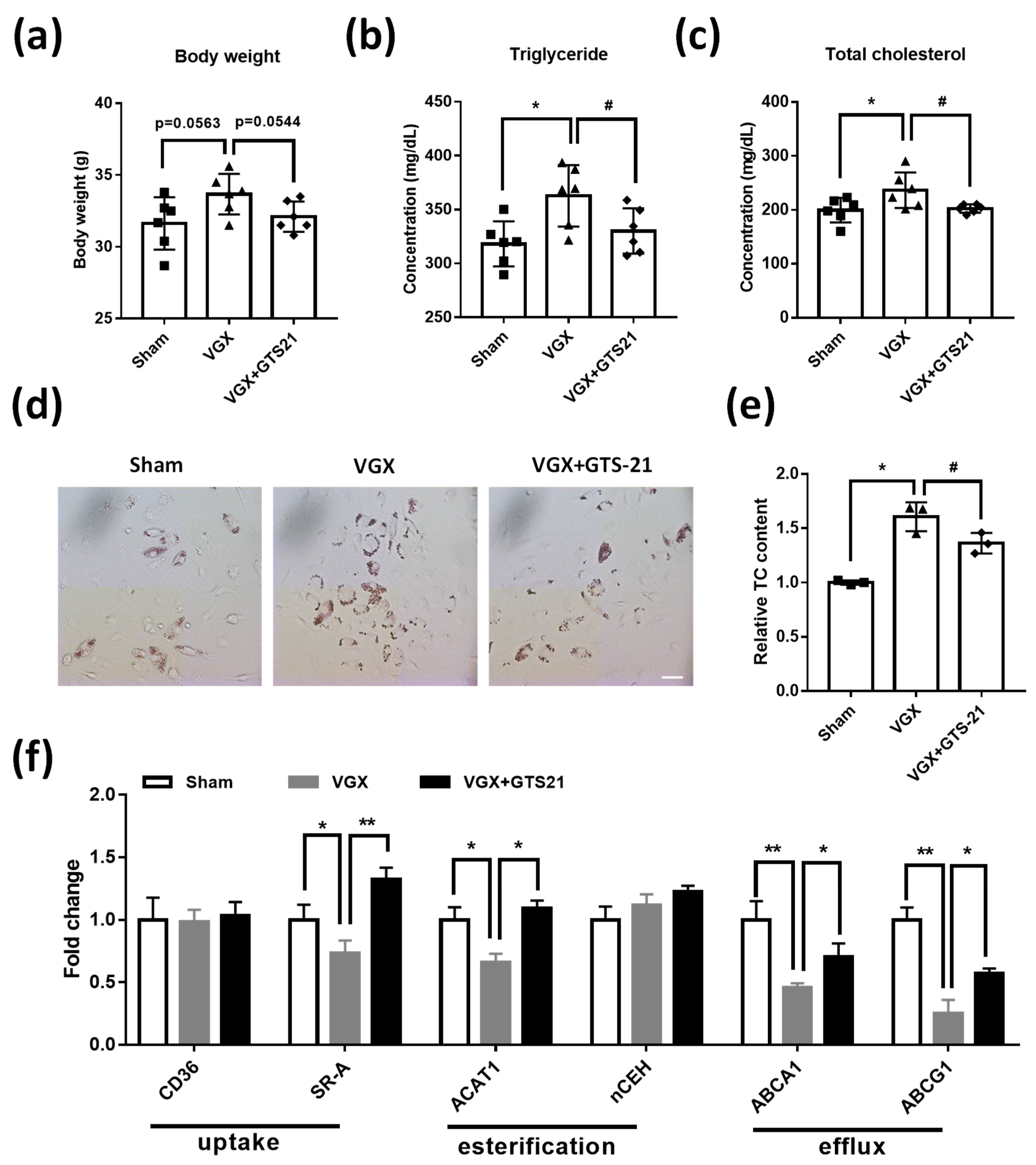
3.1. Effect of CAP Activity on Lipid Plaque Formation and Inflammatory Responses
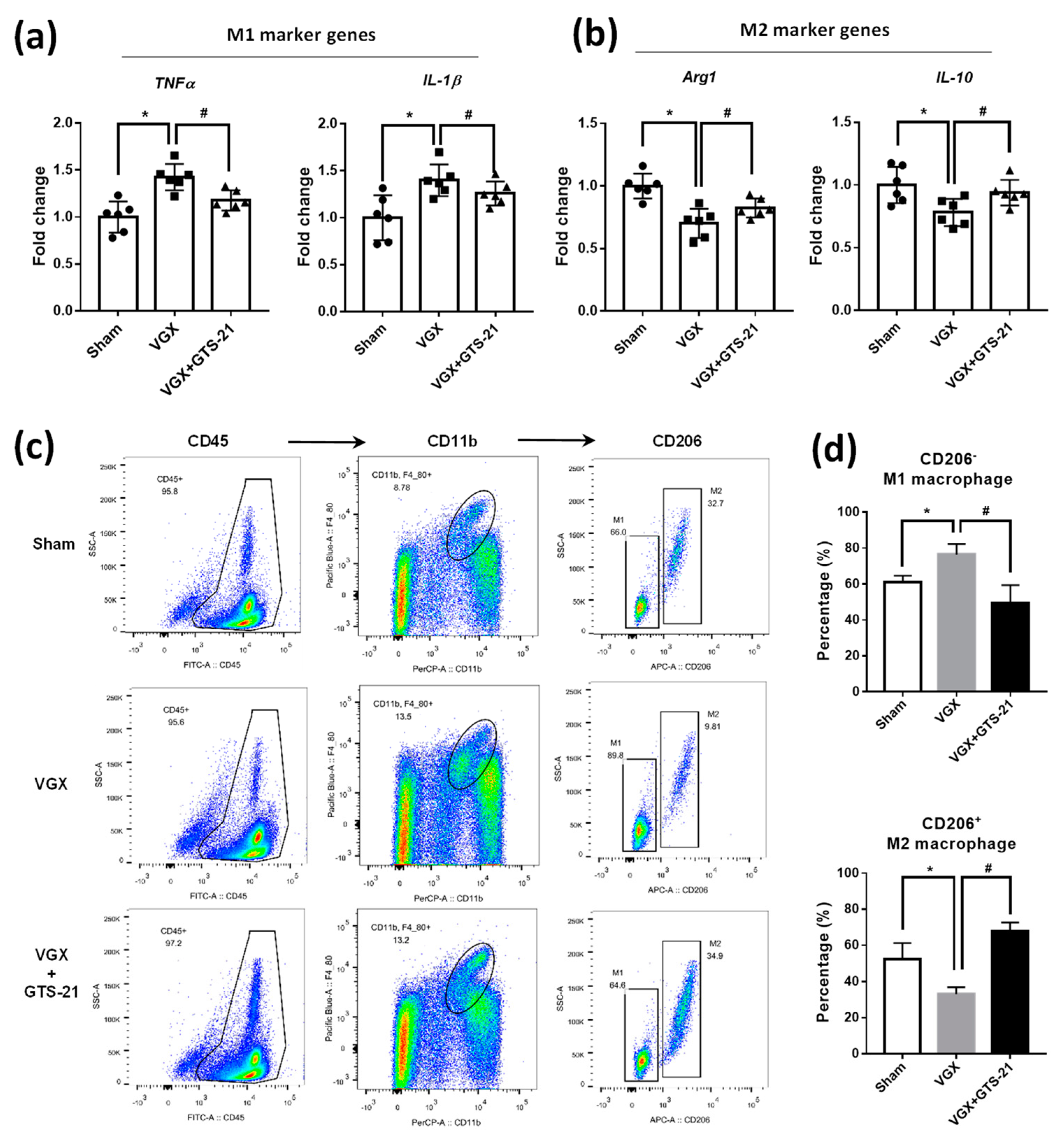
3.2. Effect of CAP Activity on the Abundance of M1 and M2 Macrophages
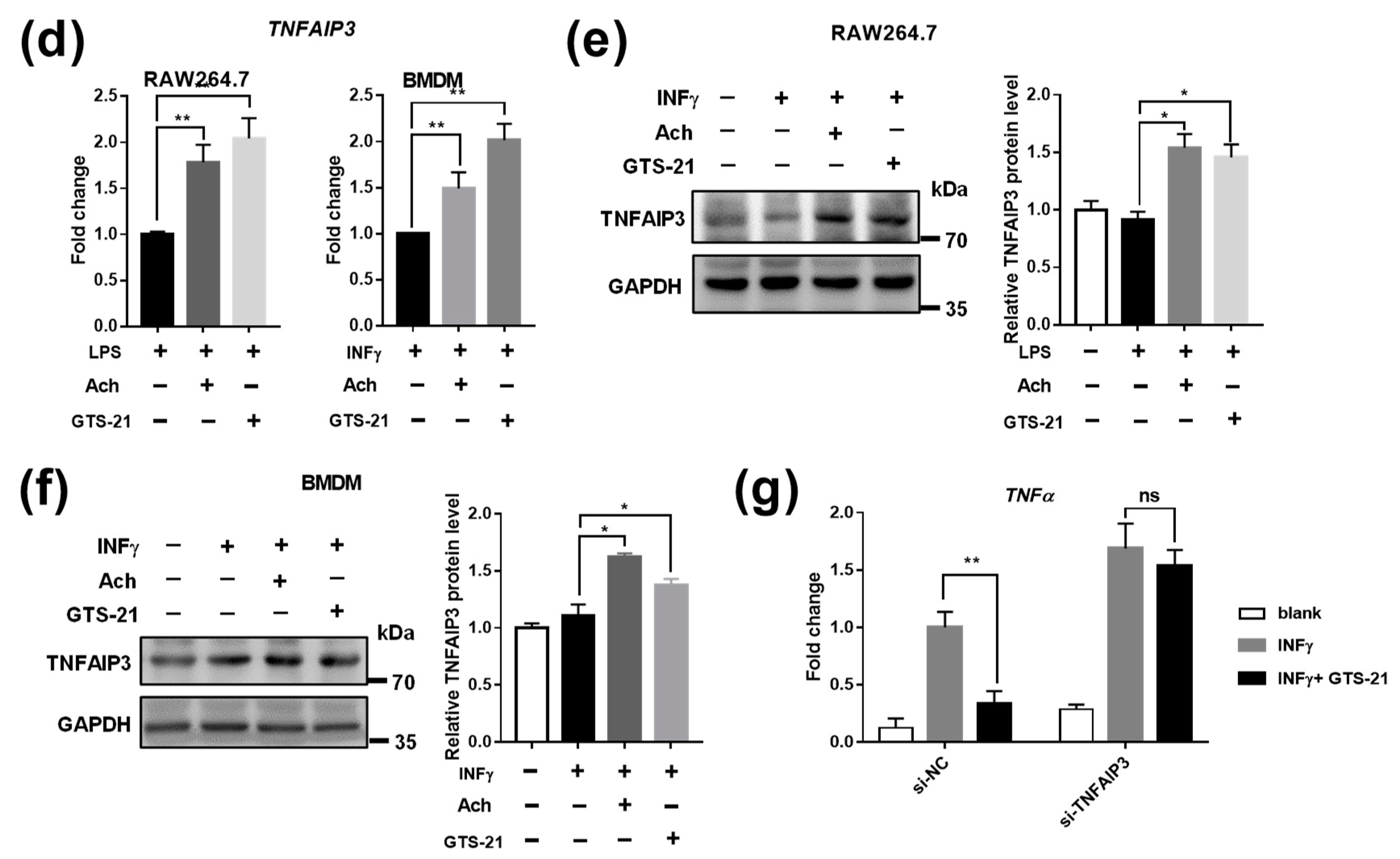
3.3. Effects of CAP Agonists on M1 Macrophage Polarization
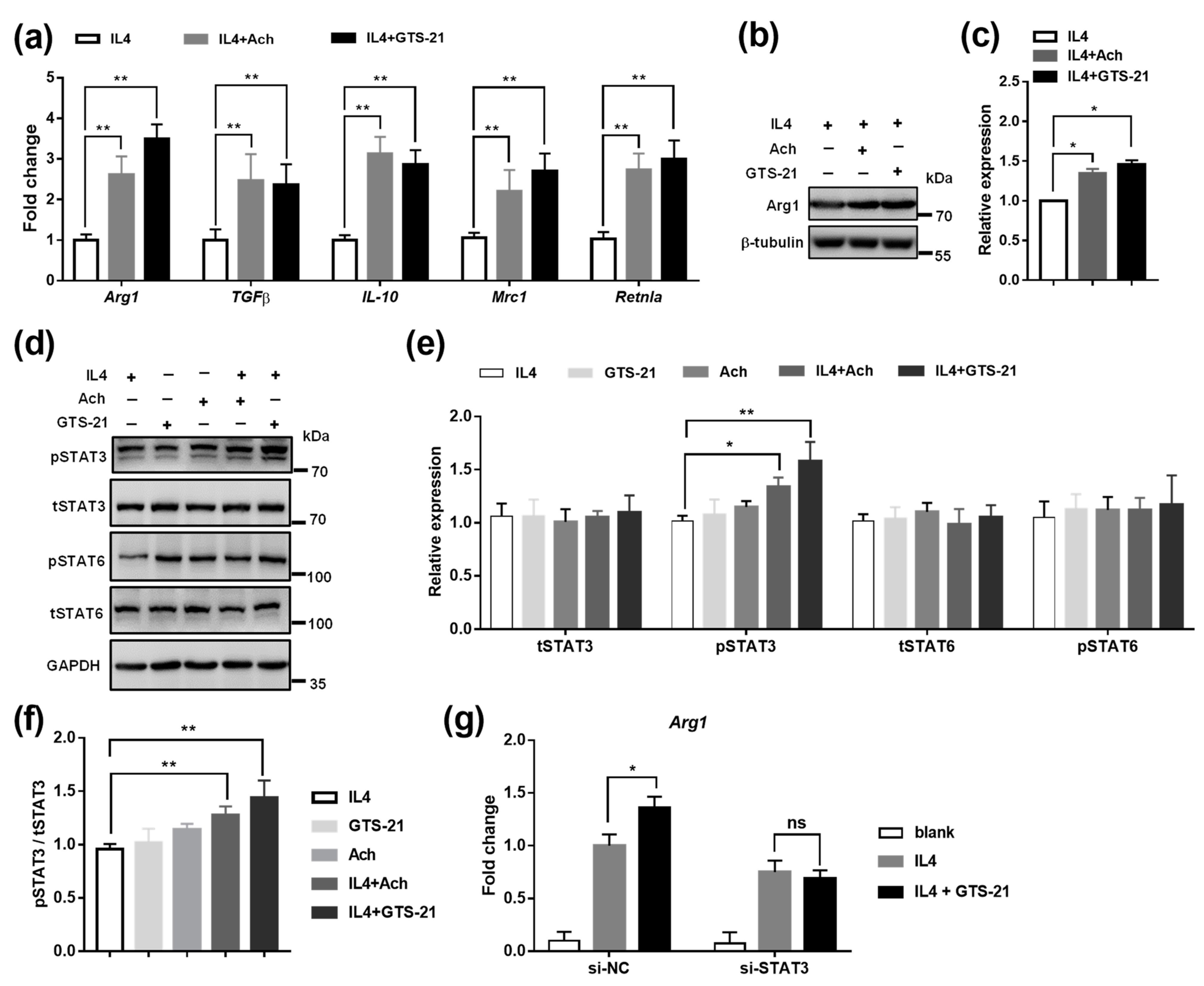
3.4. Effects of CAP Agonists on M2 Macrophage Polarization
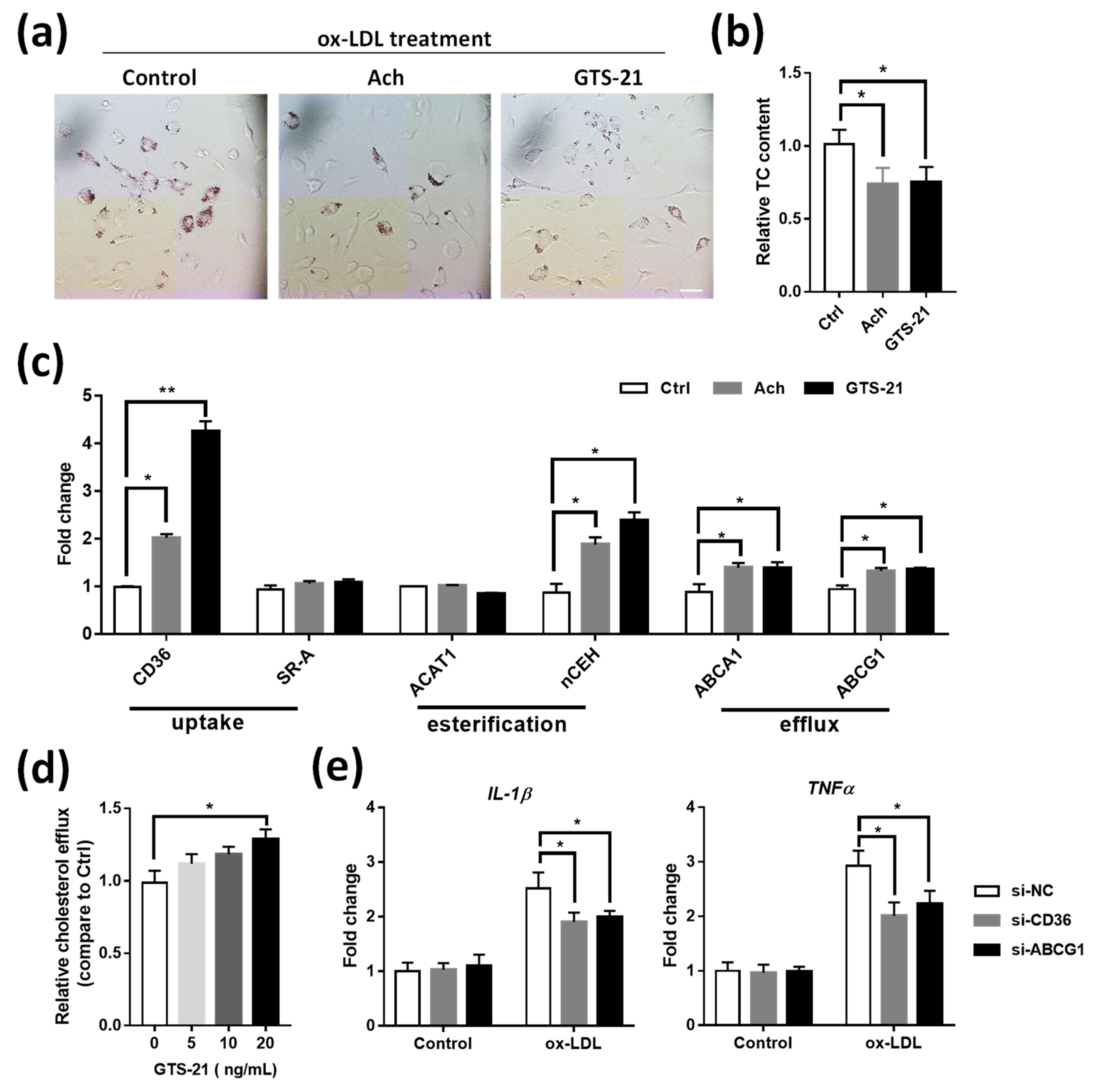
3.5. Effects of CAP Activity on Macrophage Cholesterol Metabolism
3.6. Effects of CAP Agonists on ox-LDL Induced Macrophage Foam Cell Formation
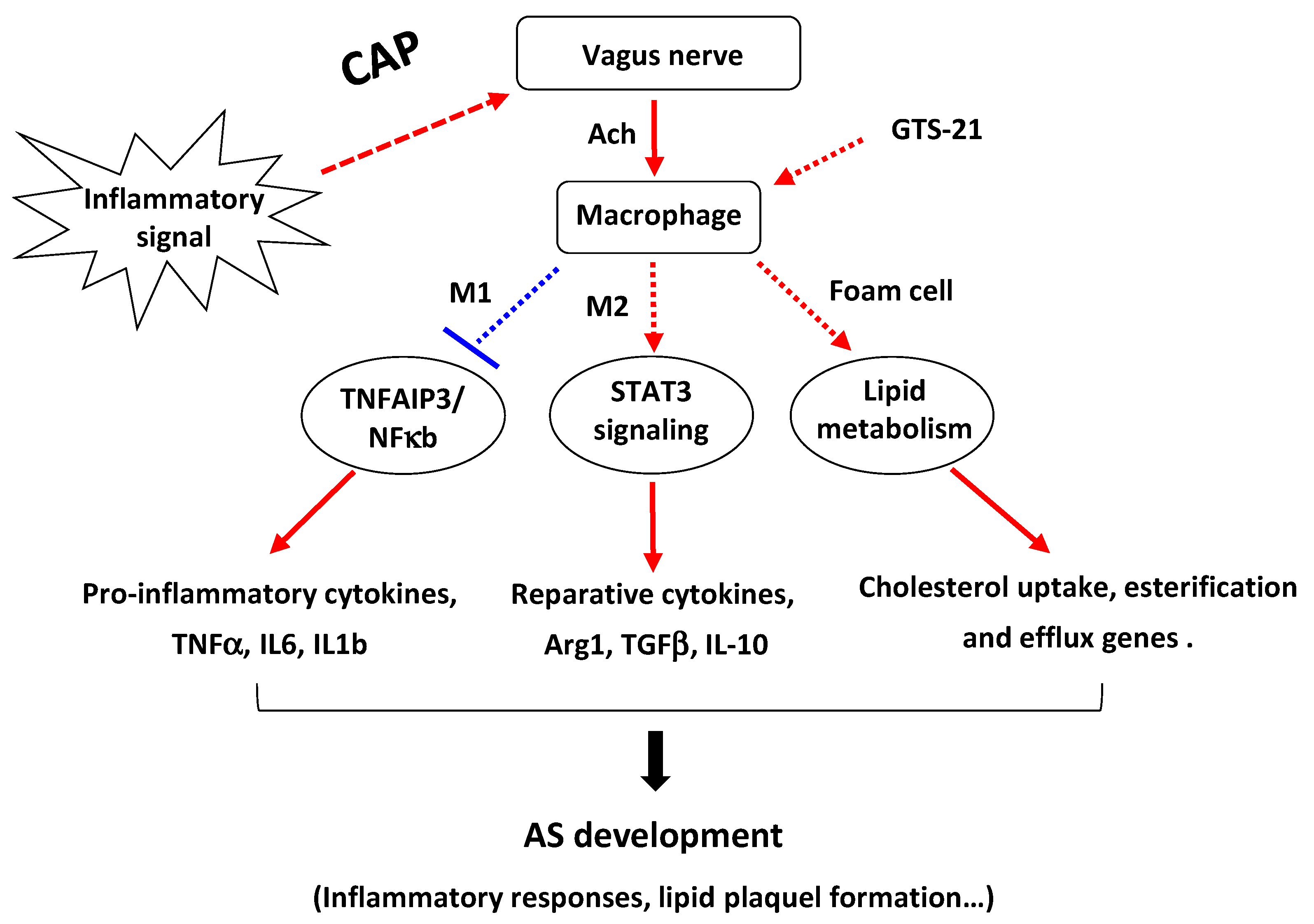
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Borovikova, L.V.; Ivanova, S.; Zhang, M.H.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.C.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. The inflammatory reflex. Nature 2002, 420, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. Reflex control of immunity. Nat. Rev. Immunol. 2009, 9, 418–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikami, Y.; Tsunoda, J.; Kiyohara, H.; Taniki, N.; Teratani, T.; Kanai, T. Vagus nerve-mediated intestinal immune regulation: Therapeutic implications for inflammatory bowel diseases. Int. Immunol. 2021, dxab039. [Google Scholar] [CrossRef] [PubMed]
- Wedn, A.M.; El-Bassossy, H.M.; Eid, A.H.; El-Mas, M.M. Modulation of preeclampsia by the cholinergic anti-inflammatory pathway: Therapeutic perspectives. Biochem. Pharmacol. 2021, 192, 114703. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Tracey, K.J. Reflex principles of immunological homeostasis. Annu. Rev. Immunol. 2012, 30, 313–335. [Google Scholar] [CrossRef] [Green Version]
- He, P.-F.; Na, A.R.; Chen, H.; Wei, H.-Y.; Cao, J.-S. Activation of alpha7 nicotinic acetylcholine receptor protects bovine endometrial tissue against LPS-induced inflammatory injury via JAK2/STAT3 pathway and COX-2 derived prostaglandin E-2. Eur. J. Pharmacol. 2021, 900, 174067. [Google Scholar] [CrossRef]
- Tracey, K.J. Reflexes in Immunity. Cell 2016, 164, 343–344. [Google Scholar] [CrossRef] [Green Version]
- Kin, I.; Sasaki, T.; Yasuhara, T.; Kameda, M.; Agari, T.; Okazaki, M.; Hosomoto, K.; Okazaki, Y.; Yabuno, S.; Kawauchi, S.; et al. Vagus Nerve Stimulation with Mild Stimulation Intensity Exerts Anti-Inflammatory and Neuroprotective Effects in Parkinson’s Disease Model Rats. Biomedicines 2021, 9, 789. [Google Scholar] [CrossRef]
- Koopman, F.A.; Chavan, S.S.; Miljko, S.; Grazio, S.; Sokolovic, S.; Schuurman, P.R.; Mehta, A.D.; Levine, Y.A.; Faltys, M.; Zitnik, R.; et al. Vagus nerve stimulation inhibits cytokine production and attenuates disease severity in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2016, 113, 8284–8289. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, R.; Peng, Z.; Zhou, W.; Hu, B.; Rao, X.; Yang, X.; Li, J. GTS-21 Reduces Inflammation in Acute Lung Injury by Regulating M1 Polarization and Function of Alveolar Macrophages. Shock 2019, 51, 389–400. [Google Scholar] [CrossRef]
- Yeboah, M.M.; Xue, X.; Duan, B.; Ochani, M.; Tracey, K.J.; Susin, M.; Metz, C.N. Cholinergic agonists attenuate renal ischemia-reperfusion injury in rats. Kidney Int. 2008, 74, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brock, C.; Rasmussen, S.E.; Drewes, A.M.; Moller, H.J.; Brock, B.; Deleuran, B.; Farmer, A.D.; Pfeiffer-Jensen, M. Vagal Nerve Stimulation-Modulation of the Anti-Inflammatory Response and Clinical Outcome in Psoriatic Arthritis or Ankylosing Spondylitis. Mediat. Inflamm. 2021, 2021, 9933532. [Google Scholar] [CrossRef]
- Wu, S.; Shi, Z.; Wang, X.; Ren, F.; Xie, Z.; Lei, L.; Chen, P. Activation of the Cholinergic Anti-inflammatory Pathway Attenuated Angiotension II-Dependent Hypertension and Renal Injury. Front. Pharmacol. 2021, 12, 276. [Google Scholar] [CrossRef]
- Hashimoto, T.; Ichiki, T.; Watanabe, A.; Hurt-Camejo, E.; Michaelsson, E.; Ikeda, J.; Inoue, E.; Matsuura, H.; Tokunou, T.; Kitamoto, S.; et al. Stimulation of alpha7 nicotinic acetylcholine receptor by AR-R17779 suppresses atherosclerosis and aortic aneurysm formation in apolipoprotein E-deficient mice. Vasc. Pharmacol. 2014, 61, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Olshansky, B. Vagus nerve modulation of inflammation: Cardiovascular implications. Trends Cardiovasc. Med. 2016, 26, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Van der Valk, F.M.; van Wijk, D.F.; Stroes, E.S. Novel anti-inflammatory strategies in atherosclerosis. Curr. Opin. Lipidol. 2012, 23, 532–539. [Google Scholar] [CrossRef]
- Vieira-Alves, I.; Coimbra-Campos, L.M.C.; Sancho, M.; da Silva, R.F.; Cortes, S.F.; Lemos, V.S. Role of the alpha 7 Nicotinic Acetylcholine Receptor in the Pathophysiology of Atherosclerosis. Front. Physiol. 2020, 11, 621769. [Google Scholar] [CrossRef]
- Inanaga, K.; Ichiki, T.; Miyazaki, R.; Takeda, K.; Hashimoto, T.; Matsuura, H.; Sunagawa, K. Acetylcholinesterase inhibitors attenuate atherogenesis in apolipoprotein E-knockout mice. Atherosclerosis 2010, 213, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Al-Sharea, A.; Lee, M.K.S.; Whillas, A.; Flynn, M.C.; Chin-Dusting, J.; Murphy, A.J. Nicotinic acetylcholine receptor alpha 7 stimulation dampens splenic myelopoiesis and inhibits atherogenesis in Apoe(-/-) mice. Atherosclerosis 2017, 265, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Ulleryd, M.A.; Bernardi, A.; Lundberg, A.M.; Andersson, A.; Folkersen, L.; Fogelstrand, L.; Islander, U.; Yan, Z.Q.; Hansson, G.K. α7 Nicotinic acetylcholine receptor is expressed in human atherosclerosis and inhibits disease in mice—Brief report. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2632–2636. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.H.; Vazquez, G. Reduced size and macrophage content of advanced atherosclerotic lesions in mice with bone marrow specific deficiency of alpha 7 nicotinic acetylcholine receptor. PLoS ONE 2015, 10, e0124584. [Google Scholar] [CrossRef]
- Kooijman, S.; Meurs, I.; van der Stoep, M.; Habets, K.L.; Lammers, B.; Berbée, J.F.; Havekes, L.M.; van Eck, M.; Romijn, J.A.; Korporaal, S.J.; et al. Hematopoietic α7 nicotinic acetylcholine receptor deficiency increases inflammation and platelet activation status, but does not aggravate atherosclerosis. J. Thromb. Haemost. JTH 2015, 13, 126–135. [Google Scholar] [CrossRef]
- Wilund, K.R.; Rosenblat, M.; Chung, H.R.; Volkova, N.; Kaplan, M.; Woods, J.A.; Aviram, M. Macrophages from alpha 7 nicotinic acetylcholine receptor knockout mice demonstrate increased cholesterol accumulation and decreased cellular paraoxonase expression: A possible link between the nervous system and atherosclerosis development. Biochem. Biophys. Res. Commun. 2009, 390, 148–154. [Google Scholar] [CrossRef]
- Moore, K.J.; Tabas, I. Macrophages in the Pathogenesis of Atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Cochain, C.; Zernecke, A. Macrophages in vascular inflammation and atherosclerosis. Pflügers Arch. Eur. J. Physiol. 2017, 469, 485–499. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, J.H. Deciphering Macrophage Phenotypes upon Lipid Uptake and Atherosclerosis. Immune Netw. 2020, 20, e22. [Google Scholar] [CrossRef] [PubMed]
- Bories, G.F.P.; Leitinger, N. Macrophage metabolism in atherosclerosis. FEBS Lett. 2017, 591, 3042–3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabas, I.; Bornfeldt, K.E. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ. Res. 2016, 118, 653–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, C.D. Anatomy of a discovery: m1 and m2 macrophages. Front. Immunol. 2015, 6, 212. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Chinetti-Gbaguidi, G.; Baron, M.; Bouhlel, M.A.; Vanhoutte, J.; Copin, C.; Sebti, Y.; Derudas, B.; Mayi, T.; Bories, G.; Tailleux, A.; et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARγ and LXRα pathways. Circ. Res. 2011, 108, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Cardilo-Reis, L.; Gruber, S.; Schreier, S.M.; Drechsler, M.; Papac-Milicevic, N.; Weber, C.; Wagner, O.; Stangl, H.; Soehnlein, O.; Binder, C.J. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol. Med. 2012, 4, 1072–1086. [Google Scholar] [CrossRef]
- Gupta, S.; Pablo, A.M.; Jiang, X.; Wang, N.; Tall, A.R.; Schindler, C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J. Clin. Investig. 1997, 99, 2752–2761. [Google Scholar] [CrossRef] [PubMed]
- De Gaetano, M.; Crean, D.; Barry, M.; Belton, O. M1- and M2-Type Macrophage Responses Are Predictive of Adverse Outcomes in Human Atherosclerosis. Front. Immunol. 2016, 7, 275. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Bobryshev, Y.V.; Nikiforov, N.G.; Elizova, N.V.; Sobenin, I.A.; Orekhov, A.N. Macrophage phenotypic plasticity in atherosclerosis: The associated features and the peculiarities of the expression of inflammatory genes. Int. J. Cardiol. 2015, 184, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Groenen, A.G.; Halmos, B.; Tall, A.R.; Westerterp, M. Cholesterol efflux pathways, inflammation, and atherosclerosis. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Remmerie, A.; Scott, C.L. Macrophages and lipid metabolism. Cell. Immunol. 2018, 330, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell. Mol. Med. 2016, 20, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.H.; Fu, Y.C.; Zhang, D.W.; Yin, K.; Tang, C.K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99. [Google Scholar] [CrossRef] [Green Version]
- Li, A.C.; Binder, C.J.; Gutierrez, A.; Brown, K.K.; Plotkin, C.R.; Pattison, J.W.; Valledor, A.F.; Davis, R.A.; Willson, T.M.; Witztum, J.L.; et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J. Clin. Investig. 2004, 114, 1564–1576. [Google Scholar] [CrossRef] [Green Version]
- Das, R.; Ganapathy, S.; Mahabeleshwar, G.H.; Drumm, C.; Febbraio, M.; Jain, M.K.; Plow, E.F. Macrophage gene expression and foam cell formation are regulated by plasminogen. Circulation 2013, 127, 1209–1218. [Google Scholar] [CrossRef] [Green Version]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vasc. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef]
- Back, M.; Yurdagul, A., Jr.; Tabas, I.; Oorni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef]
- Lin, P.; Ji, H.-H.; Li, Y.-J.; Guo, S.-D. Macrophage Plasticity and Atherosclerosis Therapy. Front. Mol. Biosci. 2021, 8, 324. [Google Scholar] [CrossRef]
- Zang, X.; Cheng, M.; Zhang, X.; Chen, X. Targeting macrophages using nanoparticles: A potential therapeutic strategy for atherosclerosis. J. Mater. Chem. B 2021, 9, 3284–3294. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.F.; El-Salhy, M.; Danielsson, A.; Shalaby, A.; Axelsson, H. Effects of unilateral cervical vagotomy on antral endocrine cells in mouse. Histol. Histopathol. 1999, 14, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Ediriweera, H.N.; Gundra, U.M.; Sheedy, F.J.; Ramkhelawon, B.; Hutchison, S.B.; Rinehold, K.; van Solingen, C.; Fullerton, M.D.; Cecchini, K.; et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J. Clin. Investig. 2015, 125, 4334–4348. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Huang, Y.; Xie, Y.; Lan, T.; Le, K.; Chen, J.; Chen, S.; Gao, S.; Xu, X.; Shen, X.; et al. Evaluation of foam cell formation in cultured macrophages: An improved method with Oil Red O staining and DiI-oxLDL uptake. Cytotechnology 2010, 62, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Z.; Li, Y.; Yang, H.; Chen, J.; Li, X.; Gou, D. PDGFBB promotes proliferation and migration via regulating miR-1181/STAT3 axis in human pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L965–L976. [Google Scholar] [CrossRef] [Green Version]
- Qian, Z.; Li, Y.; Chen, J.; Li, X.; Gou, D. miR-4632 mediates PDGF-BB-induced proliferation and antiapoptosis of human pulmonary artery smooth muscle cells via targeting cJUN. Am. J. Physiol. Cell Physiol. 2017, 313, C380–C391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Long, J.; Chen, W.; Sun, Y.; Zhou, L.; Zhang, L.; Zeng, H.; Yuan, D. Alisol B 23-acetate, a new promoter for cholesterol efflux from dendritic cells, alleviates dyslipidemia and inflammation in advanced atherosclerotic mice. Int. Immunopharmacol. 2021, 99, 107956. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Li, Q.T.; Verma, I.M. NF-kappa B regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of NF-kappa B and I kappa B proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef]
- Blackwell, T.S.; Christman, J.W. The role of nuclear factor-kappa B in cytokine gene regulation. Am. J. Respir. Cell Mol. Biol. 1997, 17, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Coornaert, B.; Carpentier, I.; Beyaert, R. A20: Central Gatekeeper in Inflammation and Immunity. J. Biol. Chem. 2009, 284, 8217–8221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.L.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappa B signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef]
- Lee, T.-M.; Chang, N.-C.; Lin, S.-Z. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic. Biol. Med. 2017, 104, 298–310. [Google Scholar] [CrossRef]
- Qin, C.; Fan, W.-H.; Liu, Q.; Shang, K.; Murugan, M.; Wu, L.-J.; Wang, W.; Tian, D.-S. Fingolimod Protects against Ischemic White Matter Damage by Modulating Microglia Toward M2 Polarization via STAT3 Pathway. Stroke 2017, 48, 3336–3346. [Google Scholar] [CrossRef]
- Liao, X.; Sharma, N.; Kapadia, F.; Zhou, G.; Lu, Y.; Hong, H.; Paruchuri, K.; Mahabeleshwar, G.H.; Dalmas, E.; Venteclef, N.; et al. Kruppel-like factor 4 regulates macrophage polarization. J. Clin. Investig. 2011, 121, 2736–2749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Sun, S.; Xu, A.; Bhargava, P.; Yang, L.; Lam, K.S.L.; Gao, B.; Lee, C.-H.; Kersten, S.; Qi, L. Activation of Natural Killer T Cells Promotes M2 Macrophage Polarization in Adipose Tissue and Improves Systemic Glucose Tolerance via Interleukin-4 (IL-4)/STAT6 Protein Signaling Axis in Obesity. J. Biol. Chem. 2012, 287, 13561–13571. [Google Scholar] [CrossRef] [Green Version]
- Huston, J.M.; Ochani, M.; Rosas-Ballina, M.; Liao, H.; Ochani, K.; Pavlov, V.A.; Gallowitsch-Puerta, M.; Ashok, M.; Czura, C.J.; Foxwell, B.; et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J. Exp. Med. 2006, 203, 1623–1628. [Google Scholar] [CrossRef] [Green Version]
- Meregnani, J.; Clarencon, D.; Vivier, M.; Peinnequin, A.; Mouret, C.; Sinniger, V.; Picq, C.; Job, A.; Canini, F.; Jacquier-Sarlin, M.; et al. Anti-inflammatory effect of vagus nerve stimulation in a rat model of inflammatory bowel disease. Auton. Neurosci. Basic Clin. 2011, 160, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Reijmen, E.; Vannucci, L.; de Couck, M.; de Grève, J.; Gidron, Y. Therapeutic potential of the vagus nerve in cancer. Immunol. Lett. 2018, 202, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zu, X.; Liu, J. The roles of immune cells in atherosclerotic calcification. Vascular 2021. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Potteaux, S.; Gautier, E.L.; Hutchison, S.B.; van Rooijen, N.; Rader, D.J.; Thomas, M.J.; Sorci-Thomas, M.G.; Randolph, G.J. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe-/- mice during disease regression. J. Clin. Investig. 2011, 121, 2025–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orekhov, A.N.; Oishi, Y.; Nikiforov, N.G.; Zhelankin, A.V.; Dubrovsky, L.; Sobenin, I.A.; Kel, A.; Stelmashenko, D.; Makeev, V.J.; Foxx, K.; et al. Modified LDL Particles Activate Inflammatory Pathways in Monocyte-derived Macrophages: Transcriptome Analysis. Curr. Pharm. Des. 2018, 24, 3143–3151. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Freeman, M.W. Scavenger receptors in atherosclerosis: Beyond lipid uptake. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1702–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchibhotla, S.; Vanegas, D.; Kennedy, D.J.; Guy, E.; Nimako, G.; Morton, R.E.; Febbraio, M. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc. Res. 2008, 78, 185–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning-Tobin, J.J.; Moore, K.J.; Seimon, T.A.; Bell, S.A.; Sharuk, M.; Alvarez-Leite, J.I.; de Winther, M.P.; Tabas, I.; Freeman, M.W. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Meir, K.S.; Leitersdorf, E. Atherosclerosis in the apolipoprotein E-deficient mouse—A decade of progress. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1006–1014. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Primer Name | Sequence (5′-3′) |
|---|---|
| TNFα-F | GCCTCTTCTCATTCCTGCTT |
| TNFα-R | TGGGAACTTCTCATCCCTTTG |
| IL-1β-F | TGGCAACTGTTCCTG |
| IL-1β-R | GGAAGCAGCCCTTCATCTTT |
| IL-6-F | CAAAGCCAGAGTCCTTCAGAG |
| IL-6-R | GTCCTTAGCCACTCCTTCTG |
| Arg1-F | AAGAATGGAAGAGTCAGTGTGG |
| Arg1-R | GGGAGTGTTGATGTCAGTGTG |
| IL10-F | AGGCGCTGTCATCGATTT |
| IL10-R | CACCTTGGTCTTGGAGCTTAT |
| TGFβ-F | CCTGAGTGGCTGTCTTTTGA |
| TGFβ-R | CGTGGAGTTTGTTATCTTTGCTG |
| Mrc1-F | CTCTGTTCAGCTATTGGACGC |
| Mrc1-R | CGGAATTTCTGGGATTCAGCTTC |
| Retnla-F | CTGGGTTCTCCACCTCTTCA |
| Retnla-R | TGCTGGGATGACTGCTACTG |
| TNFAIP3-F | ACAGGACTTTGCTACGACAC |
| TNFAIP3-R | CTGAGGATGTTGCTGAGGAC |
| CD36-F | GCGACATGATTAATGGCACAG |
| CD36-R | GATCCGAACACAGCGTAGATAG |
| SRA-F | GGGAACACTCACAGACACTG |
| SRA-R | CCCGATCACCTTTAACACCTG |
| ACAT1-F | AGCACACTGAACGATGGAG |
| ACAT1-R | CGCAAGTGGAAAATCAATGGG |
| nCEH-F | CTACGTGTACATCCCACTGC |
| nCEH-R | GATGAAATTCAGCGCGATCAG |
| ABCA1-F | TGACATGGTACATCGAAGCC |
| ABCA1-R | GATTTCTGACACTCCCTTCTGG |
| ABCG1-F | CCTTATCAATGGAATGCCCCG |
| ABCG1-R | CTGCCTTCATCCTTCTCCTG |
| GAPDH-F | AACAGCAACTCCCACTCTTC |
| GAPDH-R | CCTGTTGCTGTAGCCGTATT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, Z.; Yang, H.; Li, H.; Liu, C.; Yang, L.; Qu, Z.; Li, X. The Cholinergic Anti-Inflammatory Pathway Attenuates the Development of Atherosclerosis in Apoe-/- Mice through Modulating Macrophage Functions. Biomedicines 2021, 9, 1150. https://doi.org/10.3390/biomedicines9091150
Qian Z, Yang H, Li H, Liu C, Yang L, Qu Z, Li X. The Cholinergic Anti-Inflammatory Pathway Attenuates the Development of Atherosclerosis in Apoe-/- Mice through Modulating Macrophage Functions. Biomedicines. 2021; 9(9):1150. https://doi.org/10.3390/biomedicines9091150
Chicago/Turabian StyleQian, Zhengjiang, Haiyang Yang, Hongchao Li, Chunhua Liu, Liang Yang, Zehui Qu, and Xiang Li. 2021. "The Cholinergic Anti-Inflammatory Pathway Attenuates the Development of Atherosclerosis in Apoe-/- Mice through Modulating Macrophage Functions" Biomedicines 9, no. 9: 1150. https://doi.org/10.3390/biomedicines9091150