
Activated Alpha 2-Macroglobulin Is a Novel Mediator of Mesangial Cell Profibrotic Signaling in Diabetic Kidney Disease

 ,
,
Abstract
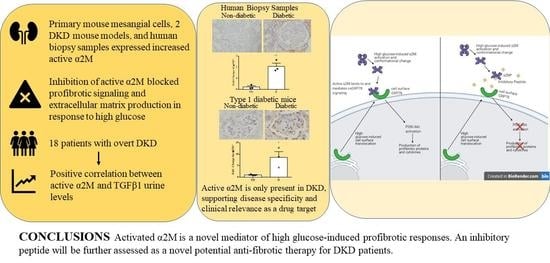
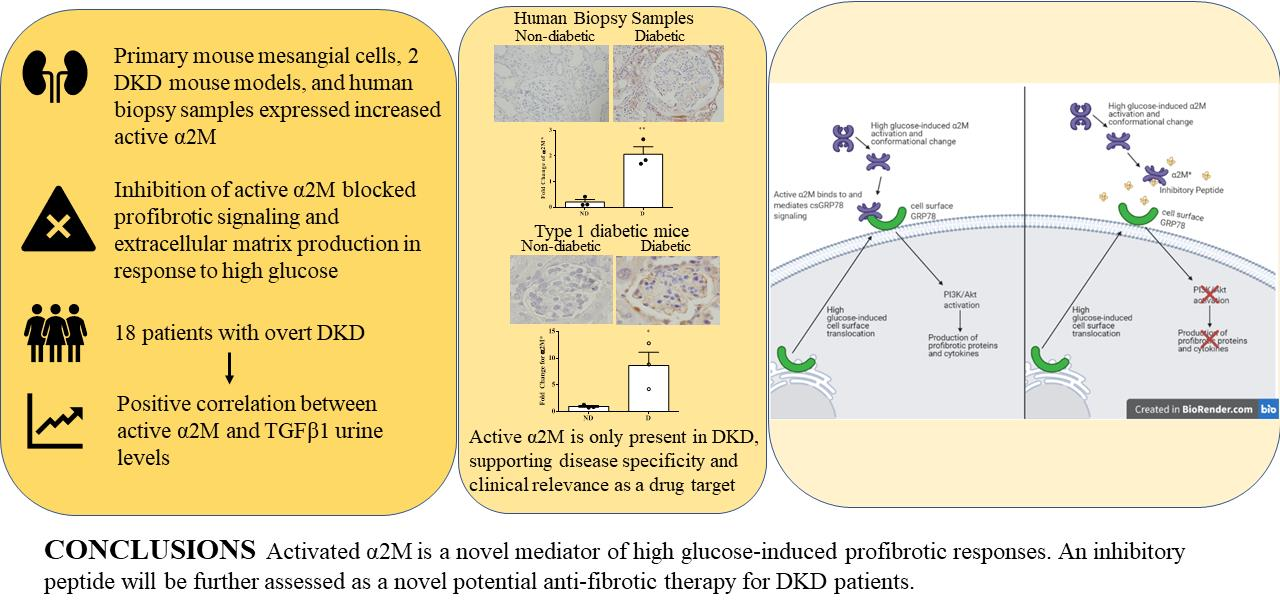
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Protein Extraction and Immunoblotting
2.3. qPCR
2.4. Experimental Animals and Tissue Processing
2.5. Transfection
2.6. Intracellular Calcium Assay
2.7. Cell Surface Protein Isolation
2.8. TGFβ1 ELISA
2.9. Recovery of α2M* from Urine Samples
2.10. Patient Cohort
2.11. Statistical Analysis
3. Results
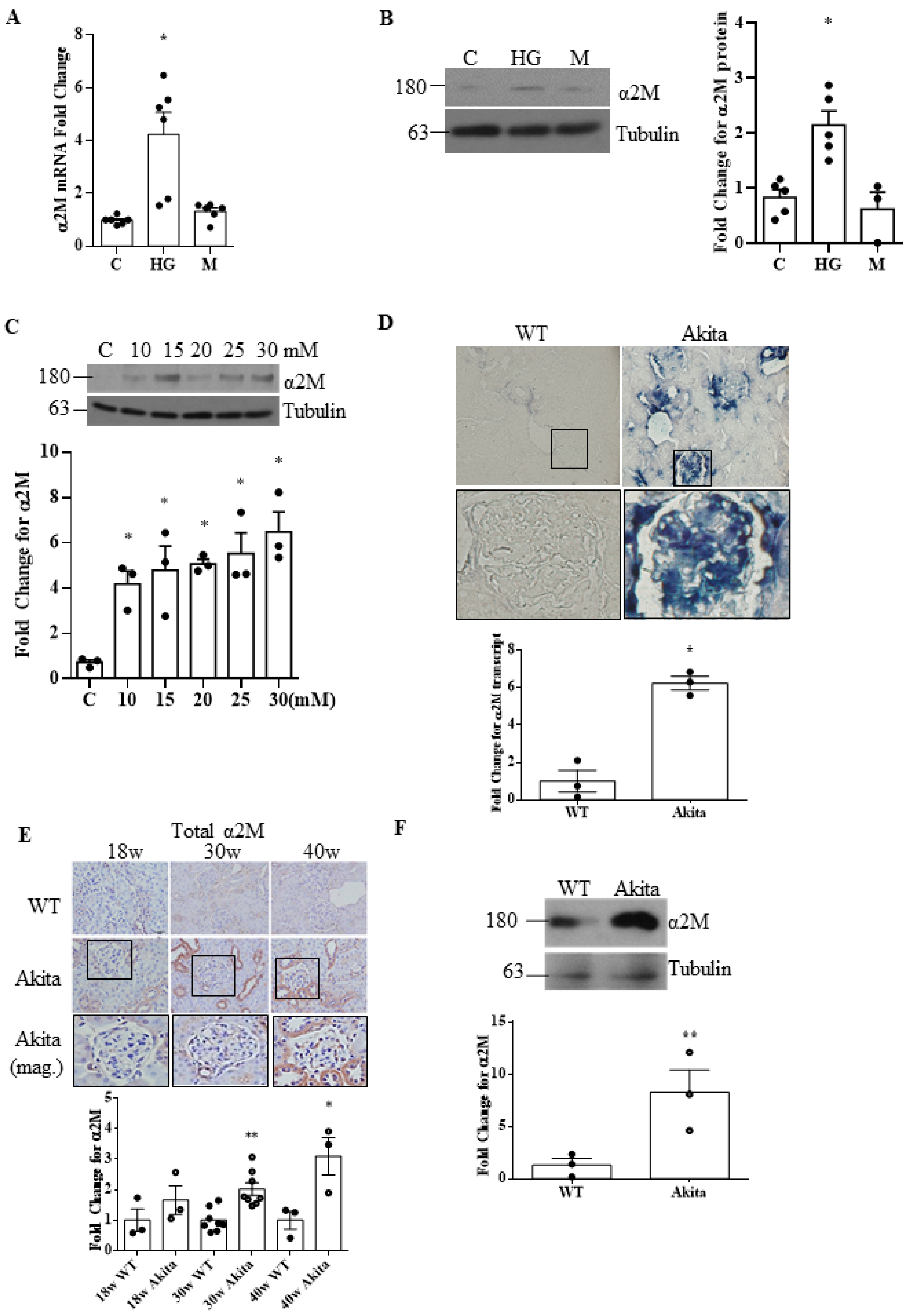
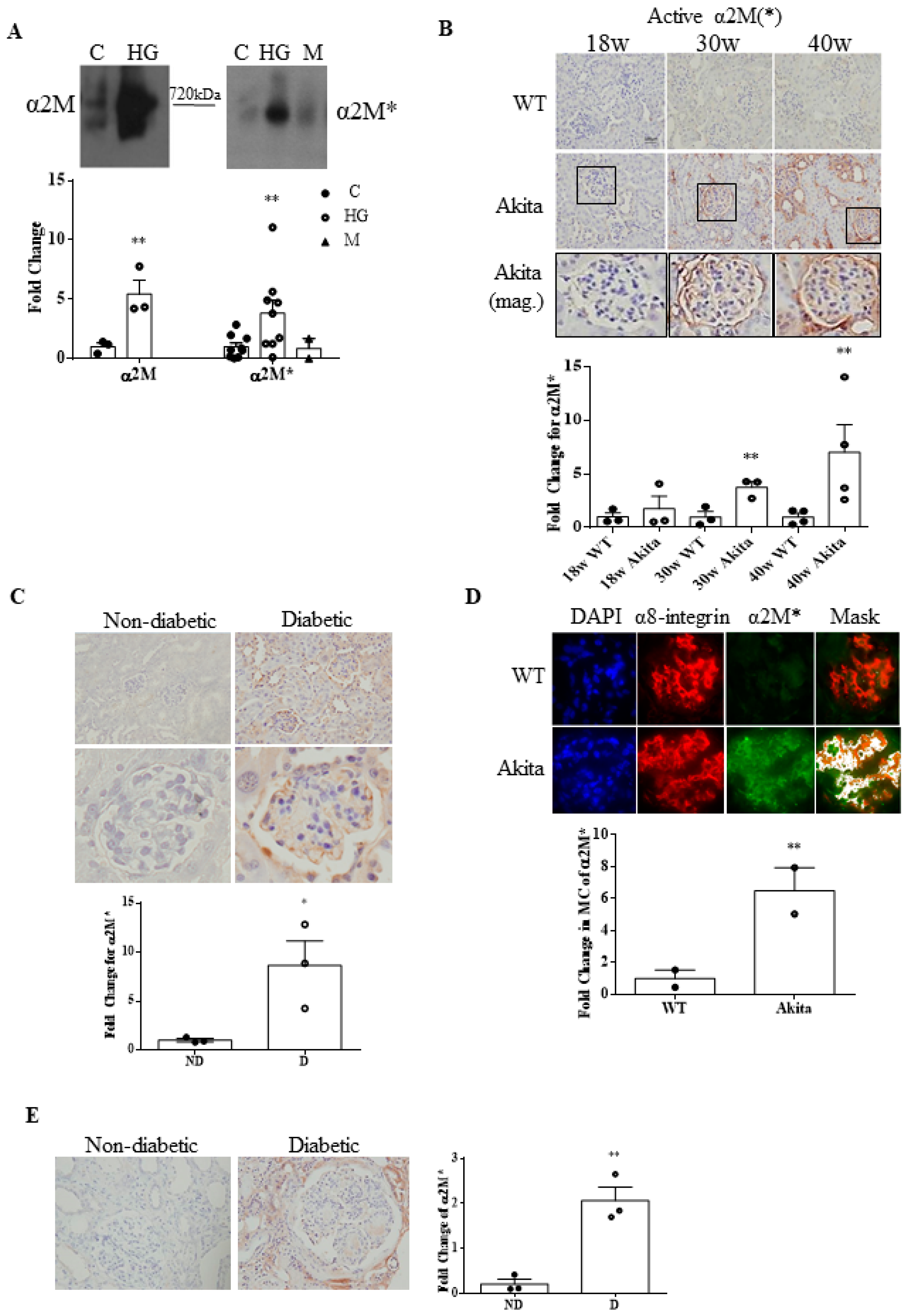
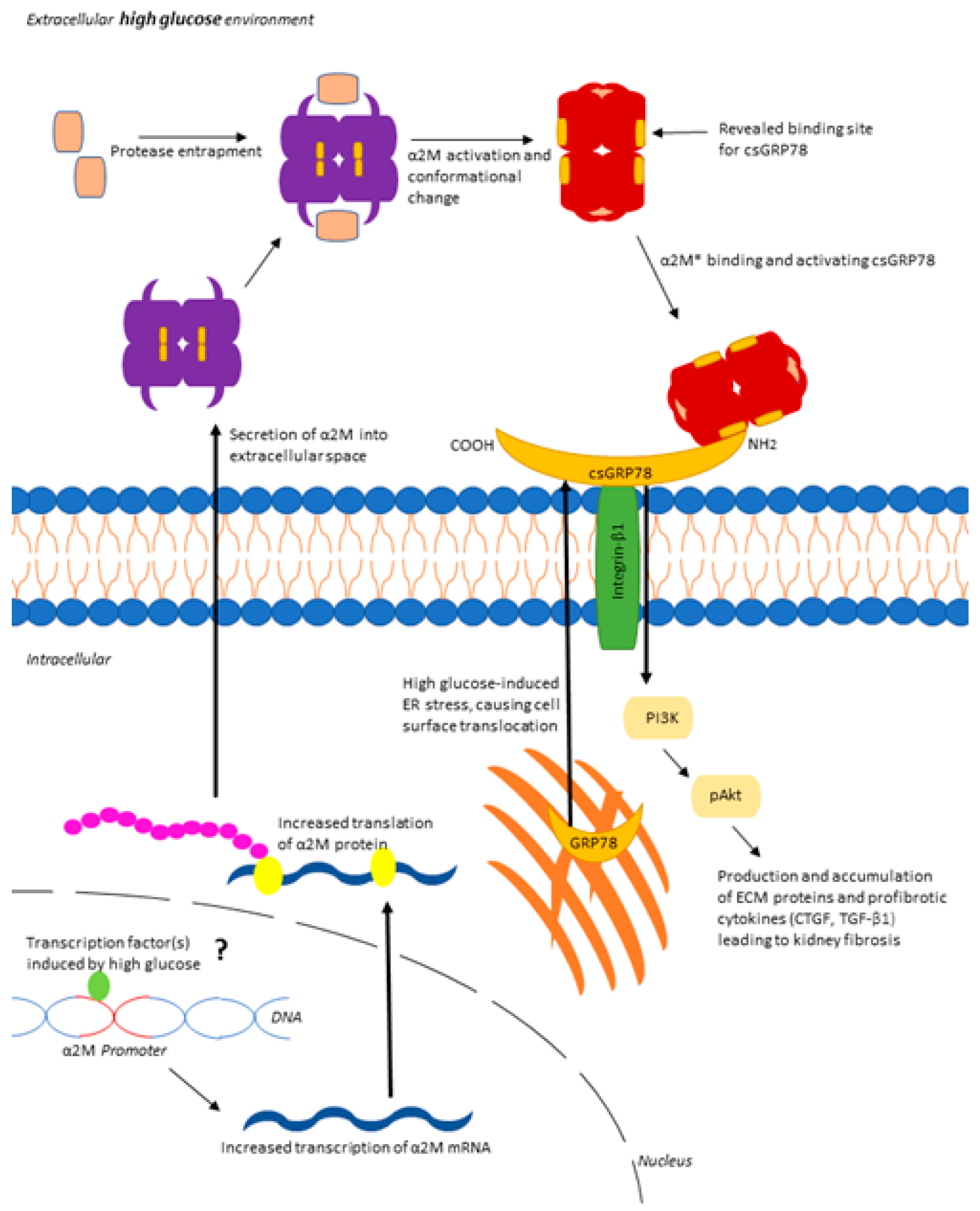
3.1. α2M Is Increased and Activated by HG in MCs and in Diabetic Kidneys
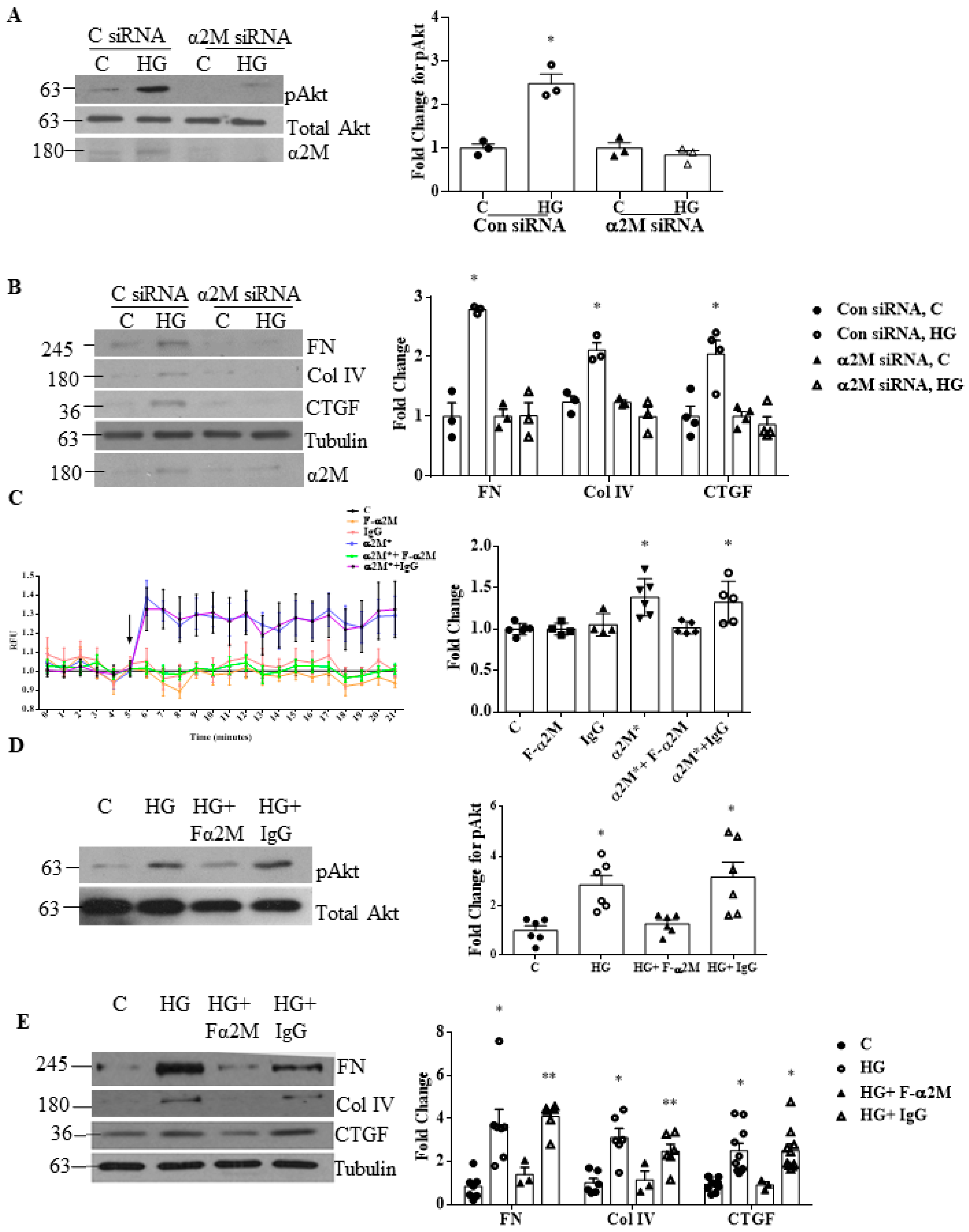
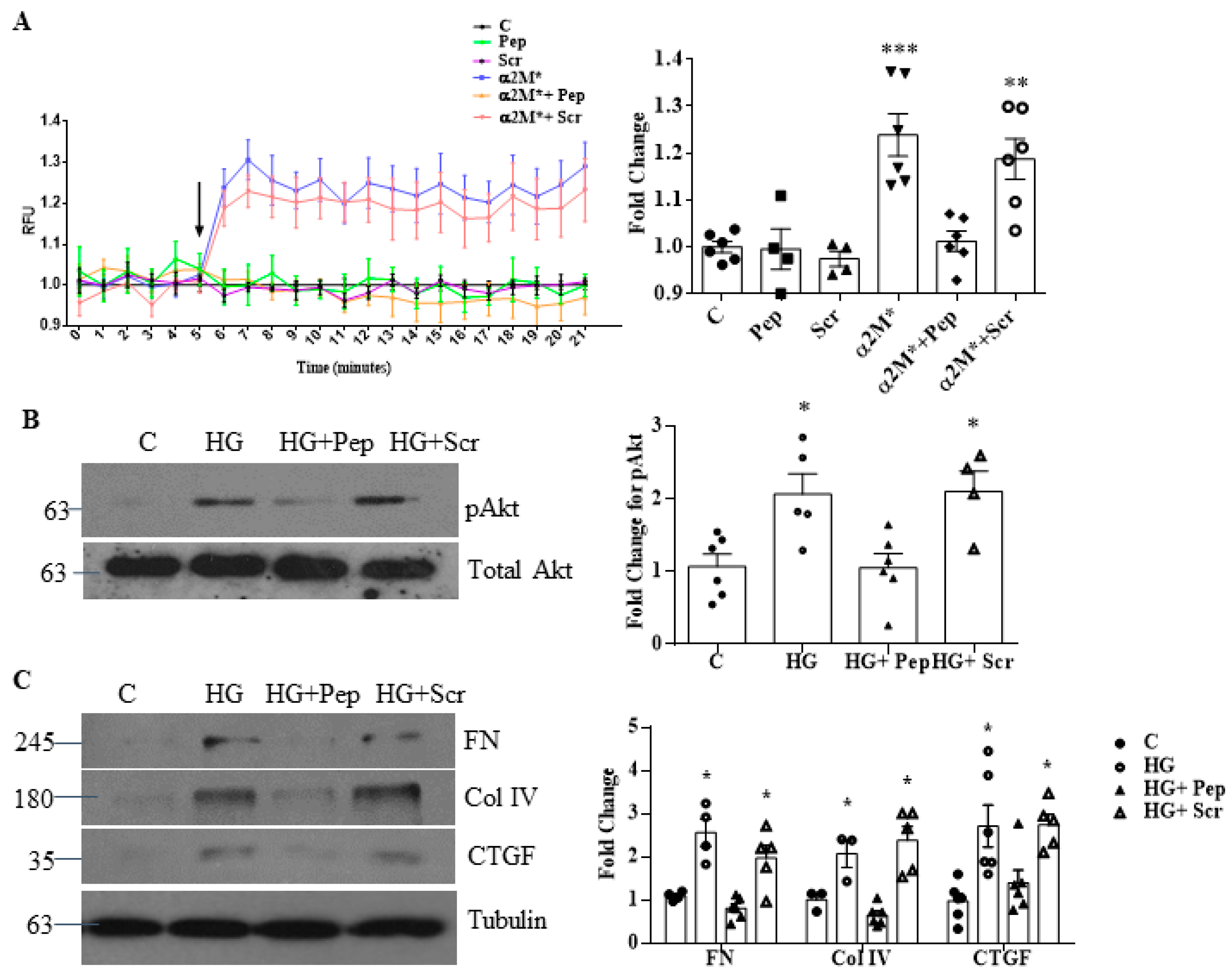
3.2. Inhibition of α2M/α2M* Inhibits HG-Induced Profibrotic Responses by MCs
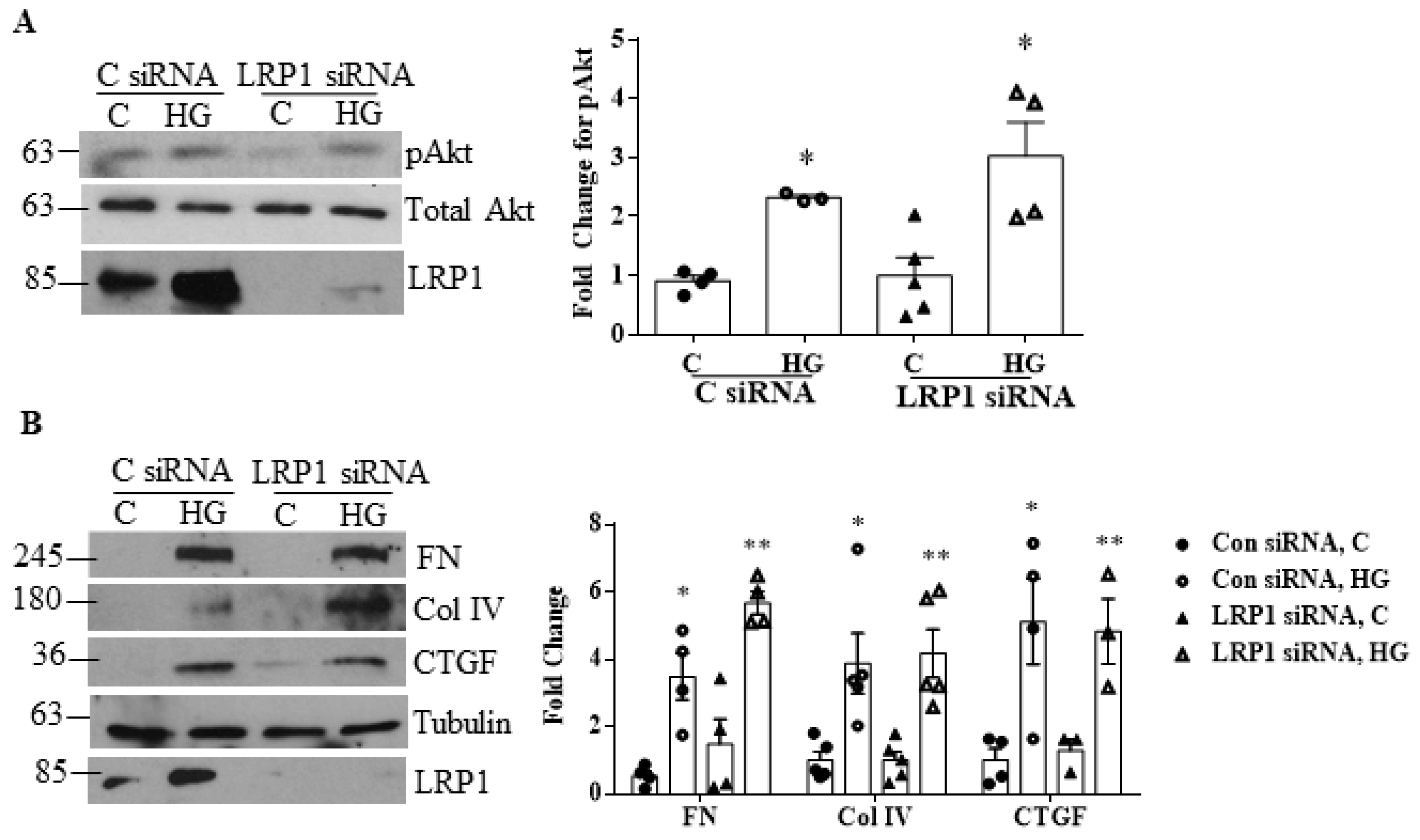
3.3. LRP1 Is Not Involved in HG-Induced Profibrotic Responses in MCs
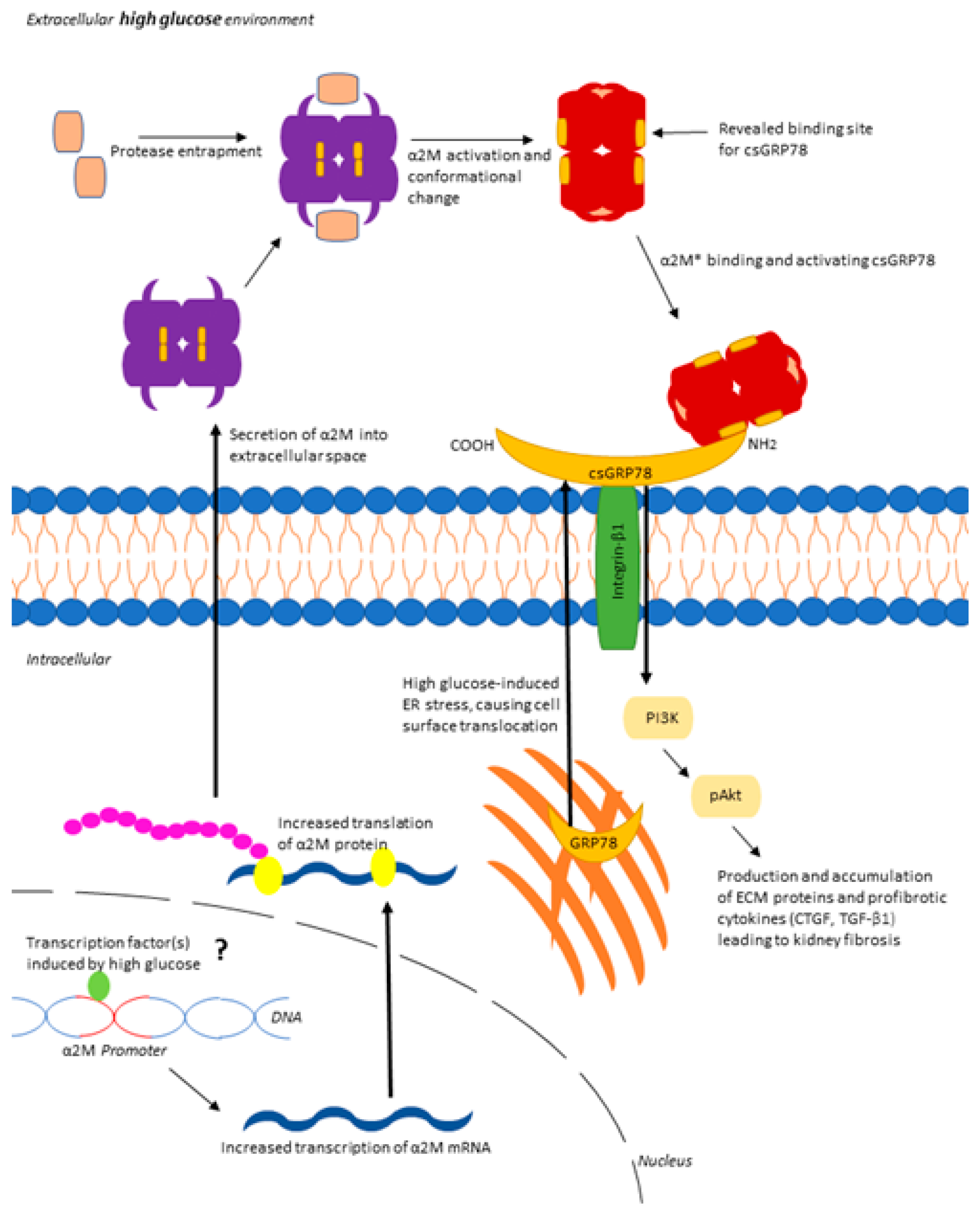
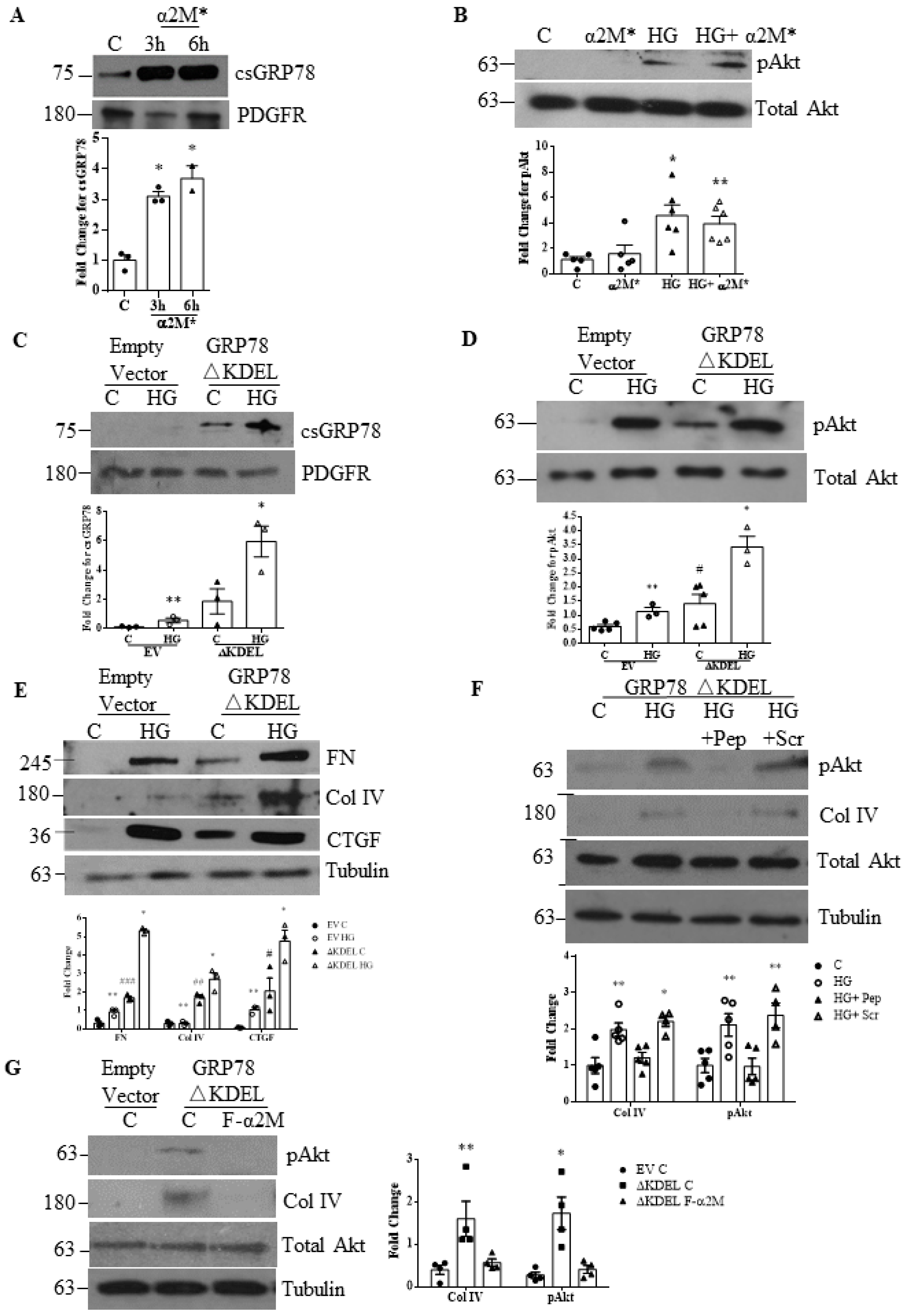
3.4. Increased Matrix Synthesis with csGRP78 Overexpression Requires α2M*
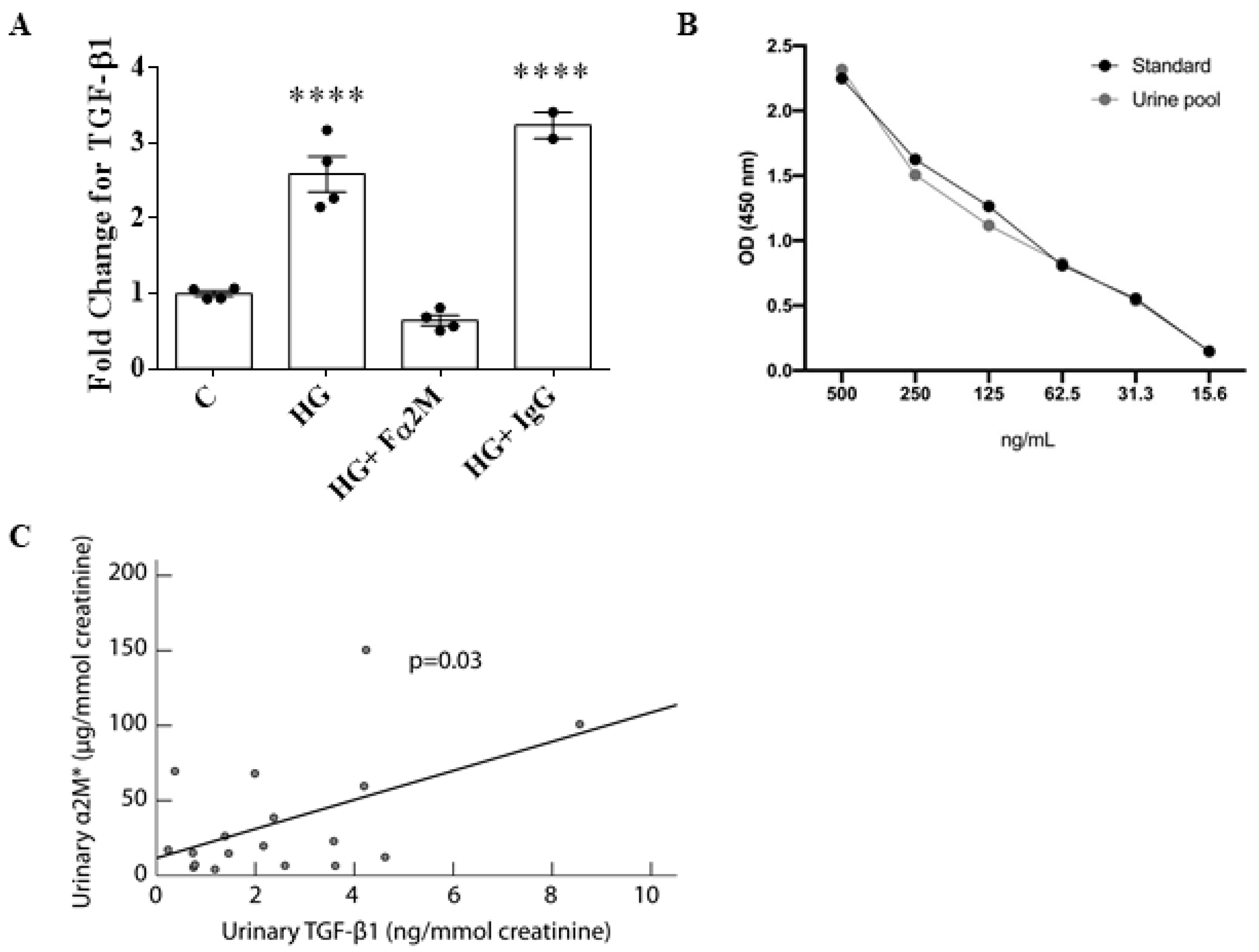
3.5. α2M* Regulates TGFβ1 Production by HG in MCs
3.6. Urinary α2M* Is Associated with Urinary TGFβ1 in Individuals with Overt DKD
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef]
- Dekkers, C.C.J.; Gansevoort, R.T.; Heerspink, H.J.L. New Diabetes Therapies and Diabetic Kidney Disease Progression: The Role of SGLT-2 Inhibitors. Curr. Diabetes Rep. 2018, 18, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sagoo, M.K.; Gnudi, L. Diabetic Nephropathy: An Overview. Methods Mol. Biol. 2019, 2067, 3–7. [Google Scholar]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef]
- Schlöndorff, D.; Banas, B. The Mesangial Cell Revisited: No Cell Is an Island. J. Am. Soc. Nephrol. 2009, 20, 1179–1187. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Lee, A.S. Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J. Cell. Physiol. 2015, 230, 1413–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Krieken, R.; Mehta, N.; Wang, T.; Zheng, M.; Li, R.; Gao, B.; Ayaub, E.; Ask, K.; Paton, J.C.; Paton, A.W.; et al. Cell surface expression of 78-kDa glucose-regulated protein (GRP78) mediates diabetic nephropathy. J. Biol. Chem. 2019, 294, 7755–7768. [Google Scholar] [CrossRef]
- Tapon-Bretaudière, J. Hemostasis and Thrombosis. Basic Principles and Clinical Practice, Fourth Edition. Robert W. Colman, Jack Hirsh, Victor J. Marder, Alexander W. Clowes, and James N. George, eds. Philadelphia, PA: Lippincott Williams & Wilkins, 2001, 1578 pp., $249.00, hardcover. ISBN 0-7817-1455-9. Clin. Chem. 2003, 49, 345–346. [Google Scholar] [CrossRef]
- Rehman, A.A.; Ahsan, H.; Khan, F.H. Alpha-2-macroglobulin: A physiological guardian. J. Cell. Physiol. 2013, 228, 1665–1675. [Google Scholar] [CrossRef]
- Cater, J.H.; Wilson, M.R.; Wyatt, A.R. Alpha-2-Macroglobulin, a Hypochlorite-Regulated Chaperone and Immune System Modulator. Oxid. Med. Cell. Longev. 2019, 2019, 5410657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, U.; Gonzalez-Gronow, M.; Pizzo, S.V. Activated α2-Macroglobulin Regulates Transcriptional Activation of c-MYC Target Genes through Cell Surface GRP78 Protein. J. Biol. Chem. 2016, 291, 10904–10915. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Gronow, M.; Cuchacovich, M.; Llanos, C.; Urzua, C.; Gawdi, G.; Pizzo, S.V. Prostate Cancer Cell ProliferationIn vitroIs Modulated by Antibodies against Glucose-Regulated Protein 78 Isolated from Patient Serum. Cancer Res. 2006, 66, 11424–11431. [Google Scholar] [CrossRef] [Green Version]
- Misra, U.K.; Gonzalez-Gronow, M.; Gawdi, G.; Hart, J.P.; Johnson, C.E.; Pizzo, S.V. The Role of Grp 78 in α2-Macroglobulin-induced Signal Transduction. J. Biol. Chem. 2002, 277, 42082–42087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, U.K.; Gonzalez-Gronow, M.; Gawdi, G.; Wang, F.; Pizzo, S.V. A novel receptor function for the heat shock protein Grp78: Silencing of Grp78 gene expression attenuates α2M*-induced signalling. Cell. Signal. 2004, 16, 929–938. [Google Scholar] [CrossRef]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef]
- Misra, U.K.; Payne, S.; Pizzo, S.V. The Monomeric Receptor Binding Domain of Tetrameric α2-Macroglobulin Binds to Cell Surface GRP78 Triggering Equivalent Activation of Signaling Cascades. Biochemistry 2013, 52, 4014–4025. [Google Scholar] [CrossRef]
- Yang, A.H.; Chen, J.Y. Glomerular deposition of alpha 2-macroglobulin in glomerular diseases. Nephrol. Dial. Transplant. 1997, 12, 465–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, K.; Merriman, J.; Gray, R.S.; Duncan, L.J.; Herd, R. Serum alpha 2-macroglobulin levels in diabetes. J. Clin. Pathol. 1980, 33, 163–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aitken, J.P.; Ortiz, C.; Morales-Bozo, I.; Rojas-Alcayaga, G.; Baeza, M.; Beltran, C.; Escobar, A. α-2-Macroglobulin in Saliva Is Associated with Glycemic Control in Patients with Type 2 Diabetes Mellitus. Dis. Markers 2015, 2015, 128653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, R.; Otto, E.A.; Hoover, P.; Eddy, S.; Mariani, L.; Godfrey, B.; Berthier, C.C.; Eichinger, F.; Subramanian, L.; Harder, J.; et al. Single cell transcriptomics identifies focal segmental glomerulosclerosis remission endothelial biomarker. JCI Insight 2020, 5, 133267. [Google Scholar] [CrossRef] [PubMed]
- Krepinsky, J.C.; Ingram, A.J.; Tang, D.; Wu, D.; Liu, L.; Scholey, J.W. Nitric Oxide Inhibits Stretch-Induced MAPK Activation in Mesangial Cells Through RhoA Inactivation. J. Am. Soc. Nephrol. 2003, 14, 2790–2800. [Google Scholar] [CrossRef] [Green Version]
- Biltoft, D.; Gram, J.B.; Larsen, A.; Münster, A.-M.B.; Sidelmann, J.J.; Skjødt, K.; Palarasah, Y. Fast form alpha-2-macroglobulin—A marker for protease activation in plasma exposed to artificial surfaces. Clin. Biochem. 2017, 50, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Van Krieken, R.; Marway, M.; Parthasarathy, P.; Mehta, N.; Ingram, A.J.; Gao, B.; Krepinsky, J.C. Inhibition of SREBP with Fatostatin Does Not Attenuate Early Diabetic Nephropathy in Male Mice. Endocrinology 2018, 159, 1479–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, R.; Ni, M.; Gill, P.; Lee, A.S. Cell Surface Relocalization of the Endoplasmic Reticulum Chaperone and Unfolded Protein Response Regulator GRP78/BiP. J. Biol. Chem. 2010, 285, 15065–15075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhave, J.C.; Bouchard, J.; Goupil, R.; Pichette, V.; Brachemi, S.; Madore, F.; Troyanov, S. Clinical value of inflammatory urinary biomarkers in overt diabetic nephropathy: A prospective study. Diabetes Res. Clin. Pract. 2013, 101, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Hartner, A.; Schöcklmann, H.; Pröls, F.; Müller, U.; Sterzel, R.B. α8 Integrin in glomerular mesangial cells and in experimental glomerulonephritis. Kidney Int. 1999, 56, 1468–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheu, M.-L.; Shen, C.-C.; Jheng, J.-R.; Chiang, C.-K. Activation of PI3K in response to high glucose leads to regulation of SOCS-3 and STAT1/3 signals and induction of glomerular mesangial extracellular matrix formation. Oncotarget 2017, 8, 16925–16938. [Google Scholar] [CrossRef] [Green Version]
- Toda, N.; Mukoyama, M.; Yanagita, M.; Yokoi, H. CTGF in kidney fibrosis and glomerulonephritis. Inflamm. Regen. 2018, 38, 1–8. [Google Scholar] [CrossRef]
- Gopal, U.; Pizzo, S.V. Cell surface GRP78 promotes tumor cell histone acetylation through metabolic reprogramming: A mechanism which modulates the Warburg effect. Oncotarget 2017, 8, 107947–107963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Li, H.; Wang, Q.; Su, C.; Wang, G.; Song, H.; Zhao, L.; Luan, Z.; Su, R. The role of c-Src in the invasion and metastasis of hepatocellular carcinoma cells induced by association of cell surface GRP78 with activated α2M. BMC Cancer 2015, 15, 389. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.S.; Hathaway, C.K.; Smithies, O.; Kakoki, M. Transforming growth factor-β1 and diabetic nephropathy. Am. J. Physiol. Physiol. 2016, 310, F689–F696. [Google Scholar] [CrossRef] [Green Version]
- Li, J.H.; Huang, X.R.; Zhu, H.-J.; Johnson, R.; Lan, H.Y. Role of TGF-β signaling in extracellular matrix production under high glucose conditions. Kidney Int. 2003, 63, 2010–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Peng, F.; Zhang, B.; Ingram, A.J.; Gao, B.; Krepinsky, J.C. Collagen I induction by high glucose levels is mediated by epidermal growth factor receptor and phosphoinositide 3-kinase/Akt signalling in mesangial cells. Diabetology 2007, 50, 2008–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, S.; Fujimoto, K.; Takada, T.; Kawamura, S.; Ogawa, J.; Kamata, Y.; Kodera, Y.; Shichiri, M. Molecular form and concentration of serum α2-macroglobulin in diabetes. Sci. Rep. 2019, 9, 12927. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, J.; Singh, M.; Saleemuddin, M. A study of plasma alpha-2-macroglobulin levels in type 2 diabetic subjects with microalbuminuria. J. Assoc. Physicians India 2001, 49, 1062–1065. [Google Scholar]
- French, K.; Yerbury, J.J.; Wilson, M.R. Protease Activation of α2-Macroglobulin Modulates a Chaperone-like Action with Broad Specificity. Biochemistry 2008, 47, 1176–1185. [Google Scholar] [CrossRef]
- Kim, K.M.; Chung, K.W.; Jeong, H.O.; Lee, B.; Kim, D.H.; Park, J.W.; Kim, S.M.; Yu, B.P.; Chung, H.Y. MMP2-A2M interaction increases ECM accumulation in aged rat kidney and its modulation by calorie restriction. Oncotarget 2017, 9, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Van Goor, H.; Diamond, J.R.; Ding, G.; Kaysen, G. Alpha Macroglobulins and the Low-Density-Lipoprotein-Related Protein/Alpha-2-Macroglobulin Receptor in Experimental Renal Fibrosis. Nephron 1999, 7, 35–43. [Google Scholar] [CrossRef]
- Mehta, N.; Gava, A.L.; Zhang, D.; Gao, B.; Krepinsky, J.C. Follistatin Protects Against Glomerular Mesangial Cell Apoptosis and Oxidative Stress to Ameliorate Chronic Kidney Disease. Antioxid. Redox Signal. 2019, 31, 551–571. [Google Scholar] [CrossRef]
- Yang, J.-K.; Wang, Y.-Y.; Liu, C.; Shi, T.-T.; Lu, J.; Cao, X.; Yang, F.-Y.; Feng, J.-P.; Chen, C.; Ji, L.-N.; et al. Urine Proteome Specific for Eye Damage Can Predict Kidney Damage in Patients with Type 2 Diabetes: A Case-Control and a 5.3-Year Prospective Cohort Study. Diabetes Care 2016, 40, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Calvo, R.; Ferrán, B.; Alonso, J.; Martí-Pàmies, I.; Aguiló, S.; Calvayrac, O.; Rodríguez, C.; Martínez-González, J. NR4A receptors up-regulate the antiproteinase alpha-2 macroglobulin (A2M) and modulate MMP-2 and MMP-9 in vascular smooth muscle cells. Thromb. Haemost. 2015, 113, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Hu, K. LRP-1: Functions, Signaling and Implications in Kidney and Other Diseases. Int. J. Mol. Sci. 2014, 15, 22887–22901. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.-Y.; Wang, W.; Desiderio, S.; Nathans, D. Synergistic Activity of STAT3 and c-Jun at a Specific Array of DNA Elements in the α2-Macroglobulin Promoter. J. Biol. Chem. 2001, 276, 26421–26429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Devi, Y.S.; Bowen-Shauver, J.; Ferguson-Gottschall, S.; Robb, L.; Gibori, G. The Role of Interleukin-11 in Pregnancy Involves Up-Regulation of α2-Macroglobulin Gene through Janus Kinase 2-Signal Transducer and Activator of Transcription 3 Pathway in the Decidua. Mol. Endocrinol. 2006, 20, 3240–3250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosius, F.C.; Tuttle, K.; Kretzler, M. JAK inhibition in the treatment of diabetic kidney disease. Diabetology 2016, 59, 1624–1627. [Google Scholar] [CrossRef]
- Weigert, C.; Sauer, U.; Brodbeck, K.; Pfeiffer, A.; Häring, H.U.; Schleicher, E.D. AP-1 Proteins Mediate Hyperglycemia-Induced Activation of the Human TGF-β1 Promoter in Mesangial Cells. J. Am. Soc. Nephrol. 2000, 11, 2007–2016. [Google Scholar] [CrossRef]
- Misra, U.K.; Payne, S.; Pizzo, S.V. Ligation of Prostate Cancer Cell Surface GRP78 Activates a Proproliferative and Antiapoptotic Feedback Loop. J. Biol. Chem. 2011, 286, 1248–1259. [Google Scholar] [CrossRef] [Green Version]
- Hinnen, D. Glucagon-Like Peptide 1 Receptor Agonists for Type 2 Diabetes. Diabetes Spectr. 2017, 30, 202–210. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | 18 |
| Male, n (%) | 16 (89) |
| Age (years) | 70 ± 8 |
| Baseline eGFR (mL/min/1.73 m²) | 25 ± 10 |
| eGFR decline rate (mL/min/1.73 m²/year) | −2 ± 2 |
| Follow-up period (year) | 2.3 ± 0.6 |
| Protein/creatinine ratio (g/g) | 1.3 ± 0.5 |
| SBP (mmHg) | 142 ± 14 |
| DBP (mmHg) | 66 ± 8 |
| RASB use, n (%) | 17 (94) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trink, J.; Li, R.; Palarasah, Y.; Troyanov, S.; Andersen, T.E.; Sidelmann, J.J.; Inman, M.D.; Pizzo, S.V.; Gao, B.; Krepinsky, J.C. Activated Alpha 2-Macroglobulin Is a Novel Mediator of Mesangial Cell Profibrotic Signaling in Diabetic Kidney Disease. Biomedicines 2021, 9, 1112. https://doi.org/10.3390/biomedicines9091112
Trink J, Li R, Palarasah Y, Troyanov S, Andersen TE, Sidelmann JJ, Inman MD, Pizzo SV, Gao B, Krepinsky JC. Activated Alpha 2-Macroglobulin Is a Novel Mediator of Mesangial Cell Profibrotic Signaling in Diabetic Kidney Disease. Biomedicines. 2021; 9(9):1112. https://doi.org/10.3390/biomedicines9091112
Chicago/Turabian StyleTrink, Jackie, Renzhong Li, Yaseelan Palarasah, Stéphan Troyanov, Thomas E. Andersen, Johannes J. Sidelmann, Mark D. Inman, Salvatore V. Pizzo, Bo Gao, and Joan C. Krepinsky. 2021. "Activated Alpha 2-Macroglobulin Is a Novel Mediator of Mesangial Cell Profibrotic Signaling in Diabetic Kidney Disease" Biomedicines 9, no. 9: 1112. https://doi.org/10.3390/biomedicines9091112
APA StyleTrink, J., Li, R., Palarasah, Y., Troyanov, S., Andersen, T. E., Sidelmann, J. J., Inman, M. D., Pizzo, S. V., Gao, B., & Krepinsky, J. C. (2021). Activated Alpha 2-Macroglobulin Is a Novel Mediator of Mesangial Cell Profibrotic Signaling in Diabetic Kidney Disease. Biomedicines, 9(9), 1112. https://doi.org/10.3390/biomedicines9091112