
Immunometabolism Modulation in Therapy

 , ,
, ,  , , and
, , and
Abstract
1. Introduction
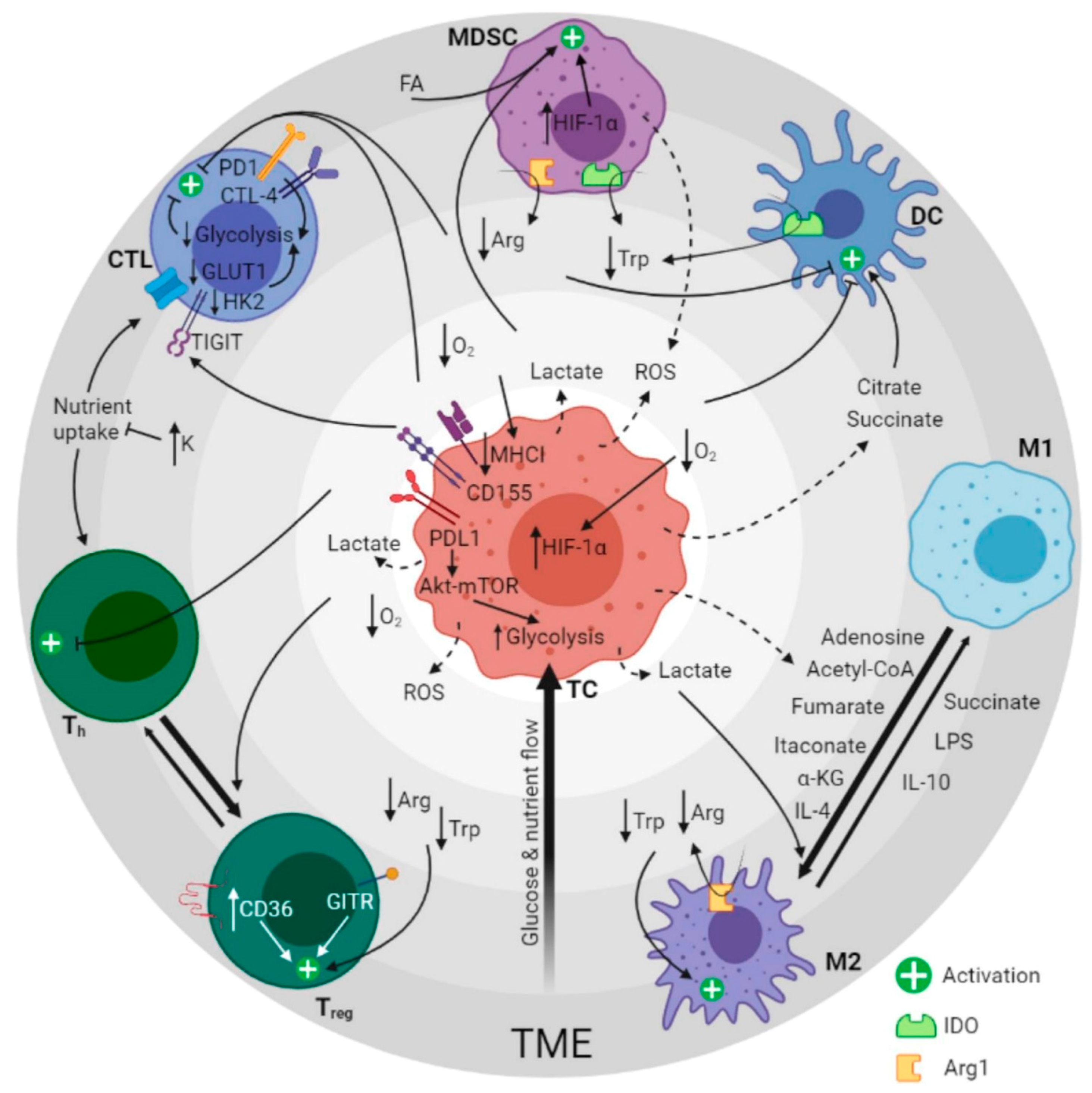
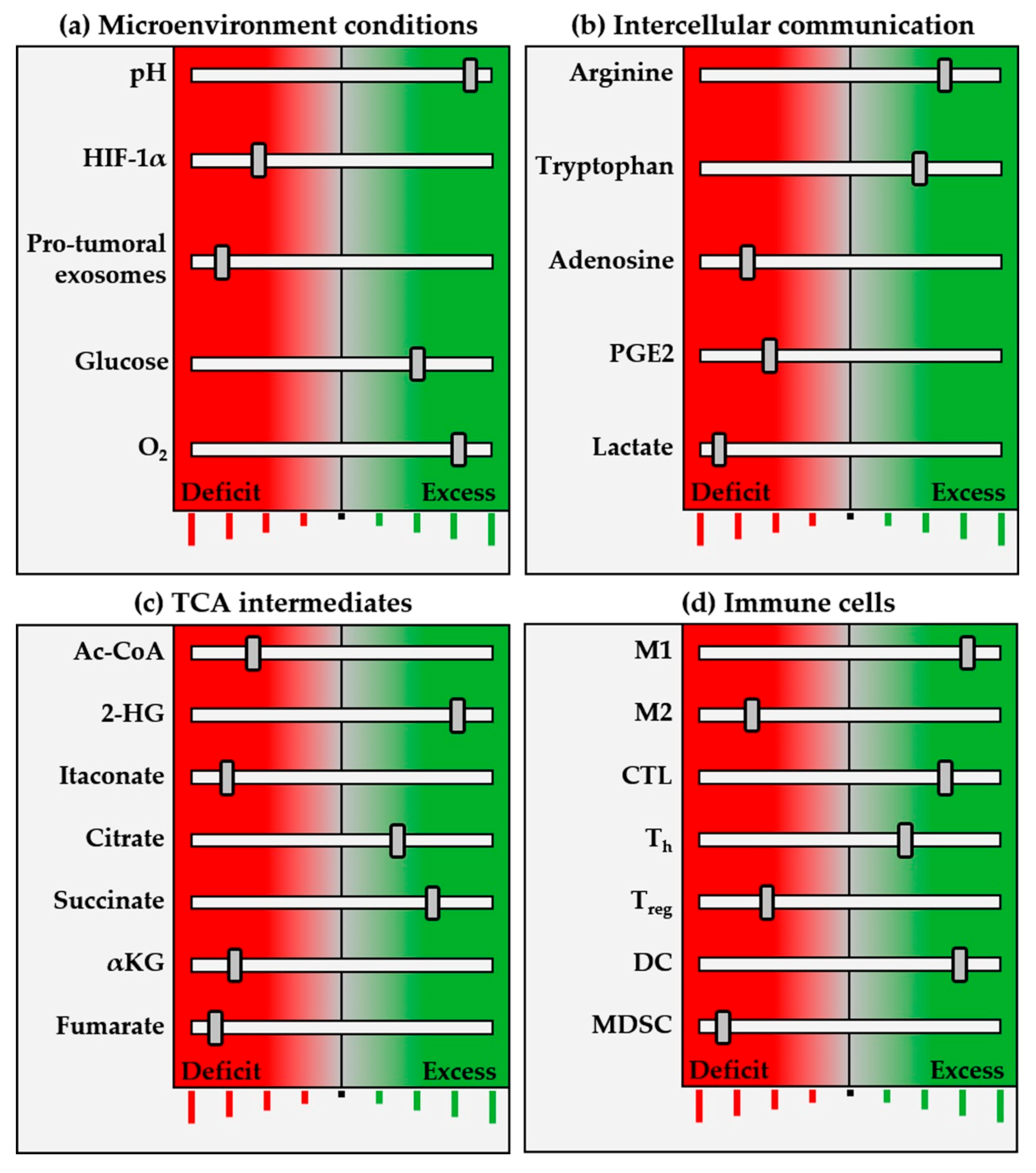
2. Metabolic Pathways of the Tumor Ecosystem
2.1. Metabolic Reprogramming
2.2. Metabolic Skewing Induced by Viral Infections
2.3. Modulation by Exosomes
3. Immunometabolites and Oncometabolites
3.1. MDSC Fate and Function
3.2. Complications of T-Cell Therapies
3.3. Polarization of Macrophages
3.4. Dendritic Cell Subsets in Immune Response Regulation
4. Therapeutic Applications of Immunometabolism Regulation
4.1. Immunotherapy Based on Exosomes
4.2. T-Cell Regulation
4.3. Macrophage Regulation with Immunometabolites
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Andrejeva, G.; Rathmell, J.C. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef] [PubMed]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.N.; Kauer, N.; Blazquez, R.; Hacker, L.; et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 2019, 29, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Tyrakis, P.A.; Palazon, A.; Macias, D.; Lee, K.L.; Phan, A.T.; Veliça, P.; You, J.; Chia, G.S.; Sim, J.; Doedens, A.; et al. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature 2016, 540, 236–241. [Google Scholar] [CrossRef]
- de Goede, K.E.; Harber, K.J.; Van den Bossche, J. Let’s Enter the Wonderful World of Immunometabolites. Trends Endocrinol. Metab. 2019, 30, 329–331. [Google Scholar] [CrossRef]
- Arnoult, D.; Soares, F.; Tattoli, I.; Girardin, S.E. Mitochondria in innate immunity. EMBO Rep. 2011, 12, 901–910. [Google Scholar] [CrossRef]
- Choi, I.; Son, H.; Baek, J.H. Tricarboxylic acid (Tca) cycle intermediates: Regulators of immune responses. Life 2021, 11, 69. [Google Scholar] [CrossRef]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab. 2019, 1, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; O’Neill, L.A.J. Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 2018, 174, 780–784. [Google Scholar] [CrossRef]
- Patil, N.K.; Bohannon, J.K.; Hernandez, A.; Patil, T.K.; Sherwood, E.R. Regulation of leukocyte function by citric acid cycle intermediates. J. Leukoc. Biol. 2019, 106, 105–117. [Google Scholar] [CrossRef]
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436. [Google Scholar] [CrossRef]
- Van Der Heijden, C.D.C.C.; Noz, M.P.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P.; Keating, S.T. Epigenetics and Trained Immunity. Antioxid. Redox Signal. 2018, 29, 1023–1040. [Google Scholar] [CrossRef]
- Monferrer, E.; Vieco-Martí, I.; López-Carrasco, A.; Fariñas, F.; Abanades, S.; de la Cruz-Merino, L.; Noguera, R.; Naranjo, T.Á. Metabolic classification and intervention opportunities for tumor energy dysfunction. Metabolites 2021, 11, 264. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Phan, L.M.; Yeung, S.C.J.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [PubMed]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell. Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Mao, Q.; Xia, W.; Xu, Y.; Wang, J.; Xu, L.; Jiang, F. PKM2 and cancer: The function of PKM2 beyond glycolysis (Review). Oncol. Lett. 2016, 11, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Iglesias, A.; Mañes, S. The importance of mitochondrial pyruvate carrier in cancer cell metabolism and tumorigenesis. Cancers 2021, 13, 1488. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef] [PubMed]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- Li, L.; Meng, Y.; Li, Z.; Dai, W.; Xu, X.; Bi, X.; Bian, J. Discovery and development of small molecule modulators targeting glutamine metabolism. Eur. J. Med. Chem. 2019, 163, 215–242. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Escoll, P.; Buchrieser, C. Metabolic reprogramming of host cells upon bacterial infection: Why shift to a Warburg-like metabolism? FEBS J. 2018, 285, 2146–2160. [Google Scholar] [CrossRef]
- Thaker, S.K.; Ch’ng, J.; Christofk, H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Dengue Virus Induces and Requires Glycolysis for Optimal Replication. J. Virol. 2015, 89, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.T.; Lee, D.Y.; Huang, Y.T.; Kou, G.H.; Wang, H.C.; Chang, G.D.; Lo, C.F. Six Hours after Infection, the Metabolic Changes Induced by WSSV Neutralize the Host’s Oxidative Stress Defenses. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Su, M.A.; Huang, Y.T.; Chen, I.T.; Lee, D.Y.; Hsieh, Y.C.; Li, C.Y.; Ng, T.H.; Liang, S.Y.; Lin, S.Y.; Huang, S.W.; et al. An Invertebrate Warburg Effect: A Shrimp Virus Achieves Successful Replication by Altering the Host Metabolome via the PI3K-Akt-mTOR Pathway. PLoS Pathog. 2014, 10, e1004196. [Google Scholar] [CrossRef]
- Luo, G.G.; Ou, J.J. Oncogenic viruses and cancer. Virol. Sin. 2015, 30, 83–84. [Google Scholar] [CrossRef]
- Black, P.H. Recent Advances in the Study of Oncogenic Viruses. N. Engl. J. Med. 1966, 275, 377–383. [Google Scholar] [CrossRef]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef]
- White, M.K.; Pagano, J.S.; Khalili, K. Viruses and human cancers: A long road of discovery of molecular paradigms. Clin. Microbiol. Rev. 2014, 27, 463–481. [Google Scholar] [CrossRef] [PubMed]
- Vastag, L.; Koyuncu, E.; Grady, S.L.; Shenk, T.E.; Rabinowitz, J.D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011, 7, e1002124. [Google Scholar] [CrossRef]
- Yu, Y.; Clippinger, A.J.; Alwine, J.C. Viral effects on metabolism: Changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol. 2011, 19, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.X.; Huang, C.J.; Yang, Z.G. Impact of hepatitis B virus infection on hepatic metabolic signaling pathway. World J. Gastroenterol. 2016, 22, 8161–8167. [Google Scholar] [CrossRef] [PubMed]
- Masson, J.J.R.; Billings, H.W.W.; Palmer, C.S. Metabolic reprogramming during hepatitis B disease progression offers novel diagnostic and therapeutic opportunities. Antivir. Chem. Chemother. 2017, 25, 53–57. [Google Scholar] [CrossRef] [PubMed]
- El-Sharkawy, A.; Al Zaidan, L.; Malki, A. Epstein-Barr virus-associated malignancies: Roles of viral oncoproteins in carcinogenesis. Front. Oncol. 2018, 8, 265. [Google Scholar] [CrossRef]
- Wang, L.W.; Jiang, S.; Gewurz, B.E. Epstein-Barr Virus LMP1-Mediated Oncogenicity. J. Virol. 2017, 91, e01718-16. [Google Scholar] [CrossRef]
- Thomas, M.; David, P.; Banks, L. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene 1999, 18, 7690–7700. [Google Scholar] [CrossRef]
- Eisenreich, W.; Rudel, T.; Heesemann, J.; Goebel, W. How viral and intracellular bacterial pathogens reprogram the metabolism of host cells to allow their intracellular replication. Front. Cell. Infect. Microbiol. 2019, 9, 42. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and Inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef]
- Zhu, C.; Zhu, Q.; Wang, C.; Zhang, L.; Wei, F.; Cai, Q. Hostile takeover: Manipulation of HIF-1 signaling in pathogen-associated cancers (Review). Int. J. Oncol. 2016, 49, 1269–1276. [Google Scholar] [CrossRef][Green Version]
- Cuninghame, S.; Jackson, R.; Zehbe, I. Hypoxia-inducible factor 1 and its role in viral carcinogenesis. Virology 2014, 456–457, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Meng, X.; Ma, J.; Zheng, Y.; Wang, Q.; Wang, Y.; Shang, H. Human papillomavirus 16 E6 contributes HIF-1α induced warburg effect by attenuating the VHL-HIF-1α interaction. Int. J. Mol. Sci. 2014, 15, 7974–7986. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, S.A.; de Andrade Júnior, D.R. HIF–1alpha and infectious diseases: A new frontier for the development of new therapies. Rev. Inst. Med. Trop. Sao Paulo 2017, 59, e92. [Google Scholar] [CrossRef] [PubMed]
- Pallett, L.J.; Schmidt, N.; Schurich, A. T cell metabolism in chronic viral infection. Clin. Exp. Immunol. 2019, 197, 143–152. [Google Scholar] [CrossRef]
- Ripoli, M.; D’Aprile, A.; Quarato, G.; Sarasin-Filipowicz, M.; Gouttenoire, J.; Scrima, R.; Cela, O.; Boffoli, D.; Heim, M.H.; Moradpour, D.; et al. Hepatitis C Virus-Linked Mitochondrial Dysfunction Promotes Hypoxia-Inducible Factor 1α-Mediated Glycolytic Adaptation. J. Virol. 2010, 84, 647–660. [Google Scholar] [CrossRef]
- Kraus, R.J.; Yu, X.; Cordes, B.L.A.; Sathiamoorthi, S.; Iempridee, T.; Nawandar, D.M.; Ma, S.; Romero-Masters, J.C.; McChesney, K.G.; Lin, Z.; et al. Hypoxia-inducible factor-1α plays roles in Epstein-Barr virus’s natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLoS Pathog. 2017, 13, e1006404. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, D. Exosomes in cancer development, metastasis, and immunity. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 455–468. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Kosaka, N.; Yoshioka, Y.; Fujita, Y.; Ochiya, T. Versatile roles of extracellular vesicles in cancer. J. Clin. Investig. 2016, 126, 1163–1172. [Google Scholar] [CrossRef]
- Yang, E.; Wang, X.; Gong, Z.; Yu, M.; Wu, H.; Zhang, D. Exosome-mediated metabolic reprogramming: The emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct. Target. Ther. 2020, 5, 1–13. [Google Scholar] [CrossRef]
- Leone, R.D.; Powell, J.D. Metabolism of immune cells in cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Xiang, K.; Jendrossek, V.; Matschke, J. Oncometabolites and the response to radiotherapy. Radiat. Oncol. 2020, 15, 1–10. [Google Scholar] [CrossRef]
- Weng, C.Y.; Kao, C.X.; Chang, T.S.; Huang, Y.H. Immuno-metabolism: The role of cancer niche in immune checkpoint inhibitor resistance. Int. J. Mol. Sci. 2021, 22, 1258. [Google Scholar] [CrossRef] [PubMed]
- Certo, M.; Tsai, C.H.; Pucino, V.; Ho, P.C.; Mauro, C. Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat. Rev. Immunol. 2021, 21, 151–161. [Google Scholar] [CrossRef]
- Kiran, D.; Basaraba, R.J. Lactate Metabolism and Signaling in Tuberculosis and Cancer: A Comparative Review. Front. Cell. Infect. Microbiol. 2021, 11, 37. [Google Scholar] [CrossRef]
- Davidov, V.; Jensen, G.; Mai, S.; Chen, S.H.; Pan, P.Y. Analyzing One Cell at a TIME: Analysis of Myeloid Cell Contributions in the Tumor Immune Microenvironment. Front. Immunol. 2020, 11, 1842. [Google Scholar] [CrossRef]
- Yan, D.; Adeshakin, A.O.; Xu, M.; Afolabi, L.O.; Zhang, G.; Chen, Y.H.; Wan, X. Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front. Immunol. 2019, 10, 1399. [Google Scholar] [CrossRef]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Del Valle, L.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Ma, E.H.; Verway, M.J.; Johnson, R.M.; Roy, D.G.; Steadman, M.; Hayes, S.; Williams, K.S.; Sheldon, R.D.; Samborska, B.; Kosinski, P.A.; et al. Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8+ T Cells. Immunity 2019, 51, 856–870. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.C.; Bihuniak, J.D.; MacIntyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Ogando, J.; Sáez, M.E.; Santos, J.; Nuevo-Tapioles, C.; Gut, M.; Esteve-Codina, A.; Heath, S.; González-Pérez, A.; Cuezva, J.M.; Lacalle, R.A.; et al. PD-1 signaling affects cristae morphology and leads to mitochondrial dysfunction in human CD8+ T lymphocytes. J. Immunother. Cancer 2019, 7, 151. [Google Scholar] [CrossRef]
- Kumar Vodnala, S.; Eil, R.; Kishton, R.J.; Sukumar, M.; Yamamoto, T.N.; Ha, N.-H.; Lee, P.-H.; Shin, M.; Patel, S.J.; Yu, Z.; et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 2019, 363, 6434. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Rosen, D.B.; Grein, J.; Tedesco, D.; Joyce-Shaikh, B.; Ueda, R.; Semana, M.; Bauer, M.; Bang, K.; Stevenson, C.; et al. GITR agonism enhances cellular metabolism to support CD8+ T-cell proliferation and effector cytokine production in a mouse tumor model. Cancer Immunol. Res. 2018, 6, 1199–1211. [Google Scholar] [CrossRef]
- Walton, Z.E.; Patel, C.H.; Brooks, R.C.; Yu, Y.; Ibrahim-Hashim, A.; Riddle, M.; Porcu, A.; Jiang, T.; Ecker, B.L.; Tameire, F.; et al. Acid Suspends the Circadian Clock in Hypoxia through Inhibition of mTOR. Cell 2018, 174, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Delgoffe, G.M.; Meyer, C.F.; Chan, W.; Powell, J.D. Anergic T Cells Are Metabolically Anergic. J. Immunol. 2009, 183, 6095–6101. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, M.; Stabilini, A.; Roncarolo, M.G. Rapamycin selectively expands CD4+CD25+FoxP3 + regulatory T cells. Blood 2005, 105, 4743–4748. [Google Scholar] [CrossRef]
- Wang, H.; Franco, F.; Tsui, Y.-C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.-H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef]
- Mougiakakos, D.; Johansson, C.C.; Kiessling, R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood 2009, 113, 3542–3545. [Google Scholar] [CrossRef]
- Marijt, K.A.; Sluijter, M.; Blijleven, L.; Tolmeijer, S.H.; Scheeren, F.A.; Van Der Burg, S.H.; Van Hall, T. Metabolic stress in cancer cells induces immune escape through a PI3K-dependent blockade of IFNγreceptor signaling. J. Immunother. Cancer 2019, 7. [Google Scholar] [CrossRef]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef]
- Batista-Gonzalez, A.; Vidal, R.; Criollo, A.; Carreño, L.J. New Insights on the Role of Lipid Metabolism in the Metabolic Reprogramming of Macrophages. Front. Immunol. 2020, 10, 2993. [Google Scholar] [CrossRef]
- Ryan, D.G.; O’Neill, L.A.J. Krebs Cycle Reborn in Macrophage Immunometabolism. Annu. Rev. Immunol. 2020, 38, 289–313. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Artyomov, M.N. Itaconate: The poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 2019, 19, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Hooftman, A.; O’Neill, L.A.J. The Immunomodulatory Potential of the Metabolite Itaconate. Trends Immunol. 2019, 40, 687–698. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-Mcdermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Hayes, C.S.; Shicora, A.C.; Keough, M.P.; Snook, A.E.; Burns, M.R.; Gilmour, S.K. Polyamine-blocking therapy reverses immunosuppression in the tumor microenvironment. Cancer Immunol. Res. 2014, 2, 274–285. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Dowling, J.K.; Afzal, R.; Gearing, L.J.; Cervantes-Silva, M.P.; Annett, S.; Davis, G.M.; De Santi, C.; Assmann, N.; Dettmer, K.; Gough, D.J.; et al. Mitochondrial arginase-2 is essential for IL-10 metabolic reprogramming of inflammatory macrophages. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Liu, P.S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; DI Conza, G.; Cheng, W.C.; Chou, C.H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Aksoylar, H.I.; Yu, J.; Snyder, N.W.; Worth, A.J.; Iyer, S.S.; Wang, J.; Ben-Sahra, I.; Byles, V.; Polynne-Stapornkul, T.; et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife 2016, 5, e11612. [Google Scholar] [CrossRef]
- Dominguez, M.; Brüne, B.; Namgaladze, D. Exploring the Role of ATP-Citrate Lyase in the Immune System. Front. Immunol. 2021, 12, 14. [Google Scholar] [CrossRef]
- Csóka, B.; Selmeczy, Z.; Koscsó, B.; Németh, Z.H.; Pacher, P.; Murray, P.J.; Kepka-Lenhart, D.; Morris, S.M., Jr.; Gause, W.C.; Leibovich, S.J.; et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012, 26, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Henze, A.T.; Mazzone, M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Investig. 2016, 126, 3672–3679. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, C.; Tewari, B.N.; Kanchan, R.K.; Baghel, K.S.; Nautiyal, N.; Shrivastava, R.; Kaur, H.; Bramha Bhatt, M.L.; Bhadauria, S. Macrophages are recruited to hypoxic tumor areas and acquire a Pro-Angiogenic M2-Polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014, 5, 5350–5368. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Nasi, A.; Fekete, T.; Krishnamurthy, A.; Snowden, S.; Rajnavölgyi, E.; Catrina, A.I.; Wheelock, C.E.; Vivar, N.; Rethi, B. Dendritic Cell Reprogramming by Endogenously Produced Lactic Acid. J. Immunol. 2013, 191, 3090–3099. [Google Scholar] [CrossRef]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting tumor microenvironment for cancer therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Chen, Y.S.; Lin, E.Y.; Chiou, T.W.; Harn, H.J. Exosomes in clinical trial and their production in compliance with good manufacturing practice. Tzu Chi Med. J. 2020, 32, 113–120. [Google Scholar]
- Rosato, P.C.; Wijeyesinghe, S.; Stolley, J.M.; Nelson, C.E.; Davis, R.L.; Manlove, L.S.; Pennell, C.A.; Blazar, B.R.; Chen, C.C.; Geller, M.A.; et al. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Farrell, P.J. Epstein-Barr Virus and Cancer. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 29–53. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Zhao, Y.Y.; Shen, J.; Sun, X.; Liu, Y.; Liu, H.; Wang, Y.; Wang, J. Nanoenabled Modulation of Acidic Tumor Microenvironment Reverses Anergy of Infiltrating T Cells and Potentiates Anti-PD-1 Therapy. Nano Lett. 2019, 19, 2774–2783. [Google Scholar] [CrossRef]
- Daneshmandi, S.; Wegiel, B.; Seth, P. Blockade of lactate dehydrogenase-A (LDH-A) improves efficacy of anti-programmed cell death-1 (PD-1) therapy in melanoma. Cancers 2019, 11, 450. [Google Scholar] [CrossRef]
- Seth, P.; Csizmadia, E.; Hedblom, A.; Vuerich, M.; Xie, H.; Li, M.; Longhi, M.S.; Wegiel, B. Deletion of lactate dehydrogenase-A in myeloid cells triggers antitumor immunity. Cancer Res. 2017, 77, 3632–3643. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.A.; Geeganage, S.; Amaladas, N.; Lu, Z.H.; Rasmussen, E.R.; Sonyi, A.; Chin, D.; Capen, A.; Li, Y.; Meyer, C.M.; et al. The folate pathway inhibitor pemetrexed pleiotropically enhances effects of cancer immunotherapy. Clin. Cancer Res. 2019, 25, 7175–7188. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Stewart, K.M.; Wang, X.; Liu, K.; Xie, M.; Kyu Ryu, J.; Li, K.; Ma, T.; Wang, H.; Ni, L.; et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature 2017, 548, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Villa, M.; Sanin, D.E.; Buck, M.D.; O’Sullivan, D.; Ching, R.; Matsushita, M.; Grzes, K.M.; Winkler, F.; Chang, C.H.; et al. Acetate Promotes T Cell Effector Function during Glucose Restriction. Cell Rep. 2019, 27, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Kilgour, M.K.; MacPherson, S.; Zacharias, L.G.; Ellis, A.E.; Sheldon, R.D.; Liu, E.Y.; Keyes, S.; Pauly, B.; Carleton, G.; Allard, B.; et al. 1-Methylnicotinamide is an immune regulatory metabolite in human ovarian cancer. Sci. Adv. 2021, 7, eabe1174. [Google Scholar] [CrossRef] [PubMed]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor–Redirected T Cells as well as Bystander Cells from Oxidative Stress–Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef]
- Jian, S.L.; Chen, W.W.; Su, Y.C.; Su, Y.W.; Chuang, T.H.; Hsu, S.C.; Huang, L.R. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ROS-mediated apoptosis. Cell Death Dis. 2017, 8. [Google Scholar] [CrossRef]
- Aricò, E.; Castiello, L.; Capone, I.; Gabriele, L.; Belardelli, F. Type i interferons and cancer: An evolving story demanding novel clinical applications. Cancers 2019, 11, 1943. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Zhang, D.W.; Zheng, X.L.; Tang, C.K. Itaconate: An emerging determinant of inflammation in activated macrophages. Immunol. Cell Biol. 2019, 97, 134–141. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study Type | Status | Study Title | Conditions | Identifier |
|---|---|---|---|---|
| Interventional (Clinical Trial) | Active, not recruiting | Gene and Vaccine Therapy in Treating Patients With Advanced Malignancies | Malignant Neoplasm | NCT01697527 |
| Interventional (Clinical Trial) | Not yet recruiting | Metformin for Chemoprevention of Lung Cancer in High Risk Obese Individuals | Lung Carcinoma | NCT04931017 |
| Interventional (Clinical Trial) | Not yet recruiting | Microenvironment and Immunity of Digestive Cancers - East Paris Multicentric Cohort (MICADO) | Colorectal Cancer Pancreas Tumor Biliary Tract Tumor Immune System and Related Disorders | NCT04707365 |
| Interventional (Clinical Trial) | Recruiting | Lower Dose Decitabine (DAC)-Primed TC (Carboplatin-Paclitaxel) Regimen in Ovary Cancer (DAC and CT) | Primary Malignant Neoplasm of Ovary FIGO Stages II to IV | NCT02159820 |
| Interventional (Clinical Trial) | Recruiting | Paclitaxel + Carboplatin + Durvalumab With or Without Oleclumab for Previously Untreated Locally Recurrent Inoperable or Metastatic TNBC (SYNERGY) | Triple-Negative Breast Cancer (TNBC) | NCT03616886 |
| Interventional (Clinical Trial) | Recruiting | Study of Sirolimus in Patients With Advanced Pancreatic Cancer | Pancreatic Cancer | NCT03662412 |
| Observational | Active, not recruiting | Myeloid Cell Reprogramming in the Context of Radioiodine Therapy in Patients With Non-Medullary Thyroid Carcinoma | Thyroid Cancer | NCT03397238 |
| Observational | Not yet recruiting | Targeting Potassium Channels to Reprogram Glioblastoma Microenvironment: In Vitro and In Vivo Studies | Cancer of Head and Neck | NCT03954691 |
| Observational | Not yet recruiting | Evaluating Immunological Parameters, Neurocognitive Changes, Activity Levels, and Driving Fitness in Patients Undergoing CAR-T Cell Therapy | Hematologic Neoplasms | NCT04275154 |
| Observational | Recruiting | The Mechanism of Enhancing the Anti-Tumor Effects of CAR-T on PC by Gut Microbiota Regulation | Pancreatic Cancer Gut Microbiota CAR-T | NCT04203459 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monferrer, E.; Sanegre, S.; Vieco-Martí, I.; López-Carrasco, A.; Fariñas, F.; Villatoro, A.; Abanades, S.; Mañes, S.; de la Cruz-Merino, L.; Noguera, R.; et al. Immunometabolism Modulation in Therapy. Biomedicines 2021, 9, 798. https://doi.org/10.3390/biomedicines9070798
Monferrer E, Sanegre S, Vieco-Martí I, López-Carrasco A, Fariñas F, Villatoro A, Abanades S, Mañes S, de la Cruz-Merino L, Noguera R, et al. Immunometabolism Modulation in Therapy. Biomedicines. 2021; 9(7):798. https://doi.org/10.3390/biomedicines9070798
Chicago/Turabian StyleMonferrer, Ezequiel, Sabina Sanegre, Isaac Vieco-Martí, Amparo López-Carrasco, Fernando Fariñas, Antonio Villatoro, Sergio Abanades, Santos Mañes, Luis de la Cruz-Merino, Rosa Noguera, and et al. 2021. "Immunometabolism Modulation in Therapy" Biomedicines 9, no. 7: 798. https://doi.org/10.3390/biomedicines9070798
APA StyleMonferrer, E., Sanegre, S., Vieco-Martí, I., López-Carrasco, A., Fariñas, F., Villatoro, A., Abanades, S., Mañes, S., de la Cruz-Merino, L., Noguera, R., & Álvaro Naranjo, T. (2021). Immunometabolism Modulation in Therapy. Biomedicines, 9(7), 798. https://doi.org/10.3390/biomedicines9070798