
Metabolic Phenotypes and Step by Step Evolution of Type 2 Diabetes: A New Paradigm

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
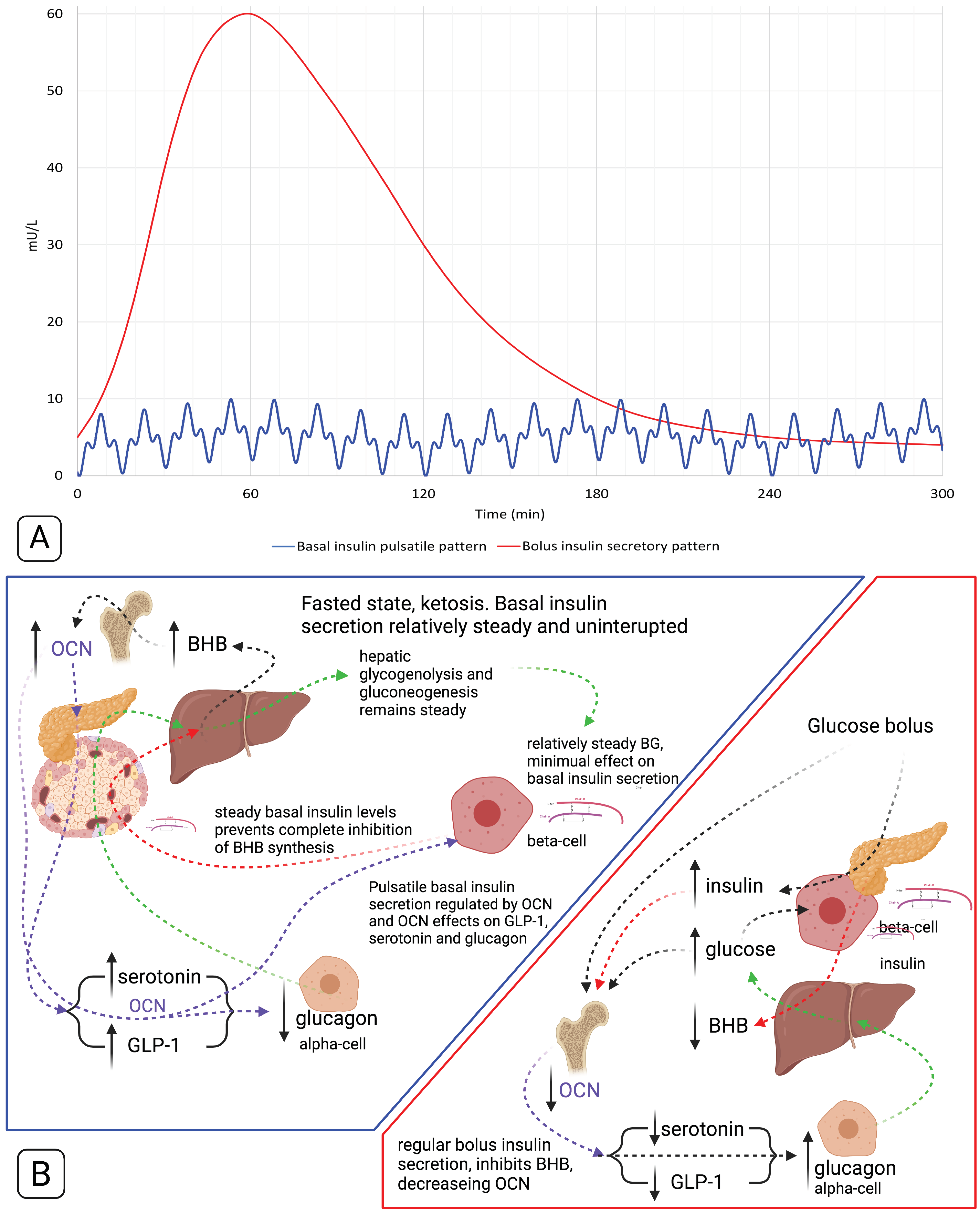
2. Bolus Insulin Secretion
3. Basal Insulin
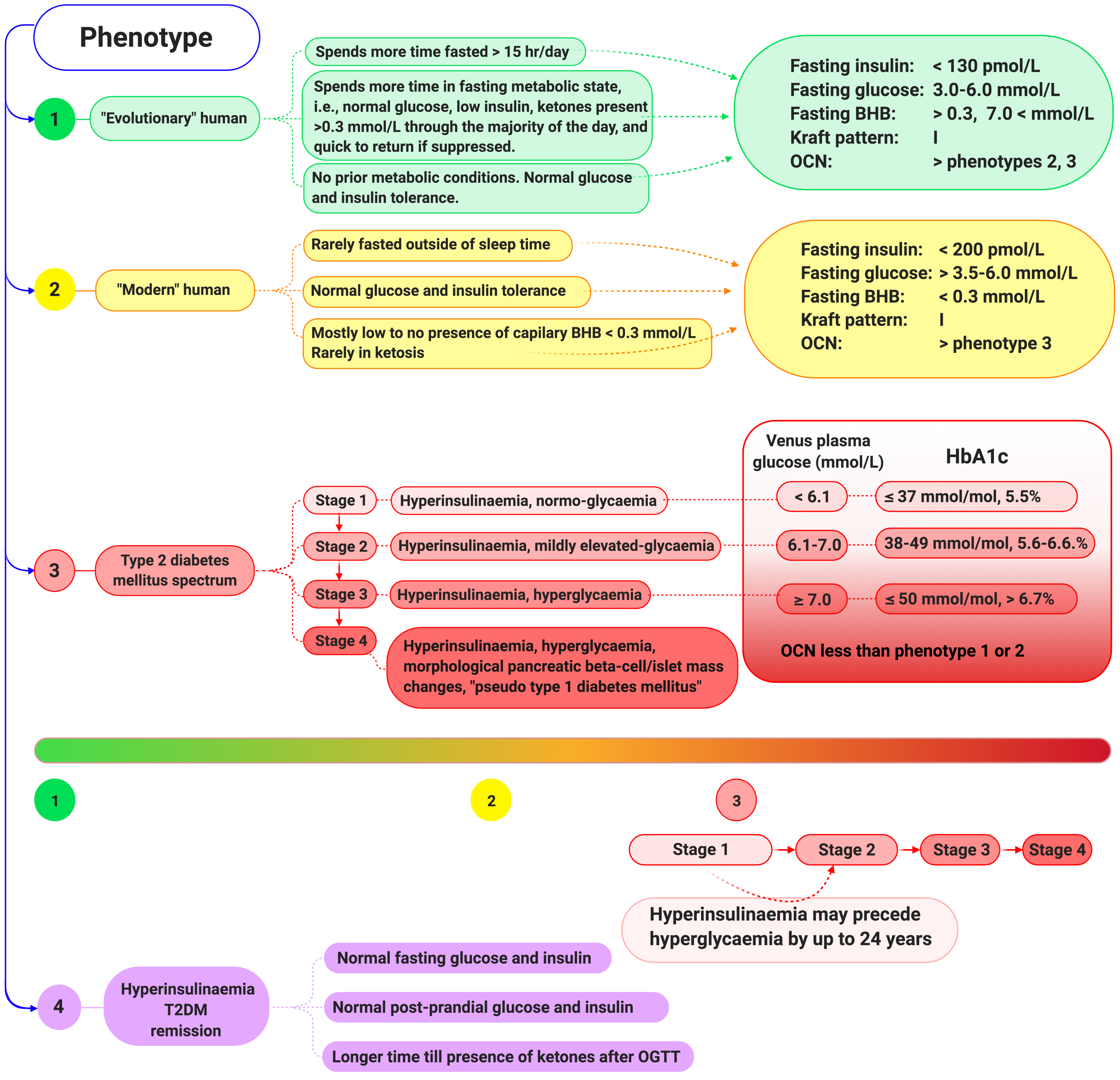
4. Insulin Secretion in the Insulin Resistant/Hyperinsulinaemic State
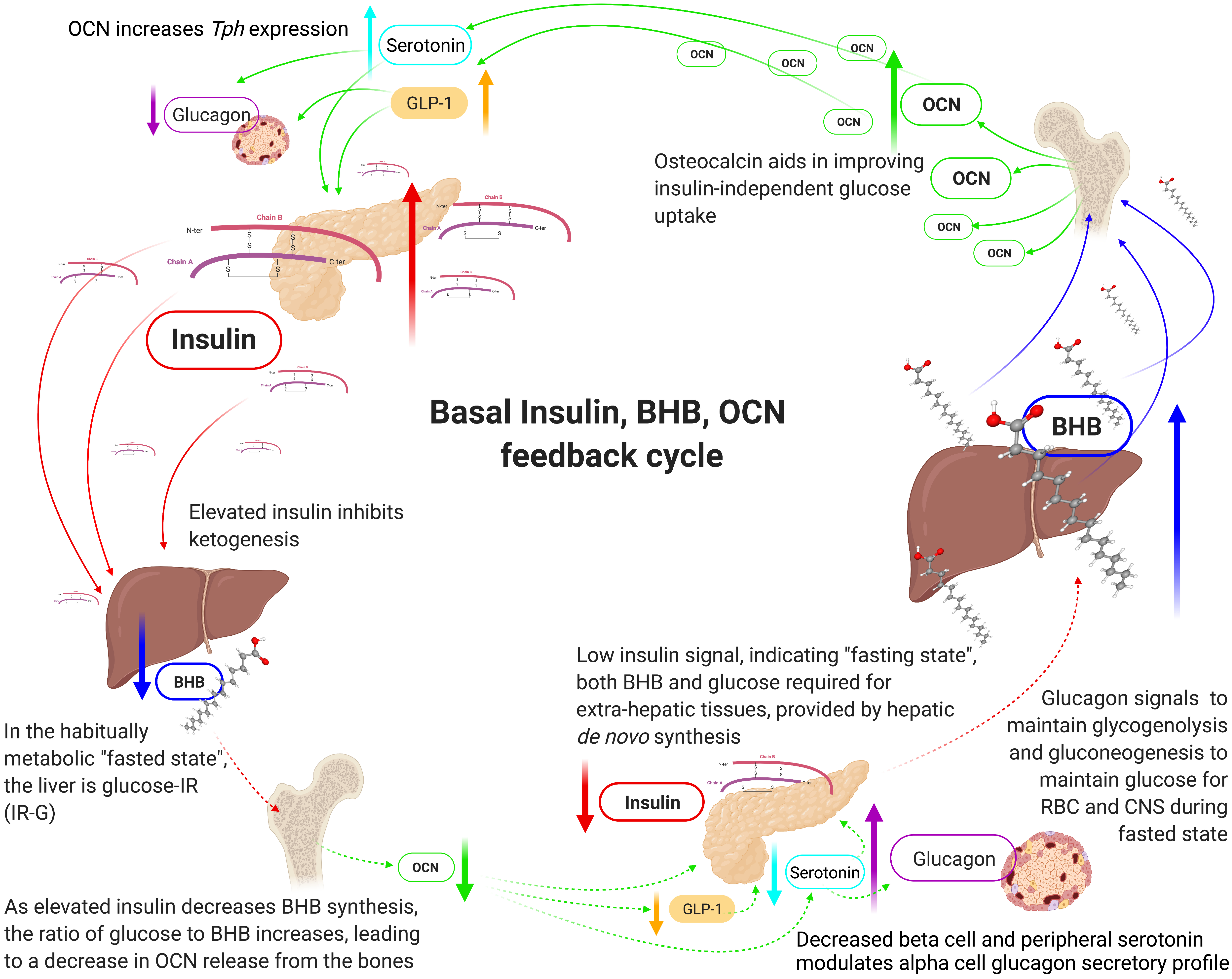
5. An Alternative Hypothesis: Insulin’s Main Role Is to Regulate Beta-Hydroxybutyrate Synthesis
6. Clinical Implications
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boland, B.B.; Brown, J.C.; Alarcon, C.; Demozay, D.; Grimsby, J.S.; Rhodes, C.J. β-Cell Control of Insulin Production During Starvation-Refeeding in Male Rats. Endocrinology 2017, 159, 895–906. [Google Scholar] [CrossRef]
- Aspinwall, C.; Lakey, J.R.T.; Kennedy, R.T. Insulin-stimulated Insulin Secretion in Single Pancreatic Beta Cells. J. Biol. Chem. 1999, 274, 6360–6365. [Google Scholar] [CrossRef]
- Keane, K.N.; Newsholme, P. Metabolic Regulation of Insulin Secretion. Vitam. Horm. 2014, 95, 1–33. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, S.; Koh, H.-C.E.; Patterson, B.W.; Yoshino, M.; LaForest, R.; Gropler, R.J.; Klein, S.; Mittendorfer, B. Obesity Is Associated With Increased Basal and Postprandial β-Cell Insulin Secretion Even in the Absence of Insulin Resistance. Diabetes 2020, 69, 2112–2119. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Braun, M. Regulation of Insulin Secretion in Human Pancreatic Islets. Annu. Rev. Physiol. 2013, 75, 155–179. [Google Scholar] [CrossRef] [PubMed]
- Crofts, C.A.P. Hyperinsulinemia: A unifying theory of chronic disease? Diabesity 2015, 1, 34. [Google Scholar] [CrossRef]
- Seino, S.; Shibasaki, T.; Minami, K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Investig. 2011, 121, 2118–2125. [Google Scholar] [CrossRef] [PubMed]
- Rachdaoui, N. Insulin: The Friend and the Foe in the Development of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 1770. [Google Scholar] [CrossRef]
- De Vos, A.; Heimberg, H.; Quartier, E.; Huypens, P.; Bouwens, L.; Pipeleers, D.; Schuit, F. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J. Clin. Investig. 1995, 96, 2489–2495. [Google Scholar] [CrossRef]
- McCulloch, L.J.; van de Bunt, M.; Braun, M.; Frayn, K.N.; Clark, A.; Gloyn, A.L. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: Implications for understanding genetic association signals at this locus. Mol. Genet. Metab. 2011, 104, 648–653. [Google Scholar] [CrossRef]
- Gould, G.; Holman, G. The glucose transporter family: Structure, function and tissue-specific expression. Biochem. J. 1993, 295, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, M.L.; Cornish-Bowden, A.; Ureta, T. Evolution and regulatory role of the hexokinases. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 1998, 1401, 242–264. [Google Scholar] [CrossRef]
- Gidh-Jain, M.; Takeda, J.; Xu, L.Z.; Lange, A.J.; Vionnet, N.; Stoffel, M.; Froguel, P.; Velho, G.; Sun, F.; Cohen, D. Glucokinase mutations associated with non-insulin-dependent (type 2) diabetes mellitus have decreased enzymatic activity: Implications for structure/function relationships. Proc. Natl. Acad. Sci. USA 1993, 90, 1932–1936. [Google Scholar] [CrossRef] [PubMed]
- Iynedjian, P. Molecular Physiology of Mammalian Glucokinase. Cell. Mol. Life Sci. 2009, 66, 27–42. [Google Scholar] [CrossRef]
- Fajans, S.S.; Bell, G.I.; Polonsky, K.S. Molecular Mechanisms and Clinical Pathophysiology of Maturity-Onset Diabetes of the Young. N. Engl. J. Med. 2001, 345, 971–980. [Google Scholar] [CrossRef]
- Matschinsky, F.M.; Wilson, D.F. The Central Role of Glucokinase in Glucose Homeostasis: A Perspective 50 Years After Demonstrating the Presence of the Enzyme in Islets of Langerhans. Front. Physiol. 2019, 10, 148. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, E.; Martínez, M.D.; Roncero, I.; Chowen, J.A.; Garcia-Cuartero, B.; Gispert, J.D.; Sanz, C.; Vázquez, P.; Maldonado, A.; De Cáceres, J.; et al. The expression of GLP-1 receptor mRNA and protein allows the effect of GLP-1 on glucose metabolism in the human hypothalamus and brainstem. J. Neurochem. 2005, 92, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Sekine, N.; Cirulli, V.; Regazzi, R.; Brown, L.J.; Gine, E.; Tamarit-Rodriguez, J.; Girotti, M.; Marie, S.; Macdonald, M.J.; Wollheim, C.B. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J. Biol. Chem. 1994, 269, 4895–4902. [Google Scholar] [CrossRef]
- Alcazar, O.; Tiedge, M.; Lenzen, S. Importance of lactate dehydrogenase for the regulation of glycolytic flux and insulin secretion in insulin-producing cells. Biochem. J. 2000, 352, 373–380. [Google Scholar] [CrossRef]
- Ritzel, R.A.; Michael, D.J.; Butler, P.C. Insulin Secretion. In Encyclopedia of Hormones; Elsevier: Amsterdam, The Netherlands, 2003; pp. 384–390. [Google Scholar]
- Henquin, J.-C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 2000, 49, 1751–1760. [Google Scholar] [CrossRef]
- Griesche, N.; Sanchez, G.; Hermans, C.; Idevall-Hagren, O. Cortical mitochondria regulate insulin secretion by local Ca2+ buffering in rodent beta cells. J. Cell Sci. 2019, 132, jcs.228544. [Google Scholar] [CrossRef]
- Cook, D.L.; Hales, N. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature 1984, 311, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Dusonchet, J.; Ashcroft, F. Metabolic regulation of the pancreatic β-cell ATP-sensitive K+ channel: A pas de deux. Diabetes 2004, 53, S113–S122. [Google Scholar] [CrossRef]
- Fridlyand, L.E.; Jacobson, D.A.; Philipson, L. Ion channels and regulation of insulin secretion in human β-cells: A computational systems analysis. Islets 2013, 5, 1–15. [Google Scholar] [CrossRef]
- Braun, M.; Ramracheya, R.; Bengtsson, M.; Zhang, Q.; Karanauskaite, J.; Partridge, C.; Johnson, P.R.; Rorsman, P. Voltage-Gated Ion Channels in Human Pancreatic β-Cells: Electrophysiological Characterization and Role in Insulin Secretion. Diabetes 2008, 57, 1618–1628. [Google Scholar] [CrossRef]
- Rorsman, P.; Renström, E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003, 46, 1029–1045. [Google Scholar] [CrossRef] [PubMed]
- Wollheim, C.; Sharp, G.W. Regulation of insulin release by calcium. Physiol. Rev. 1981, 61, 914–973. [Google Scholar] [CrossRef]
- Georgiadou, E.; Rutter, G.A. Control by Ca2+ of mitochondrial structure and function in pancreatic β-cells. Cell Calcium 2020, 91, 102282. [Google Scholar] [CrossRef]
- Marliss, E.B.; Aoki, T.T.; Unger, R.H.; Soeldner, J.S.; Cahill, G.F. Glucagon levels and metabolic effects in fasting man. J. Clin. Investig. 1970, 49, 2256–2270. [Google Scholar] [CrossRef] [PubMed]
- Færch, K.; Vistisen, D.; Pacini, G.; Torekov, S.; Johansen, N.B.; Witte, D.R.; Jonsson, A.; Pedersen, O.; Hansen, T.; Lauritzen, T.; et al. Insulin Resistance Is Accompanied by Increased Fasting Glucagon and Delayed Glucagon Suppression in Individuals With Normal and Impaired Glucose Regulation. Diabetes 2016, 65, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Gershuni, V.M.; Yan, S.L.; Medici, V. Nutritional Ketosis for Weight Management and Reversal of Metabolic Syndrome. Curr. Nutr. Rep. 2018, 7, 97–106. [Google Scholar] [CrossRef]
- Shannahoff-Khalsa, D.S.; Kennedy, B.; Yates, F.E.; Ziegler, M.G. Low-frequency ultradian insulin rhythms are coupled to cardiovascular, autonomic, and neuroendocrine rhythms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1997, 272, R962–R968. [Google Scholar] [CrossRef] [PubMed]
- Satin, L.S.; Butler, P.C.; Ha, J.; Sherman, A.S. Pulsatile insulin secretion, impaired glucose tolerance and type 2 diabetes. Mol. Asp. Med. 2015, 42, 61–77. [Google Scholar] [CrossRef]
- Franco, M.C.D.; De Leon, R.F.; Villafan-Bernal, J.R. Osteocalcin-GPRC6A: An update of its clinical and biological multi-organic interactions (Review). Mol. Med. Rep. 2019, 19, 15–22. [Google Scholar] [CrossRef]
- Crofts, C.; Schofield, G.; Zinn, C.; Wheldon, M.; Kraft, J. Identifying hyperinsulinaemia in the absence of impaired glucose tolerance: An examination of the Kraft database. Diabetes Res. Clin. Pract. 2016, 118, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Crofts, C.; Neill, A.; Campbell, A.; Bartley, J.; White, D.E. Sleep architecture, insulin resistance and the nasal cycle: Implications for positive airway pressure therapy. J. Insul. Resist. 2018, 3. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Choi, T.G.; Kim, S.S. Physiological Functions of Mitochondrial Reactive Oxygen Species. Free Radic. Med. Biol. 2019. [Google Scholar] [CrossRef]
- Kowalska, M.; Piekut, T.; Prendecki, M.; Sodel, A.; Kozubski, W.; Dorszewska, J. Mitochondrial and Nuclear DNA Oxidative Damage in Physiological and Pathological Aging. DNA Cell Biol. 2020, 39, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef] [PubMed]
- Cooper, I.D.; Crofts, C.A.P.; DiNicolantonio, J.J.; Malhotra, A.; Elliott, B.; Kyriakidou, Y.; Brookler, K.H. Relationships between hyperinsulinaemia, magnesium, vitamin D, thrombosis and COVID-19: Rationale for clinical management. Open Hear. 2020, 7, e001356. [Google Scholar] [CrossRef]
- Sack, M.N.; Finkel, T. Mitochondrial Metabolism, Sirtuins, and Aging. Cold Spring Harb. Perspect. Biol. 2012, 4, a013102. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef]
- Hansen, M.E.; Tippetts, T.S.; Anderson, M.C.; Holub, Z.E.; Moulton, E.R.; Swensen, A.C.; Prince, J.T.; Bikman, B.T. Insulin Increases Ceramide Synthesis in Skeletal Muscle. J. Diabetes Res. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Krauss, S.; Zhang, C.-Y.; Scorrano, L.; Dalgaard, L.T.; St-Pierre, J.; Grey, S.T.; Lowell, B.B. Superoxide-mediated activation of uncoupling protein 2 causes pancreatic β cell dysfunction. J. Clin. Investig. 2003, 112, 1831–1842. [Google Scholar] [CrossRef]
- Anello, M.; Lupi, R.; Spampinato, D.; Piro, S.; Masini, M.; Boggi, U.; Del Prato, S.; Rabuazzo, A.M.; Purrello, F.; Marchetti, P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005, 48, 282–289. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Menezes-Filho, S.L.; Assali, E.A.; Gonçalves, I.G.; Cabral-Costa, J.V.; Abreu, P.; Miller, N.; Nolasco, P.; Laurindo, F.R.M.; Bruni-Cardoso, A.; et al. Mitochondrial morphology regulates organellar Ca2+ uptake and changes cellular Ca2+ homeostasis. FASEB J. 2019, 33, 13176–13188. [Google Scholar] [CrossRef]
- De la Fuente, S.; Lambert, J.; Nichtova, Z.; Sanz, C.F.; Elrod, J.; Sheu, S.-S.; Csordás, G. Spatial Separation of Mitochondrial Calcium Uptake and Extrusion for Energy-Efficient Mitochondrial Calcium Signaling in the Heart. Cell Rep. 2018, 24, 3099–3107.e4. [Google Scholar] [CrossRef]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal. 2011, 9, 19. [Google Scholar] [CrossRef]
- Maechler, P.; Li, N.; Casimir, M.; Vetterli, L.; Frigerio, F.; Brun, T. Role of Mitochondria in β-Cell Function and Dysfunction. In Islets of Langerhans, 2nd ed.; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2015; pp. 633–657. [Google Scholar] [CrossRef]
- Georgiadou, E.; Haythorne, E.; Dickerson, M.T.; Lopez-Noriega, L.; Pullen, T.J.; Xavier, G.D.S.; Davis, S.P.X.; Martinez-Sanchez, A.; Semplici, F.; Rizzuto, R.; et al. The pore-forming subunit MCU of the mitochondrial Ca2+ uniporter is required for normal glucose-stimulated insulin secretion in vitro and in vivo in mice. Diabetologia 2020, 63, 1368–1381. [Google Scholar] [CrossRef]
- Contreras, L.; Drago, I.; Zampese, E.; Pozzan, T. Mitochondria: The calcium connection. Biochim. Biophys. Acta (BBA) Bioenerg. 2010, 1797, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J.; Malhotra, J.D. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2014, 1843, 2233–2239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, I.X.; Ren, J.; Vadrevu, S.; Raghavan, M.; Satin, L.S. ER stress increases store-operated Ca2+ entry (SOCE) and augments basal insulin secretion in pancreatic beta cells. J. Biol. Chem. 2020, 295, 5685–5700. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Koh, D.-S.; Hille, B. Dynamics of Calcium Clearance in Mouse Pancreatic β-Cells. Diabetes 2003, 52, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Ashcroft, F. Pancreatic β-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef]
- Lee, K.-U.; Harris, R.A. Mitochondria and Endoplasmic Reticulum in Diabetes and Its Complications. Exp. Diabetes Res. 2012, 2012, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Di Giovanni, J.; Iborra, C.; Maulet, Y.; Lévêque, C.; El Far, O.; Seagar, M. Calcium-dependent Regulation of SNARE-mediated Membrane Fusion by Calmodulin. J. Biol. Chem. 2010, 285, 23665–23675. [Google Scholar] [CrossRef]
- Thurmond, D.C. Regulation of Insulin Action and Insulin Secretion by SNARE-Mediated Vesicle Exocytosis. In Mechanisms of Insulin Action; Springer: New York, NY, USA, 2007; pp. 52–70. [Google Scholar]
- Bello, O.; Jouannot, O.; Chaudhuri, A.; Stroeva, E.; Coleman, J.; Volynski, K.E.; Rothman, J.E.; Krishnakumar, S.S. Synaptotagmin oligomerization is essential for calcium control of regulated exocytosis. Proc. Natl. Acad. Sci. USA 2018, 115, E7624–E7631. [Google Scholar] [CrossRef]
- Babbar, M.; Sheikh, M.S. Metabolic Stress and Disorders Related to Alterations in Mitochondrial Fission or Fusion. Mol. Cell. Pharmacol. 2013, 5, 109–133. [Google Scholar] [CrossRef]
- Parker, B.A.; Walton, C.M.; Carr, S.T.; Andrus, J.L.; Cheung, E.C.K.; Duplisea, M.J.; Wilson, E.K.; Draney, C.; Lathen, D.R.; Kenner, K.B.; et al. β-Hydroxybutyrate Elicits Favorable Mitochondrial Changes in Skeletal Muscle. Int. J. Mol. Sci. 2018, 19, 2247. [Google Scholar] [CrossRef]
- Gao, J.; Qin, A.; Liu, D.; Ruan, R.; Wang, Q.; Yuan, J.; Cheng, T.S.; Filipovska, A.; Papadimitriou, J.M.; Dai, K.; et al. Endoplasmic reticulum mediates mitochondrial transfer within the osteocyte dendritic network. Sci. Adv. 2019, 5, eaaw7215. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Pullen, T.; Hodson, D.; Sanchez, A.M. Pancreatic β-cell identity, glucose sensing and the control of insulin secretion. Biochem. J. 2015, 466, 203–218. [Google Scholar] [CrossRef]
- Ma, Z.A.; Zhao, Z.; Turk, J. Mitochondrial Dysfunction and β-Cell Failure in Type 2 Diabetes Mellitus. Exp. Diabetes Res. 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- Fex, M.; Nicholas, L.M.; Vishnu, N.; Medina, A.; Sharoyko, V.V.; Nicholls, D.G.; Spégel, P.; Mulder, H. The pathogenetic role of β-cell mitochondria in type 2 diabetes. J. Endocrinol. 2018, 236, R145–R159. [Google Scholar] [CrossRef]
- Matveyenko, A.V.; Butler, P.C. Relationship between β-cell mass and diabetes onset. Diabetes Obes. Metab. 2008, 10, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Cohrs, C.M.; Stertmann, J.; Bozsak, R.; Speier, S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol. Metab. 2017, 6, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Mari, A. β-Cell function in type 2 diabetes. Metabolism 2014, 63, 1217–1227. [Google Scholar] [CrossRef]
- Ogilvie, R.F. The islands of langerhans in 19 cases of obesity. J. Pathol. Bacteriol. 1933, 37, 473–481. [Google Scholar] [CrossRef]
- Cho, J.-H.; Kim, J.-W.; Shin, J.-A.; Shin, J.; Yoon, K.-H. β-cell mass in people with type 2 diabetes. J. Diabetes Investig. 2011, 2, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Pontzer, H.; Wood, B.M.; Raichlen, D.A. Hunter-gatherers as models in public health. Obes. Rev. 2018, 19, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Lougheed, T. The Changing Landscape of Arctic Traditional Food. Environ. Health Perspect. 2010, 118, A386. [Google Scholar] [CrossRef][Green Version]
- Anton, S.D.; Moehl, K.; Donahoo, W.; Marosi, K.; Lee, S.; Mainous, A.G.; Leeuwenburgh, C.; Mattson, M.P. Flipping the Metabolic Switch: Understanding and Applying the Health Benefits of Fasting. Obesity 2018, 26, 254–268. [Google Scholar] [CrossRef]
- Owen, O.E.; Morgan, A.P.; Kemp, H.G.; Sullivan, J.M.; Herrera, M.G.; Cahill, G.F. Brain Metabolism during Fasting. J. Clin. Investig. 1967, 46, 1589–1595. [Google Scholar] [CrossRef]
- Cahill, G.F.; Herrera, M.G.; Morgan, A.P.; Soeldner, J.S.; Steinke, J.; Levy, P.L.; Reichard, G.A.; Kipnis, D.M. Hormone-fuel interrelationships during fasting. J. Clin. Investig. 1966, 45, 1751–1769. [Google Scholar] [CrossRef]
- Dhatariya, K.; Savage, M.W.; Claydon, A. The Management of Diabetic Ketoacidosis in Adults *. Jt. Br. Diabetes Soc. Inpatient Care Gr. NHS. 2021. Available online: https://www.bsped.org.uk/media/1798/bsped-dka-guideline-2020.pdf (accessed on 1 July 2021).
- Seetho, I.W.; Wilding, J. The clinical management of diabetes mellitus. In Clinical Biochemistry: Metabolic and Clinical Aspects, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 305–332. [Google Scholar] [CrossRef]
- Gosmanov, A.R.; Nematollahi, L.R. Diabetic Ketoacidosis—Symptoms, Diagnosis and Treatment. BMJ Best Pract. 2020. Available online: https://bestpractice.bmj.com/topics/en-us/162 (accessed on 4 August 2020).
- Elzouki, A.-N.; Eledrisi, M.S. Management of diabetic ketoacidosis in adults: A narrative review. Saudi J. Med. Med Sci. 2020, 8, 165–173. [Google Scholar] [CrossRef]
- Min, S.; Oh, T.; Baek, S.-I.; Lee, D.-H.; Kim, K.; Moon, J.; Choi, S.H.; Park, K.; Jang, H.; Lim, S. Degree of ketonaemia and its association with insulin resistance after dapagliflozin treatment in type 2 diabetes. Diabetes Metab. 2018, 44, 73–76. [Google Scholar] [CrossRef]
- Ebeling, P.; Koistinen, H.A.; Koivisto, V.A. Insulin-independent glucose transport regulates insulin sensitivity. FEBS Lett. 1998, 436, 301–303. [Google Scholar] [CrossRef]
- Sowers, J.R.; Frohlich, E.D. Insulin and insulin resistance: Impact on blood pressure and cardiovascular disease. Med. Clin. N. Am. 2004, 88, 63–82. [Google Scholar] [CrossRef]
- Marliss, E.; Aoki, T.; Felig, P.; Pozefsky, T.; Cahill, G. Hormones and substrates in the regulation of gluconeogenesis in fasting man. Adv. Enzym. Regul. 1970, 8, 3–11. [Google Scholar] [CrossRef]
- Robinson, A.M.; Williamson, D.H. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of Ketone Body Metabolism and the Role of PPARα. Int. J. Mol. Sci. 2016, 17, 2093. [Google Scholar] [CrossRef] [PubMed]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. β-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Grandl, G.; Straub, L.; Rudigier, C.; Arnold, M.; Wueest, S.; Konrad, D.; Wolfrum, C. Short-term feeding of a ketogenic diet induces more severe hepatic insulin resistance than an obesogenic high-fat diet. J. Physiol. 2018, 596, 4597–4609. [Google Scholar] [CrossRef]
- Santoleri, D.; Titchenell, P.M. Resolving the Paradox of Hepatic Insulin Resistance. CMGH 2019, 7, 447–456. [Google Scholar] [CrossRef]
- Reaven, G.M.; Chen, Y.-D.I.; Golay, A.; Swislocki, A.L.M.; Jaspan, J.B. Documentation of Hyperglucagonemia Throughout the Day in Nonobese and Obese Patients with Noninsulin-Dependent Diabetes Mellitus. J. Clin. Endocrinol. Metab. 1987, 64, 106–110. [Google Scholar] [CrossRef]
- Unger, R.H.; Orci, L. Physiology and pathophysiology of glucagon. Physiol. Rev. 1976, 56, 778–826. [Google Scholar] [CrossRef] [PubMed]
- Ferron, M.; Wei, J.; Yoshizawa, T.; Del Fattore, A.; DePinho, R.; Teti, A.M.; Ducy, P.; Karsenty, G. Insulin Signaling in Osteoblasts Integrates Bone Remodeling and Energy Metabolism. Cell 2010, 142, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Saleem, U.; Mosley, J.T.H.; Kullo, I.J. Serum Osteocalcin Is Associated With Measures of Insulin Resistance, Adipokine Levels, and the Presence of Metabolic Syndrome. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Yoshimura, K.; Miyamoto, Y.; Kaneko, K.; Chikazu, D.; Yamamoto, M.; Kamijo, R. Enhanced and suppressed mineralization by acetoacetate and β-hydroxybutyrate in osteoblast cultures. Biochem. Biophys. Res. Commun. 2016, 473, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Ferron, M.; Hinoi, E.; Karsenty, G.; Ducy, P. Osteocalcin differentially regulates β cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc. Natl. Acad. Sci. USA 2008, 105, 5266–5270. [Google Scholar] [CrossRef]
- Ferron, M.; Lacombe, J. Regulation of energy metabolism by the skeleton: Osteocalcin and beyond. Arch. Biochem. Biophys. 2014, 561, 137–146. [Google Scholar] [CrossRef]
- Ferron, M.; McKee, M.D.; Levine, R.L.; Ducy, P.; Karsenty, G. Intermittent injections of osteocalcin improve glucose metabolism and prevent type 2 diabetes in mice. Bone 2012, 50, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Hinoi, E.; Gao, N.; Jung, D.Y.; Yadav, V.K.; Yoshizawa, T.; Myers, M.G.; Chua, S.C.; Kim, J.; Kaestner, K.H.; Karsenty, G. The sympathetic tone mediates leptin’s inhibition of insulin secretion by modulating osteocalcin bioactivity. J. Cell Biol. 2008, 183, 1235–1242. [Google Scholar] [CrossRef]
- Mizokami, A.; Yasutake, Y.; Gao, J.; Matsuda, M.; Takahashi, I.; Takeuchi, H.; Hirata, M. Osteocalcin Induces Release of Glucagon-Like Peptide-1 and Thereby Stimulates Insulin Secretion in Mice. PLoS ONE 2013, 8, e57375. [Google Scholar] [CrossRef]
- Nauck, M.A.; Niedereichholz, U.; Ettler, R.; Holst, J.J.; Ørskov, C.; Ritzel, R.; Schmiegel, W.H. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am. J. Physiol. Endocrinol. Metab. 1997, 273, E981–E988. [Google Scholar] [CrossRef]
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef] [PubMed]
- Hui, H.; Farilla, L.; Merkel, P.; Perfetti, R. The short half-life of glucagon-like peptide-1 in plasma does not reflect its long-lasting beneficial effects. Eur. J. Endocrinol. 2002, 146, 863–869. [Google Scholar] [CrossRef]
- Brown, J.P.; Albert, C.; Nassar, B.A.; Adachi, J.D.; Cole, D.; Davison, K.S.; Dooley, K.C.; Don-Wauchope, A.; Douville, P.; Hanley, D.A.; et al. Bone turnover markers in the management of postmenopausal osteoporosis. Clin. Biochem. 2009, 42, 929–942. [Google Scholar] [CrossRef]
- Macdonald, M.J.; Longacre, M.J.; Stoker, S.W.; Brown, L.J.; Hasan, N.M.; Kendrick, M.A. Acetoacetate and β-hydroxybutyrate in combination with other metabolites release insulin from INS-1 cells and provide clues about pathways in insulin secretion. Am. J. Physiol. Physiol. 2008, 294, C442–C450. [Google Scholar] [CrossRef]
- Oury, F.; Khrimian, L.; Denny, C.A.; Gardin, A.; Chamouni, A.; Goeden, N.; Huang, Y.-Y.; Lee, H.; Srinivas, P.; Gao, X.-B.; et al. Maternal and Offspring Pools of Osteocalcin Influence Brain Development and Functions. Cell 2013, 155, 228–241. [Google Scholar] [CrossRef]
- Almaça, J.; Molina, J.; Menegaz, D.; Pronin, A.N.; Tamayo, A.; Slepak, V.; Berggren, P.-O.; Caicedo, A. Human Beta Cells Produce and Release Serotonin to Inhibit Glucagon Secretion from Alpha Cells. Cell Rep. 2016, 17, 3281–3291. [Google Scholar] [CrossRef]
- Ebou, M.H.; Singh-Estivalet, A.; Launay, J.-M.; Callebert, J.; Tronche, F.; Ferre, P.; Gautier, J.-F.; Guillemain, G.; Bréant, B.; Blondeau, B.; et al. Glucocorticoids Inhibit Basal and Hormone-Induced Serotonin Synthesis in Pancreatic Beta Cells. PLoS ONE 2016, 11, e0149343. [Google Scholar] [CrossRef]
- Paulmann, N.; Grohmann, M.; Voigt, J.-P.; Bert, B.; Vowinckel, J.; Bader, M.; Skelin, M.; Jevšek, M.; Fink, H.; Rupnik, M.S.; et al. Intracellular Serotonin Modulates Insulin Secretion from Pancreatic β-Cells by Protein Serotonylation. PLoS Biol. 2009, 7, e1000229. [Google Scholar] [CrossRef]
- Ramracheya, R.; Chapman, C.; Chibalina, M.; Dou, H.; Miranda, C.; Gonzalez, A.; Moritoh, Y.; Shigeto, M.; Zhang, Q.; Braun, M.; et al. GLP-1 suppresses glucagon secretion in human pancreatic alpha-cells by inhibition of P/Q-type Ca2+ channels. Physiol. Rep. 2018, 6, e13852. [Google Scholar] [CrossRef] [PubMed]
- Razny, U.; Fedak, D.; Kiec-Wilk, B.; Góralska, J.; Gruca, A.; Zdzienicka, A.; Kiec-Klimczak, M.; Solnica, B.; Hubalewska-Dydejczyk, A.; Malczewska-Malec, M. Carboxylated and undercarboxylated osteocalcin in metabolic complications of human obesity and prediabetes. Diabetes Metab. Res. Rev. 2016, 33, e2862. [Google Scholar] [CrossRef] [PubMed]
- Guney, G.; Sener-Simsek, B.; Tokmak, A.; Yucel, A.; Buyukkagnici, U.; Yilmaz, N.; Engin-Ustun, Y.; Ozgu-Erdinc, A.S. Assessment of the Relationship between Serum Vitamin D and Osteocalcin Levels with Metabolic Syndrome in Non-Osteoporotic Postmenopausal Women. Geburtshilfe Frauenheilkd. 2019, 79, 293–299. [Google Scholar] [CrossRef]
- Gallego, B.R.; García-Molina, L.; Cano-Ibáñez, N.; Sanchez-Delgado, G.; Andújar-Vera, F.; García-Fontana, C.; González-Salvatierra, S.; García-Recio, E.; Martínez-Ruiz, V.; Bueno-Cavanillas, A.; et al. Circulating Undercarboxylated Osteocalcin as Estimator of Cardiovascular and Type 2 Diabetes Risk in Metabolic Syndrome Patients. Sci. Rep. 2020, 10, 1840. [Google Scholar] [CrossRef]
- Sánchez-de-Diego, C.; Artigas, N.; Pimenta-Lopes, C.; Valer, J.A.; Torrejon, B.; Gama-Pérez, P.; Villena, J.A.; Garcia-Roves, P.M.; Rosa, J.L.; Ventura, F. Glucose Restriction Promotes Osteocyte Specification by Activating a PGC-1α-Dependent Transcriptional Program. iScience 2019, 15, 79–94. [Google Scholar] [CrossRef]
- Berger, J.M.; Singh, P.; Khrimian, L.; Morgan, D.A.; Chowdhury, S.; Arteaga-Solis, E.; Horvath, T.L.; Domingos, A.; Marsland, A.L.; Yadav, V.K.; et al. Mediation of the Acute Stress Response by the Skeleton. Cell Metab. 2019, 30, 890–902.e8. [Google Scholar] [CrossRef] [PubMed]
- Stegenga, M.E.; Van Der Crabben, S.N.; Levi, M.; De Vos, A.F.; Tanck, M.; Sauerwein, H.P.; Van Der Poll, T. Hyperglycemia Stimulates Coagulation, Whereas Hyperinsulinemia Impairs Fibrinolysis in Healthy Humans. Diabetes 2006, 55, 1807–1812. [Google Scholar] [CrossRef]
- Perkins, J.M.; Joy, N.G.; Tate, D.B.; Davis, S.N. Acute effects of hyperinsulinemia and hyperglycemia on vascular inflammatory biomarkers and endothelial function in overweight and obese humans. Am. J. Physiol. Metab. 2015, 309, E168–E176. [Google Scholar] [CrossRef] [PubMed]
- Somogyi, M. Effects of insulin upon the production of ketone bodies. J. Biol. Chem. 1941, 141, 219–227. [Google Scholar] [CrossRef]
- Wolfrum, C.; Besser, D.; Luca, E.; Stoffel, M. Insulin regulates the activity of forkhead transcription factor Hnf-3β/Foxa-2 by Akt-mediated phosphorylation and nuclear/cytosolic localization. Proc. Natl. Acad. Sci. USA 2003, 100, 11624–11629. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L. The therapeutic implications of ketone bodies: The effects of ketone bodies in pathological conditions: Ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 309–319. [Google Scholar] [CrossRef]
- Sampson, M.; Lathen, D.R.; Dallon, B.W.; Draney, C.; Ray, J.D.; Kener, K.B.; Parker, B.A.; Gibbs, J.L.; Gropp, J.S.; Tessem, J.S.; et al. β-Hydroxybutyrate improves β-cell mitochondrial function and survival. J. Insul. Resist. 2017, 1, 8. [Google Scholar] [CrossRef]
- Hayashi, T.; Boyko, E.J.; Sato, K.K.; McNeely, M.J.; Leonetti, D.L.; Kahn, S.E.; Fujimoto, W.Y. Patterns of Insulin Concentration During the OGTT Predict the Risk of Type 2 Diabetes in Japanese Americans. Diabetes Care 2013, 36, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.; Unwin, D.; Finucane, F. Low-Carbohydrate Diets in the Management of Obesity and Type 2 Diabetes: A Review from Clinicians Using the Approach in Practice. Int. J. Environ. Res. Public Health 2020, 17, 2557. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Unwin, D.; Cavan, D.; Cucuzzella, M.; Patel, M. Adapting diabetes medication for low carbohydrate management of type 2 diabetes: A practical guide. Br. J. Gen. Pract. 2019, 69, 360–361. [Google Scholar] [CrossRef]
- Athinarayanan, S.J.; Adams, R.N.; Hallberg, S.J.; McKenzie, A.L.; Bhanpuri, N.H.; Campbell, W.W.; Volek, J.S.; Phinney, S.D.; McCarter, J.P. Long-Term Effects of a Novel Continuous Remote Care Intervention Including Nutritional Ketosis for the Management of Type 2 Diabetes: A 2-Year Non-randomized Clinical Trial. Front. Endocrinol. 2019, 10, 348. [Google Scholar] [CrossRef] [PubMed]
- Al-Mrabeh, A.; Hollingsworth, K.G.; Shaw, J.A.M.; McConnachie, A.; Sattar, N.; Lean, M.E.J.; Taylor, R. 2-year remission of type 2 diabetes and pancreas morphology: A post-hoc analysis of the DiRECT open-label, cluster-randomised trial. Lancet Diabetes Endocrinol. 2020, 8, 939–948. [Google Scholar] [CrossRef]
- Gerstein, H.; Miller, M.E.; Byington, R.P.; Goff, D.C.; Bigger, J.T.; Buse, J.; Cushman, W.C.; Genuth, S.; Ismail-Beigi, F.; Grimm, R.H.; et al. Effects of Intensive Glucose Lowering in Type 2 Diabetes. N. Engl. J. Med. 2008, 358, 2545–2559. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cooper, I.D.; Brookler, K.H.; Kyriakidou, Y.; Elliott, B.T.; Crofts, C.A.P. Metabolic Phenotypes and Step by Step Evolution of Type 2 Diabetes: A New Paradigm. Biomedicines 2021, 9, 800. https://doi.org/10.3390/biomedicines9070800
Cooper ID, Brookler KH, Kyriakidou Y, Elliott BT, Crofts CAP. Metabolic Phenotypes and Step by Step Evolution of Type 2 Diabetes: A New Paradigm. Biomedicines. 2021; 9(7):800. https://doi.org/10.3390/biomedicines9070800
Chicago/Turabian StyleCooper, Isabella D., Kenneth H. Brookler, Yvoni Kyriakidou, Bradley T. Elliott, and Catherine A. P. Crofts. 2021. "Metabolic Phenotypes and Step by Step Evolution of Type 2 Diabetes: A New Paradigm" Biomedicines 9, no. 7: 800. https://doi.org/10.3390/biomedicines9070800
APA StyleCooper, I. D., Brookler, K. H., Kyriakidou, Y., Elliott, B. T., & Crofts, C. A. P. (2021). Metabolic Phenotypes and Step by Step Evolution of Type 2 Diabetes: A New Paradigm. Biomedicines, 9(7), 800. https://doi.org/10.3390/biomedicines9070800