Abstract
The study of cancer biology should be based around a comprehensive vision of the entire tumor ecosystem, considering the functional, bioenergetic and metabolic state of tumor cells and those of their microenvironment, and placing particular importance on immune system cells. Enhanced understanding of the molecular bases that give rise to alterations of pathways related to tumor development can open up new therapeutic intervention opportunities, such as metabolic regulation applied to immunotherapy. This review outlines the role of various oncometabolites and immunometabolites, such as TCA intermediates, in shaping pro/anti-inflammatory activity of immune cells such as MDSCs, T lymphocytes, TAMs and DCs in cancer. We also discuss the extraordinary plasticity of the immune response and its implication in immunotherapy efficacy, and highlight different therapeutic intervention possibilities based on controlling the balanced systems of specific metabolites with antagonistic functions.
1. Introduction
The metabolic approach in cancer biology examines the structure and metabolic pathways of the tumor ecosystem, encompassing both tumor cellularity and microenvironment (TME). The functional and bioenergetic characteristics of tumors depend on their alterations, which are linked to tumor etiopathogenesis, initial development, progression and metastasis. Specifically, tumor cells share metabolic pathways with immune cells [1], establishing resource competition systems, regulating tumor progression, immune response polarization and sensitivity to oncological treatments [2]. Therefore, therapeutic approaches from the metabolic point of view consider different intervention opportunities based on host immune response, metabolic remodeling of immune cell infiltration, tumor cellularity and other TME elements. Specifically, different tumor metabolites (immunometabolites and oncometabolites) participate in the humoral and cellular immune response, establishing the pro/anti-inflammatory balance, determining the activation of different immune cell subpopulations and regulating immune checkpoint mechanisms. Consequently, metabolic reprogramming of the tumor immune response has acquired special importance in immunotherapy approaches.
Immune system plasticity and capacity for metabolic reprogramming [3] opens up a vast field of research on the metabolic pathways involved in its physiology and pathology, an emerging area of immunometabolism knowledge. The metabolites produced by immune cells, known as immunometabolites [4] determine cell phenotype and function, act as cofactors of metabolic enzymes and mediate post-translational modifications [5]. Immunometabolites generated inside the cell can be released, acting on their surrounding environment like cytosines or at a systemic level, regulating different pro/anti-inflammatory mechanisms. Furthermore, immunometabolites influence disease progression and treatment response, thus constituting an emerging focus of interest and knowledge [5].
Tricarboxylic acid (TCA) cycle intermediate metabolites (TCAis) (acting as oncometabolites in cancer) can trigger processes of programmed cell death, autophagy, inflammation and immune signaling in response to cellular stress, pathogens, toxins or cancer [6]. TCAis are produced in the mitochondria and are distributed within the mitochondrial membranes due to their polarity and electrophilic properties. Under physiological conditions, they exert their function inside the mitochondria and can be released in a controlled way outside the cells [7]. Thus, TCAis such as succinate, fumarate, itaconate, 2-hydroxyglutarate (D-2-HG and L-2-HG) and acetyl-CoA express a wide range of non-metabolic signaling functions in physiological and pathological immunological contexts [8], especially activating the innate immune response on myeloid cells [9,10]. In turn, activation of these immune cells regulates the TCA cycle through an immune Warburg effect. In addition, TCAis such as succinate, fumarate, itaconate, citrate and α-ketoglutarate (α-KG) can also regulate activation of the inflammatory process [11] through epigenetic mechanisms [12], reactive oxygen species (ROS) modulation or post-translational modification of other proteins.
This review is focused on the regulatory metabolic pathways of both immune and tumor cells, the metabolic differences between stromal and tumor cells and especially the effector and immunosuppressive metabolic pathways of T effectors (Teff) and memory lymphocytes, M1 and M2 macrophages and myeloid-derived suppressor cells (MDSCs), as well as the role of acidosis and hypoxia in all of these factors. This approach consolidates the hypothesis of cancer as an energy dysfunction [13], where alterations can be found in the metabolic pathways of fatty acids (FAs), amino acids, nucleic acids and carbohydrates that affect immune dysfunction, allowing clinical tumor development. We conclude by discussing the extraordinary plasticity of the immune response, its role in immunotherapy efficacy and measures to modulate energy metabolism that should be implemented in cancer treatment.
2. Metabolic Pathways of the Tumor Ecosystem
2.1. Metabolic Reprogramming
The most widely known metabolic reprogramming process in cancer cells is the Warburg effect [14]. Compared to normal cells, tumor cells prefer using glycolysis rather than the mitochondrial oxidative phosphorylation system (OXPHOS), even in oxygen abundance states [15]. Although the glycolysis efficiency of ATP production is low, its yield is much faster, providing energy to tumor cells for growth and proliferation. Furthermore, glycolysis allows tumor cells to obtain several building blocks for biomass synthesis [16]. The oncogene c-MYC and hypoxia inducible factor-1 (HIF-1α) activate the expression of key enzymes which enhance aerobic glycolysis, the most important of which are glucose transporter 1 (GLUT1), hexokinase 2 (HK2), pyruvate kinase 2 (PKM2) and lactate dehydrogenase A (LDHA) [3,15,16]. GLUT1 overexpression increases glucose uptake by tumor cells. HK2 overexpression transforms glucose into glucose-6-phosphate (p), the first step in glycolysis, and enhances its flow to the pentose phosphate pathway (PPP), generating nicotinamide adenine dinucleotide phosphate oxidase (NADPH). NADPH is essential to anaerobic processes such as nucleotide synthesis, and also to protect the cell against ROS [15,16].
The regulation of pyruvate metabolism is central in the oncogenic metabolic program since this metabolite is at the crossroads between OXPHOS and lactic acid fermentation. Many cancer cells upregulate the expression of the less active PKM2 isoform, which slows down pyruvate synthesis and permits the diversion of glycolytic intermediates to other anabolic pathways, such as the serine synthesis pathway (SSP), further activating PKM2 [17]. In addition, cancer cells also downregulate the mitochondrial pyruvate carrier (MPC) complex, in charge of transporting pyruvate from the cytosol into the mitochondrial matrix. Reduced expression of MPC subunits causes the accumulation of pyruvate in the cytosol, thus favoring its conversion to lactate by lactate dehydrogenase (LDHA) [18]. Lactate is released to the TME via monocarboxylate transporter (MCT), and can be used as fuel by the cells. This is called the lactate shuttle, in which lactate is a linking vehicle between glycolytic and oxidative metabolism [19]. SSP is enhanced by the oncogene c-MYC which mediates the overexpression of several enzymes involved in this metabolic pathway [16,20]. Serine is the precursor for glycine, which itself is a precursor of glutathione. Therefore, tumor cells use SSP to increase glutathione and protect themselves against ROS. Serine is also necessary to initiate the one carbon metabolism (folate cycle and methionine synthesis pathway) essential for synthesis of nucleotides and many other biomolecules [20,21].
Turning to amino acid metabolic reprogramming, glutaminolysis is enhanced in many cancers [15] since cells can use glutamine as a glucose alternative to obtain energy, and it can also be converted into glutamate and then α-KG. The former is obtained in glutamine lysis by glutaminase and is necessary for nucleotide production and glutathione synthesis [22]. The latter, α-KG, is a TCA metabolite obtained from glutamate in the mitochondria via different pathways [23]. α-KG is considered to be an oncometabolite since it can be used to obtain energy, as well as in amino acid and lipid synthesis [15,16].
Lipid pathways are also involved in tumor cell metabolic reprogramming. Sterol regulatory element-binding transcription factor 1 (SREBP1), which upregulates the transcription of many enzymes of the FA synthesis pathway, is overexpressed in several cancers and has an important role in cell survival [15,16]. FA biosynthesis is frequently increased in cancer cells to meet lipid requirements for membrane synthesis, although in many cancer types fatty acid oxidation (FAO) is also enhanced in the mitochondria. The acetyl-CoA carboxylase generated by FAO enters into the TCA cycle for citrate synthesis and ATP energy can be produced by the electron transport chain [15].
Mitochondria therefore connects many essential metabolic pathways such as the folate cycle, glutamine lysis and FAO. Consequently, tumor cells increase mitochondrial biogenesis through the c-MYC oncogene, which transactivates mitochondrial transcription factor A [15]. Lastly, tumor metabolic reprogramming modifies the TME, regulating the immune response alongside the metabolic reprogramming capacity of immune cells themselves, and thus determining the extent of tumor progression and aggressiveness.
2.2. Metabolic Skewing Induced by Viral Infections
Different in vitro and in vivo studies show that many human intracellular viral, bacterial and protozoal pathogens can induce a Warburg-like metabolic state [24,25]. These pathogens hijack the host cell metabolism to redirect glycolysis and TCAis towards amino acid, lipid and nucleotide biosynthesis, which they need for their own nutritional and survival needs. Many intermediaries in the glycolytic pathway are significantly increased after viral infection. Some viruses induce glycolysis to aid replication [26], or induce metabolic change to counteract the ROS produced by the host during infection response [27,28].
Oncogenic viruses, such as human papillomavirus (HPV), hepatitis B and C (HBV and HCV), Epstein–Barr (EBV or HHV4), cytomegalovirus (CMV or HHV5), Kaposi’s sarcoma-associated herpesvirus (KSHV) and human herpesvirus 8 (HHV8) [29,30,31,32], can induce a Warburg-like effect or altered metabolic status in tumors. For example, human fibroblasts infected with CMV increase glucose consumption and lactate production, typical of Warburg metabolism [33,34]. Proteins formed during HBV replication are capable of manipulating the glucose, lipids and metabolism of nucleic acids, amino acids, vitamins and bile acids [35]. HBV has been shown to induce hepatocyte damage through dysregulation of aerobic glycolysis and lipid metabolism in a Warburg phenotype [36]. EBV stimulates oncogenesis and B-lymphocyte proliferation, hijacking mitochondrial metabolic pathways with a Warburg-like profile. As an example of this, in the study by Wang et al., human B lymphocytes were infected with EBV, revealing that shortly after infection EBV promoted oncogenesis by altering mitochondrial metabolism. Culture in a medium rich in galactose instead of glucose significantly affected transformation and proliferation of these cells, showing that glucose is a key carbon source in the transformation of B-lymphocyte metabolism when infected by EBV. EBV also expresses latent membrane protein 1 (LMP1), an oncoprotein that mimics cell CD40 signaling to activate multiple growth pathways [37]. Activation of B-lymphocyte proliferation by LMP1 has been shown to coincide with aerobic glycolysis induction [38]. Viruses also lead to production of proteins such as HPV E6 [39], which modulate central carbon metabolism in infected cells through inactivation or degradation of tumor suppressor genes such as p53 [40].
Importantly, many intracellular pathogens also exert different mechanisms which can improve the stability and activity of proteins such as HIF-1α [41,42]. HIF-1α regulates aerobic glycolysis, whose role in carcinogenesis is well established [43]. HPV E6 protein increases cellular HIF-1α levels, promoting a Warburg effect [44]. HCV and HPV also can manipulate infected cell metabolism through HIF-1α activation [45,46]. Specifically, HCV infection stabilizes HIF-1α under normoxic conditions, facilitating glycolytic enzyme expression, while HCV-associated mitochondrial dysfunction promotes HIF-1α-mediated glycolytic adaptation. This provides crucial insight into the pathogenesis of chronic hepatitis C and possibly HCV-related hepatocellular carcinoma [47]. Similarly, a hypoxic TME can promote the activity and survival of intracellular pathogens. For example, hypoxia can induce EBV reactivation when HIF-1α binds to the BZLF1 gene for the EBV primary latent-lytic switch [48].
2.3. Modulation by Exosomes
Exosomes have recently been shown to play a fundamental role in communication between cancer cells and TME cells, influencing cancer initiation, progression and metastasis [49]. Exosomes are endocytic nanovesicles, homogeneous in size (between 40–100 nm), which carry a variety of small molecules (cargo) essential for cell communication. Their cargo includes nucleic acids, proteins and lipids, and their double membrane encapsulation allows them to travel to tissues far away from their origin. Protected from degradation, they can stimulate specific receptors of target cells and horizontally transfer genetic material, triggering a pleiotropic effect [50].
Different microRNA (miRNA) have been isolated in the exosome cargo of different cancer cells. miRNA are non-coding RNA, strategic in the different stages of tumor development and expansion by allowing adaptation to a hostile environment. In early stages, primary tumor exosomes interact with contiguous cells, promoting epithelial transition (miR-200 family), fibroblast conversion into tumor cells (miR-1247-3p, miR-27a, miR-10b, miR-125b), extracellular matrix remodeling (miR-150, miR-23b) and immune system evasion (miR-197, miR-200, miR-203, miR-23a, miR-1246). As the tumor grows, its energy requirements increase beyond its blood supply, and it becomes more hypoxic, enhancing exosome production to promote angiogenesis (miR-9, miR-21, miR-210). This facilitates tumor cell release to the circulation (miR-1227) and spread to distant locations (miR-181c, miR-105, which alter the blood–brain barrier) and creating resistance mechanisms against different drugs (miR-21, miR-155, miR-222, miR-30a, miR-100-5p, miR-196a) [51].
Exosomes also play a strategic role in the metabolic reprogramming exerted in the TME and pre-metastatic niches. This allows cancer cells to adapt to a nutrient-deficient environment, modulating the stromal cells of the tumor niche towards profiles that favor the Warburg effect, promoting more aggressive and invasive phenotypes. For instance, exosomal miR-122 of some tumor types intervenes in glucose metabolism reprogramming by reducing its consumption in healthy cells surrounding the pre-metastatic niche, thereby favoring tumor development [52].
3. Immunometabolites and Oncometabolites
Immune cell function, activation, cytokine secretion and antitumor or antiviral effect depend on cellular metabolism [53]. Detailed knowledge of the metabolic pathways involved reveals functional differences between resting and activated immune cells, immune cells with homeostatic or altered functions, and permanent and transit TME immune cells. In general, CD8+ cytotoxic T lymphocytes (CTL), CD4+ helper T lymphocytes (Th), type M1 tumor-associated macrophages (TAMs) and natural killer cells play an antitumor role, while type M2 TAMs, mast cells, neutrophils and certain T- and B-lymphocyte subtypes promote cancer. However, these cells can be affected by metabolites (self-produced or immunometabolites), tumor cells or oncometabolites [54] as well as by TME conditions (Figure 1).
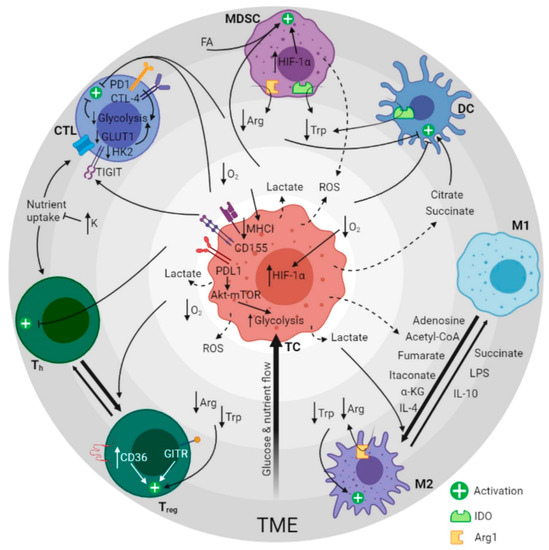
Figure 1.
Graphical summary of the metabolic regulation of the immune response in cancer. The image depicts the crosstalking between tumor cells and immune cells by altering TME composition. Dashed arrows represent secretion of molecules, solid arrows indicate the effect of the molecules and bold arrows show immune cell type polarization direction. Concentric gray circles represent glucose and nutrient availability (dark gray = low, light gray = high) and define groups of related molecules participating synergistically in specific metabolic regulation pathways (i.e., lactate, low O2 and ROS). TME: tumor microenvironment. MDSC: myeloid-derived stem cells. CTL: cytotoxic T cells. Th: T helpers. Treg: T regulatory cells. M2: macrophages type 2. M1: macrophages type 1. DC: dendritic cells. FA: fatty acids. Arg: arginine. Trp: tryptophan. ROS: reactive oxygen species. α-KG: alpha-ketoglutarate. LPS: lipopolysaccharide. IDO: indoleamine-pyrrole 2,3-dioxygenase. Arg1: arginase-1. Created with BioRender.com (accessed date: 13 June 2021).
TCAis lead to metabolic reprogramming, which determines the functional balance of immune cells. These products possess bioenergetic, biosynthetic, immune and oncogenic actions [8] and can regulate expression of inflammatory genes [55]. In general, succinate and citrate show pro-inflammatory properties, while fumarate, itaconate and α-KG are more related to immunosuppressive functions [8]. Many of these metabolites increase during immune activation, modulating the immune activation/suppression balance. Somatic mutations in cytosolic isocitrate dehydrogenase 1 (IDH1) can lead to production of oncometabolite 2-HG, although elevated levels of 2-HG have been observed in cytogenetically normal tumors [54]. In fact, less than half the elevated 2-HG cases had IDH1 mutations, while the remaining cases had mutations in IDH2, the mitochondrial homologue of IDH1. Succinate, 2-HG and fumarate promote cancer progression, also acquiring the ability to modulate cell signaling and affect chemotherapy and radiotherapy response through epigenetic mechanisms [56,57].
Prostaglandin E2 (PGE2), produced by cyclooxygenase-2 enzyme (COX-2), is involved in anti-inflammatory cytokine generation in cancer, promoting MDSC, regulatory T lymphocyte (Treg) and M2 TAM accumulation. TME glucose availability is another key modulator in immune cell activation. Glucose is captured mainly by tumor cells to feed the exacerbated aerobic glycolysis (Warburg effect). This entails several phenomena, such as increased lactate production, TME acidosis and subsequent immune response regulation [58], an effect that also occurs in other, non-cancer-related inflammatory conditions [59]. Additionally, ROS produced by tumor cells participate in oxidative stress encountered by TME immune cells, while reduced blood flow in certain tumor areas results in hypoxia, which leads to HIF-1α stabilization. The HIF-1α pathway provides a metabolic switch through c-Myc or Ras oncogenes, and is therefore a critical transcriptional regulator of immunity and cancer inflammation. This metabolic reprogramming with increased glycolysis and coordinated TCA cycle rearrangement, together with reduced mitochondrial OXPHOS, enhances chronic tumor-related inflammation.
3.1. MDSC Fate and Function
MDSCs are heterogeneous populations of tumor-associated innate immunosuppressive cells. In MDSCs, tryptophan is involved in tumor progression. MDSCs that synthesize tryptophan-degrading enzyme indoleamine 2,3-dioxygenase (IDO) are thought to protect tumors from specific T-cell attack by inducing tolerance during the priming phase or directly in the TME through tryptophan catabolism. Moreover, MDSCs can deplete amino acids by several mechanisms, and thus determine CTL fate, growth and immune functions in the TME, such as L-arginine metabolism by arginase-1 (Arg1) activity. Besides this, MDSC-generated arginine and tryptophan depletion also facilitates TAM and Treg immunosuppressive activity and hinders dendritic cell (DC) maturation.
MDSCs also sense lipid metabolites produced by the TME, which particularly enhance MDSC immunosuppressive function. In fact, some studies have suggested that tumor-associated MDSCs reprogram their metabolic pathway to adapt to a particular TME, such as one with limited O2 and glucose but high FA levels, and thus prefer to use lipids or FAs as an alternative energy source [60,61].
Nonetheless, MDSCs tend to activate their metabolism and function through aerobic glycolysis and OXPHOS [62]. The polarization of metabolism towards glycolysis generates lactate, which stimulates generation of MDSCs and phosphoenolpyruvate, an antioxidant agent that prevents ROS overproduction, contributing to MDSC survival by protecting them from apoptosis. ROS not only activate anti-oxidative pathways but also induce transcriptional programs that regulate the fate and function of MDSCs. Furthermore, MDSCs release ROS molecules as part of a major mechanism to suppress T-cell responses and modulate TAM functions, whereas hypoxia contributes to the immunosuppressive phenotype of MDSC through a mechanism linked to HIF-1α [63].
3.2. Complications of T-Cell Therapies
Tumor glucose uptake limits nutritional resources and IFN-γ expression in CTLs, reducing their functional response capacity [64], as also occurs in antitumor Th cells [65]. PD-L1 expression may contribute to this effect by driving Akt-mTOR activation and glycolysis in cancer cells [66]. Similarly, PD-1 and CTLA-4 expression in T cells suppress aerobic glycolysis, necessary for T-cell activation [67,68], while CD155 signaling reduces T-cell glucose uptake, lactate production and GLUT1 and HK2 expression. In addition, elevated potassium levels within the TME have been recognized to disrupt T-cell nutrient uptake, leading to a stemness state, further limiting the acquisition of Teff metabolism [69]. In contrast, the GITR costimulatory pathway increases T-cell proliferation and metabolic activity [70].
Among the accumulated glycolysis products generated by tumor cells, lactic acid impairs immune effector cells by directly inhibiting T-cell cytolytic functions. Likewise, high acidosis levels in a hypoxic TME result in mTOR signaling inhibition in T lymphocytes, producing energy in these cells [71,72] and promoting Treg activation [73]. Interestingly, Treg lymphocytes easily adapt to a lactic acid-enriched TME by CD36 upregulation [74], and are more resistant to oxidative stress-induced cell death than other T-lymphocyte subtypes [75], which would imply greater tumor tolerance in these TMEs. Taken together, these mechanisms induce Teff lymphocyte inhibition and encourage tumor growth-favoring Treg lymphocytes. A recent publication showed that hypoxia and glucose deprivation lead to decreased expression of major histocompatibility complex (MHC) class I molecules on tumor cells, facilitating their immune escape. Furthermore, tumor cells lose their sensitivity to IFN-γ induction, mediated by increased MHC [76]. As a consequence, tumor cells evade death by IFN-γ-producing T cells, creating another obstacle to T-cell therapies.
3.3. Polarization of Macrophages
Like T cells, macrophages also have a regulatory balance for their activation. The dual role of TAMs within the tumor, whether cytotoxic (pro-inflammatory, M1) or immunosuppressive (anti-inflammatory, M2), depends on the metabolic stimuli of the environment. In general, M1 metabolism usually resembles that of tumor cells, with a Warburg effect and aerobic glycolysis, while M2 metabolism tends to be based on FAO, although this might be overly simplistic [77,78]. Succinate, itaconate, fumarate and α-KG levels in macrophages and other immune cells have a profound impact on TAM polarization and innate immune memory [5,79]. Macrophage lipopolysaccharide (LPS) stimulation induces M1 macrophages, while stimulation with interleukin (IL)-4 induces M2 macrophages. TCA reprogramming in LPS-treated macrophages induces the Irg1 gene and causes succinate and itaconate accumulation [80]. These two immunometabolites form an essential immunomodulatory system [81], succinate having an important role in inflammatory signaling [82] by IL-1β, hypoxia by HIF-1α and metabolism through ROS, while itaconate shows an anti-inflammatory role [9], inhibiting succinate dehydrogenase [83] and mediating antioxidant/anti-inflammatory pathway Nrf2 activation and modulation of IFN type I [84].
Regulation of mitochondrial respiration and FAO of TAMs are determined by their metabolic programming [85]. In TAMs, FA synthesis is considered pro-tumorigenic, unlike Teff, Treg and T memory cells which oxidize FA for fuel.
M2 macrophages metabolize amino acids expressing high levels of Arg1, which depletes arginine and generates highly immunosuppressive polyamines [86]. Lactic acid produced by tumor cells acts as a signaler through HIF-1α and induces VEGF expression and particularly Arg1 production and M2 polarization [87]. Upregulated Arg1 in M2 TAMs also leads to immunosuppression in T cells. Mitochondrial localization of Arg2 is a central regulator of oxidative phosphorylation and macrophage polarization towards the M1 phenotype, a process controlled by miR-155 and IL-10. Arg2 increases complex II (succinate dehydrogenase) activity and downregulates succinate inflammatory mediators such as IL-10, HIF-1α and IL-1β [88].
Other mechanisms of IL-4-induced M2 TAMs regulation require the glutaminolysis-mediated production of α-KG, a cofactor of the epigenetic enzyme Jmjd3, which promotes IL-4 response in macrophages and inhibits pro-inflammatory signals [89]; this establishes a new system to balance macrophage function through the succinate/α-KG ratio. Finally, ATP citrate lyase (ACLY) of TCA cycle metabolite acetyl-CoA supports the anti-inflammatory responses of macrophages by promoting histone acetylation and IL-4-induced gene transcription [90]. ACLY is one of the main enzymes that catalyze acetyl-CoA formation; its functions include acetyl-CoA provision for lipogenesis, epigenetic regulation through histone acetylation and mediation of innate and adaptive immune responses [91].
Adenosine is a metabolite generated as a result of hypoxia inside the tumor [92] which promotes alternative macrophage activation towards M2 accumulation and expression of checkpoint inhibitors with immunosuppressive results. Furthermore, this hypoxic environment contributes to angiogenesis factor and cytosine production, also favoring accumulation of immunosuppressive M2 TAMs [93,94].
3.4. Dendritic Cell Subsets in Immune Response Regulation
Like macrophages, DCs undergo intense metabolic reprogramming in response to hypoxia, nutrient availability, growth factors, cytokines and other environmental signals. These include immunometabolites such as succinate and citrate [95], which regulate immunogenic/tolerogenic levels of DC. Moreover, IDO activation in DC has been shown to be involved in tumor immune evasion. Similarly, lactate produced through glycolysis can reduce DC activation and antigen presentation, facilitating tumor cell escape from immune attack [96,97].
4. Therapeutic Applications of Immunometabolism Regulation
In cancer, metabolites have an especially significant effect on immune cells, hence cancer immunotherapy aims to act at the metabolic level (Figure 2). For example, PGE2’s effect can be countered by COX-2 inhibitors such as acetylsalicylic acid or celecoxib, as established in colorectal cancer, mainly in the context of chemoprevention strategies; clinical research in this field is, however, still ongoing. Regarding 2-HG oncometabolite production, glutamine and glutamate pathways can be inhibited, providing alternative potential targets. Furthermore, the exceptional glucose metabolism of cancer cells (Warburg effect) implies TME acidification and lower glucose availability. These events seem key in explaining the wide range of immunosuppressive effects associated with cancer cell-induced biochemical reactions. Thus, regarding the metabolic switch to glycolysis mediated by metformin, dependent suppression of the mitochondrial complex followed by glucose deprivation could be used in combination with different chemotherapy schedules or immune checkpoint inhibitors as new cancer therapy approaches.
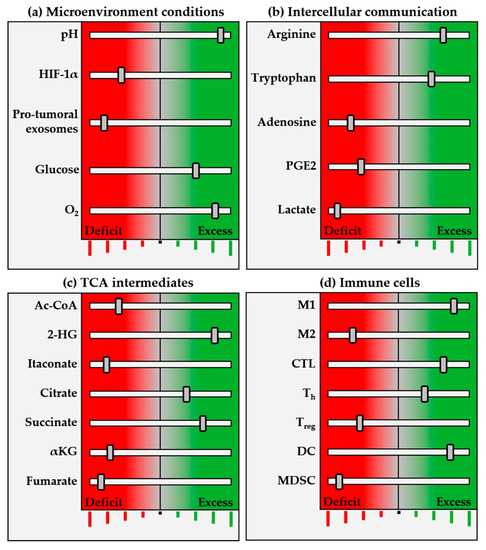
Figure 2.
Schematic representation of pro-inflammatory (antitumor) immunometabolic and cellular profile. Immune system programming begins at the perinatal stage, matures through environmental stimulation and is regulated by a balanced network that includes genetic and epigenetic profile, microbiota, hypothalamic–pituitary–adrenal (H–P–A) axis, sleep, diet and many other elements. Within this network, metabolism amplifies or inhibits immune function, determining physiological, adaptive or altered responses by (a) microenvironmental conditions, such as pH, oxygenation, etc.; (b) intercellular communication with chemokines, lactate, PGE2, etc.; and (c) TCAis, such as succinate, α-KG and others. The figure shown represents different frequency (inhibition/activation) bands, with an equilibrium position in the center, decreased activity towards the left (red), and increased towards the right (green). Like an equalization system, this network selects the immune “frequency band” determined by fine-tuning the elements making up the expanded system. This determines the immune response pattern, defined by cell subpopulations (d), their functionality, cytokine secretion and intercellular communication relationships within the TME. This schema introduces a concept that must be investigated and determined separately for each type of immune response, depending on specific physiological or pathophysiological situations, thus providing a spectrum of frequencies in which the greater the number of immune equalizer bands, the finer the tuning and function. Accumulated knowledge in oncological biology has brought into vision an immunometabolic equalizer with pro-/anti-tumor, pro-/anti-inflammatory, hot/cold tumor and pro-/anti-therapeutic-related functions. The precise immunometabolic alterations of each patient and tumor emerge as promising biological markers to guide immunotherapy treatments. The proposed immunometabolic equalizer schema represents crosstalk between the balance of different metabolic regulation levels (a–c), capable of determining the balance between pro-/anti-inflammatory cell populations, and unblocking immune checkpoints. The equalizer is not autonomous, but is connected to an amplifier in the form of macroenvironment elements such as the H–P–A axis, microbiota, stress, diet, exercise and medications, which in turn have their own equalization controls, in a fractal-like manner.
Immunometabolism therefore represents an emerging new target for cancer immunotherapy, and new lines of research are focused on directly or indirectly regulating different metabolites to improve current immunotherapeutic approaches (Table 1).

Table 1.
Therapeutic application of immunometabolism regulation. Forty studies related to immunometabolism regulation in cancer were found in clinicaltrials.gov database (accessed on 15 June 2021) using the following Boolean search string: “(immune AND reprogramming) OR (regulation AND metabolism AND immune) OR (immunotherapy AND metabolism AND modulation)”. Here are summarized 10 of the most representative current studies in the field.
4.1. Immunotherapy Based on Exosomes
Tumor exosomes modify immune cell metabolism, helping the tumor evade the immune response, so different strategies against exosome biogenesis and their mechanism of action make interesting targets against cancer [51,52]. Among the alternatives are use of exosomes released by immune cells to suppress tumor proliferation, inhibiting tumor exosome production or blocking their uptake by receptor cells, as well as designing bioengineered exosomes as transporters of anti-cancer products (miRNA, siRNA, proteins, drugs or vaccine) [98]. In addition to their cargo specificity, exosomes are released in greater quantities by cancer cells than healthy cells, so they can be isolated from body fluids (liquid biopsy), giving them great potential as biomarkers.
Consequently, to date there have been around 50 clinical studies related to exosomes and cancer (www.clinicaltrials.gov, accessed on 15 June 2021), most focused on diagnostic/prognostic or drug response biomarkers, while others are centered on using exosomes as antitumor therapeutic elements [99].
4.2. T-Cell Regulation
Although viruses modify the immune response, antiviral responses often successfully eliminate infectious agents or keep them under control throughout life, as evidenced by the infrequent occurrence of CMV- or EBV-mediated disease in healthy individuals despite persistent infection in up to 90% of the human population [46]. This has led to the interesting proposition of repurposing antiviral T cells against tumors. In a recent study, Rosato et al. demonstrated that antiviral T cells can target tumors when loaded with exogenous viral peptide. This strategy became even more efficient when combined with checkpoint blocking [100], potentially opening up new therapeutic avenues. It remains to be determined whether such a strategy influences anti-viral control or whether use of antiviral T cells could eventually lead to their depletion and/or reprogramming to Treg lymphocytes in a suppressor TME. Therefore, the choice of viral target peptides and combination with other strategies is crucial, especially considering that common persistent viruses such as EBV are oncogenic if uncontrolled [101].
Another therapeutic approach consists of intratumoral administration, through lipid nanoparticles, of an interfering RNA that silences lactate dehydrogenase A (LDH-A) production and activity [102]. In animal models of melanoma, anti-PD1 treatment has been shown to improve mice with LDH-A-deficient tumors [103]. One study also reported that deletion of LDH-A in myeloid cells can induce antitumor immunity of T lymphocytes against lung carcinoma [104]. In addition, the combination of pemetrexed (antifolate antineoplastic agent) with anti-PD-L1 shows direct antitumor effects together with improved CTL metabolism and immune function, by stimulating mitochondrial biogenesis and thus facilitating their activation and antitumor effect [105].
The metabolic differences observed between tumor cells and immune cells invite an exploration of opportunities for T-cell metabolic reprogramming prior to starting other cancer treatments, especially immunotherapy. Among the metabolic targets to improve immune response are Arg1, IDO, lactate, CD36, CD73 and D-2-HG [53,74]. The study of these and other immunometabolites creates a need to identify specific metabolic determinants of response, which help gain objective insight into the metabolic consequences of checkpoint therapy.
As an illustration, CD36 ablation in Treg reduced its survival in TME conditions, leading to tumor growth suppression, antitumor activity enhancement of T cells and additive antitumor responses with anti-PD1 therapy [74]. Alternatively, one strategy to develop therapeutic agents against autoimmune diseases uses the immunometabolite 2-HG, which can epigenetically regulate the balance between pro-inflammatory Th17 cells and induced Treg (iTreg) cells towards Th17 differentiation [106]. Increased transamination, catalyzed by GOT1, produces increased 2-HG levels in Th17 cells, hypermethylation of the Foxp3 gene locus and inhibition of Foxp3 transcription, determining Th17 differentiation. When glutamate−α-KG conversion is inhibited, 2-HG production decreases, methylation of the Foxp3 gene locus is reduced and Foxp3 expression increases, resulting in Th17 cell differentiation blockade and iTreg cell development. Selective inhibition of GOT1 with (aminooxy) acetic acid has been shown to improve autoimmunity by regulating Th17/iTreg fate, conforming to a regulatory system based on a 2-HG/(aminooxy) acetic acid balance.
Alternatively, considering that tumor glucose consumption restricts the glycolytic capacity and IFN-γ production of T cells, the checkpoint blockade can be mitigated with antibodies against PD-1, its ligands and CTLA-4 [66]. In addition, glucose availability in the TME can be enhanced, which allows improved CTL cytokine expression and avoids toxic concentrations of certain metabolites, such as adenosine, kynurenine and ornithine, ROS and increased acidosis, thus preventing antitumor immune response suppression. Likewise, a recent report showed that acetate could be used as an alternative carbon source and rescue the functions (such as IFN-γ production) of exhausted T lymphocytes infiltrating the tumor [107]. It has been described that high potassium reduces T-cell nutrient uptake, which results in T-cell stemness and exhaustion. Interestingly, treatment of antitumor T cells with elevated extracellular potassium, as well as pharmacologic or gene therapies, enables enhanced tumor destruction during immunotherapy performance [69]. Other actions in the TME are inhibition of the regulatory immunometabolite 1-methylnicotinamide, which is noticeably increased in tumor-infiltrating T cells [108], and expression of the enzyme catalase by CAR- (chimeric antigen receptor) T cells, through which these cells are better protected from ROS-induced oxidative stress [109].
4.3. Macrophage Regulation with Immunometabolites
Highly immunosuppressive phenotypes have been used as targets in immunotherapeutic treatment, with the aim of reverting their function to an immunoprotective role. MDSC enhances crosstalk with TAMs through an IL10- and cell contact-dependent mechanism, and skews them towards an M2 phenotype, leading to an immunosuppressive environment. Therefore, immunotherapies aiming to polarize TAMs into M1-like macrophages have been suggested as a therapeutic approach against cancer. MDSC function can be regulated by therapeutically altering its metabolism, as achieved with D-2-HG by blocking the glycolytic pathway [110] or using Arg1 and IDO inhibitors, thus hindering TAM−M2 polarization. Moreover, as CD73 is a cell-surface glycoprotein essential for extracellular adenosine generation, CD73 inhibition combined with other strategies induces antitumor effects in preclinical mouse models of cancer. In fact, several anti-CD73 antibodies are under clinical research in phase I–II trials, mostly combined with immune checkpoint inhibitors.
Since Nrf2 activation is necessary for the anti-inflammatory action of itaconate in TAMs, another therapeutic alternative has sought itaconate derivatives to reproduce this action at a therapeutic level, such as 4-octyl itaconate, which protects against LPS-induced lethality and decreases cytokine production [84]. Likewise, IFN type I is frequently used in immunotherapy [111], due to its ability to increase Arg1 expression and itaconate production, which forms a new negative feedback loop through the IFN/itaconate system [84] mediated by inflammatory macrophages.
5. Discussion
Reprogramming tumor cell metabolism allows cells to acquire growth and survival advantages, while tumor ecosystem changes in the form of acidosis, hypoxia, nutrient depletion and cellular waste accumulation affect the action of the cellular elements responsible for an effective immune response, promoting, for example, tumor immune evasion.
Different metabolism types and possible tumor and stromal cell abnormalities play key roles in shaping the degree of morphological, phenotypic and molecular heterogeneity within the tumor, and the extent of response or resistance to oncological, cytotoxic, immunotherapeutic or other treatments. This pinpoints the metabolism of tumor and immune cells as a preferred target for new research lines and treatment development, due to the characteristic metabolic plasticity of these cells. This forms the basis for intervening in immunometabolism to reverse immune escape and favor the anti-tumor immune response, an approach that involves combining metabolic inhibitors with immune checkpoint inhibitors, considering immune-regulatory metabolites on the TME as immune targets, and taking tumor and immune cell metabolism into account to improve the efficacy of immunotherapy.
Accordingly, the importance of studying TCA intermediates and other metabolites in immunotherapy arises from their regulatory role in cancer, which can be identified at various levels. At one level is the immune balance that determines the pro-inflammatory versus anti-inflammatory effect, such as Th17/Treg regulated by 2-HG/(aminooxy) acetic acid balance, or M1/M2 TAMs regulated by succinate/itaconate, succinate/α-KG and IFN/itaconate balancing systems. At another lies energy balance, which regulates CTL and Teff function via glucose availability for tumor cells versus antitumor immune cells. These regulatory systems are not exclusive to oncological pathophysiology, but are available as immune and inflammatory control systems in different pathological situations. Thus, the balance between Th17/Treg establishes the degree and rhythm of both physiological and pathological autoimmunity. The shift from Th17 to Treg protects against autoimmune inflammation, and this fine balance is regulated by 2-HG inhibition [106].
On another plane, itaconate, derived from the TCA cycle, is an immunometabolite with a direct antimicrobial effect, which by inhibiting isocitrate lyase reduces the production of pro-inflammatory mediators in macrophages and improves sepsis and psoriasis in animal models [112]. This action, combined with succinate, offers a balanced macrophage control system, which can be reprogrammed from a pro-inflammatory to anti-inflammatory state, with the aim of limiting the damage of the inflammatory process, facilitating tissue repair and preventing inflammatory diseases and tumors from becoming unhealable wounds.
Combinations of new metabolic modulators with immune checkpoint inhibitors are a common strategy currently in clinical research, as it seems anti PD1/PD-L1 antibodies may restore the metabolic fitness of T lymphocytes. In addition, the favorable safety profile of immune checkpoint inhibitors (particularly against PD1 and PD-L1) has paved the way for extensive testing of combined immunometabolic approaches. Unfortunately, the phase III results of the epacadostat (IDO inhibitor) plus pembrolizumab (anti-PD1/PD-L1 monoclonal antibody) combination in metastatic melanoma were disappointing, highlighting the fact that this type of interventional target is not easily modulated; they represent a major challenge requiring finely tuned approaches, preferably based on reliable biomarkers and new technical strategies selectively targeting TME.
In summary, the entire tumor ecosystem is subject to metabolic functional regulation mechanisms, which shape the polarization of the immune response, in both a cytotoxic and immunosuppressive sense, and modulating these mechanisms opens the door to complementary therapies in different pathologies, with special importance in oncology. New clinical and translational data hopefully available in the near future will elucidate the true value of the aforementioned therapies.
Author Contributions
T.Á.N. and R.N. conceived and designed the review. All authors participated in manuscript redaction. E.M. unified the revision content and with S.S. and I.V.-M. designed the figures. A.L.-C., F.F., A.V., S.A., S.M. and L.d.l.C.-M. revised and edited the review. T.Á.N. and R.N. supervised the work. All authors have read and agreed to the published version of the manuscript.
Funding
E.M. is supported by the Asociación Fundación Española contra el Cáncer, JAP-AECC(2018/150). This work was supported by grants from ISCIII (FIS) and FEDER (European Regional Development Fund) PI20/01107; Cris Cancer Foundation (contract 2020/066); and Comunidad de Madrid (B2017/BMD-3733/IMMUNOTHERCAN-CM).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Andrejeva, G.; Rathmell, J.C. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef] [PubMed]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.N.; Kauer, N.; Blazquez, R.; Hacker, L.; et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 2019, 29, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Tyrakis, P.A.; Palazon, A.; Macias, D.; Lee, K.L.; Phan, A.T.; Veliça, P.; You, J.; Chia, G.S.; Sim, J.; Doedens, A.; et al. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature 2016, 540, 236–241. [Google Scholar] [CrossRef]
- de Goede, K.E.; Harber, K.J.; Van den Bossche, J. Let’s Enter the Wonderful World of Immunometabolites. Trends Endocrinol. Metab. 2019, 30, 329–331. [Google Scholar] [CrossRef]
- Arnoult, D.; Soares, F.; Tattoli, I.; Girardin, S.E. Mitochondria in innate immunity. EMBO Rep. 2011, 12, 901–910. [Google Scholar] [CrossRef]
- Choi, I.; Son, H.; Baek, J.H. Tricarboxylic acid (Tca) cycle intermediates: Regulators of immune responses. Life 2021, 11, 69. [Google Scholar] [CrossRef]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab. 2019, 1, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; O’Neill, L.A.J. Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 2018, 174, 780–784. [Google Scholar] [CrossRef]
- Patil, N.K.; Bohannon, J.K.; Hernandez, A.; Patil, T.K.; Sherwood, E.R. Regulation of leukocyte function by citric acid cycle intermediates. J. Leukoc. Biol. 2019, 106, 105–117. [Google Scholar] [CrossRef]
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436. [Google Scholar] [CrossRef]
- Van Der Heijden, C.D.C.C.; Noz, M.P.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P.; Keating, S.T. Epigenetics and Trained Immunity. Antioxid. Redox Signal. 2018, 29, 1023–1040. [Google Scholar] [CrossRef]
- Monferrer, E.; Vieco-Martí, I.; López-Carrasco, A.; Fariñas, F.; Abanades, S.; de la Cruz-Merino, L.; Noguera, R.; Naranjo, T.Á. Metabolic classification and intervention opportunities for tumor energy dysfunction. Metabolites 2021, 11, 264. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Phan, L.M.; Yeung, S.C.J.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [PubMed]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell. Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Mao, Q.; Xia, W.; Xu, Y.; Wang, J.; Xu, L.; Jiang, F. PKM2 and cancer: The function of PKM2 beyond glycolysis (Review). Oncol. Lett. 2016, 11, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Iglesias, A.; Mañes, S. The importance of mitochondrial pyruvate carrier in cancer cell metabolism and tumorigenesis. Cancers 2021, 13, 1488. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef] [PubMed]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- Li, L.; Meng, Y.; Li, Z.; Dai, W.; Xu, X.; Bi, X.; Bian, J. Discovery and development of small molecule modulators targeting glutamine metabolism. Eur. J. Med. Chem. 2019, 163, 215–242. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Escoll, P.; Buchrieser, C. Metabolic reprogramming of host cells upon bacterial infection: Why shift to a Warburg-like metabolism? FEBS J. 2018, 285, 2146–2160. [Google Scholar] [CrossRef]
- Thaker, S.K.; Ch’ng, J.; Christofk, H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Dengue Virus Induces and Requires Glycolysis for Optimal Replication. J. Virol. 2015, 89, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.T.; Lee, D.Y.; Huang, Y.T.; Kou, G.H.; Wang, H.C.; Chang, G.D.; Lo, C.F. Six Hours after Infection, the Metabolic Changes Induced by WSSV Neutralize the Host’s Oxidative Stress Defenses. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Su, M.A.; Huang, Y.T.; Chen, I.T.; Lee, D.Y.; Hsieh, Y.C.; Li, C.Y.; Ng, T.H.; Liang, S.Y.; Lin, S.Y.; Huang, S.W.; et al. An Invertebrate Warburg Effect: A Shrimp Virus Achieves Successful Replication by Altering the Host Metabolome via the PI3K-Akt-mTOR Pathway. PLoS Pathog. 2014, 10, e1004196. [Google Scholar] [CrossRef]
- Luo, G.G.; Ou, J.J. Oncogenic viruses and cancer. Virol. Sin. 2015, 30, 83–84. [Google Scholar] [CrossRef]
- Black, P.H. Recent Advances in the Study of Oncogenic Viruses. N. Engl. J. Med. 1966, 275, 377–383. [Google Scholar] [CrossRef]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef]
- White, M.K.; Pagano, J.S.; Khalili, K. Viruses and human cancers: A long road of discovery of molecular paradigms. Clin. Microbiol. Rev. 2014, 27, 463–481. [Google Scholar] [CrossRef] [PubMed]
- Vastag, L.; Koyuncu, E.; Grady, S.L.; Shenk, T.E.; Rabinowitz, J.D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011, 7, e1002124. [Google Scholar] [CrossRef]
- Yu, Y.; Clippinger, A.J.; Alwine, J.C. Viral effects on metabolism: Changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol. 2011, 19, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.X.; Huang, C.J.; Yang, Z.G. Impact of hepatitis B virus infection on hepatic metabolic signaling pathway. World J. Gastroenterol. 2016, 22, 8161–8167. [Google Scholar] [CrossRef] [PubMed]
- Masson, J.J.R.; Billings, H.W.W.; Palmer, C.S. Metabolic reprogramming during hepatitis B disease progression offers novel diagnostic and therapeutic opportunities. Antivir. Chem. Chemother. 2017, 25, 53–57. [Google Scholar] [CrossRef] [PubMed]
- El-Sharkawy, A.; Al Zaidan, L.; Malki, A. Epstein-Barr virus-associated malignancies: Roles of viral oncoproteins in carcinogenesis. Front. Oncol. 2018, 8, 265. [Google Scholar] [CrossRef]
- Wang, L.W.; Jiang, S.; Gewurz, B.E. Epstein-Barr Virus LMP1-Mediated Oncogenicity. J. Virol. 2017, 91, e01718-16. [Google Scholar] [CrossRef]
- Thomas, M.; David, P.; Banks, L. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene 1999, 18, 7690–7700. [Google Scholar] [CrossRef]
- Eisenreich, W.; Rudel, T.; Heesemann, J.; Goebel, W. How viral and intracellular bacterial pathogens reprogram the metabolism of host cells to allow their intracellular replication. Front. Cell. Infect. Microbiol. 2019, 9, 42. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and Inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef]
- Zhu, C.; Zhu, Q.; Wang, C.; Zhang, L.; Wei, F.; Cai, Q. Hostile takeover: Manipulation of HIF-1 signaling in pathogen-associated cancers (Review). Int. J. Oncol. 2016, 49, 1269–1276. [Google Scholar] [CrossRef][Green Version]
- Cuninghame, S.; Jackson, R.; Zehbe, I. Hypoxia-inducible factor 1 and its role in viral carcinogenesis. Virology 2014, 456–457, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Meng, X.; Ma, J.; Zheng, Y.; Wang, Q.; Wang, Y.; Shang, H. Human papillomavirus 16 E6 contributes HIF-1α induced warburg effect by attenuating the VHL-HIF-1α interaction. Int. J. Mol. Sci. 2014, 15, 7974–7986. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, S.A.; de Andrade Júnior, D.R. HIF–1alpha and infectious diseases: A new frontier for the development of new therapies. Rev. Inst. Med. Trop. Sao Paulo 2017, 59, e92. [Google Scholar] [CrossRef] [PubMed]
- Pallett, L.J.; Schmidt, N.; Schurich, A. T cell metabolism in chronic viral infection. Clin. Exp. Immunol. 2019, 197, 143–152. [Google Scholar] [CrossRef]
- Ripoli, M.; D’Aprile, A.; Quarato, G.; Sarasin-Filipowicz, M.; Gouttenoire, J.; Scrima, R.; Cela, O.; Boffoli, D.; Heim, M.H.; Moradpour, D.; et al. Hepatitis C Virus-Linked Mitochondrial Dysfunction Promotes Hypoxia-Inducible Factor 1α-Mediated Glycolytic Adaptation. J. Virol. 2010, 84, 647–660. [Google Scholar] [CrossRef]
- Kraus, R.J.; Yu, X.; Cordes, B.L.A.; Sathiamoorthi, S.; Iempridee, T.; Nawandar, D.M.; Ma, S.; Romero-Masters, J.C.; McChesney, K.G.; Lin, Z.; et al. Hypoxia-inducible factor-1α plays roles in Epstein-Barr virus’s natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLoS Pathog. 2017, 13, e1006404. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, D. Exosomes in cancer development, metastasis, and immunity. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 455–468. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Kosaka, N.; Yoshioka, Y.; Fujita, Y.; Ochiya, T. Versatile roles of extracellular vesicles in cancer. J. Clin. Investig. 2016, 126, 1163–1172. [Google Scholar] [CrossRef]
- Yang, E.; Wang, X.; Gong, Z.; Yu, M.; Wu, H.; Zhang, D. Exosome-mediated metabolic reprogramming: The emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct. Target. Ther. 2020, 5, 1–13. [Google Scholar] [CrossRef]
- Leone, R.D.; Powell, J.D. Metabolism of immune cells in cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Xiang, K.; Jendrossek, V.; Matschke, J. Oncometabolites and the response to radiotherapy. Radiat. Oncol. 2020, 15, 1–10. [Google Scholar] [CrossRef]
- Weng, C.Y.; Kao, C.X.; Chang, T.S.; Huang, Y.H. Immuno-metabolism: The role of cancer niche in immune checkpoint inhibitor resistance. Int. J. Mol. Sci. 2021, 22, 1258. [Google Scholar] [CrossRef] [PubMed]
- Certo, M.; Tsai, C.H.; Pucino, V.; Ho, P.C.; Mauro, C. Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat. Rev. Immunol. 2021, 21, 151–161. [Google Scholar] [CrossRef]
- Kiran, D.; Basaraba, R.J. Lactate Metabolism and Signaling in Tuberculosis and Cancer: A Comparative Review. Front. Cell. Infect. Microbiol. 2021, 11, 37. [Google Scholar] [CrossRef]
- Davidov, V.; Jensen, G.; Mai, S.; Chen, S.H.; Pan, P.Y. Analyzing One Cell at a TIME: Analysis of Myeloid Cell Contributions in the Tumor Immune Microenvironment. Front. Immunol. 2020, 11, 1842. [Google Scholar] [CrossRef]
- Yan, D.; Adeshakin, A.O.; Xu, M.; Afolabi, L.O.; Zhang, G.; Chen, Y.H.; Wan, X. Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front. Immunol. 2019, 10, 1399. [Google Scholar] [CrossRef]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Del Valle, L.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Ma, E.H.; Verway, M.J.; Johnson, R.M.; Roy, D.G.; Steadman, M.; Hayes, S.; Williams, K.S.; Sheldon, R.D.; Samborska, B.; Kosinski, P.A.; et al. Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8+ T Cells. Immunity 2019, 51, 856–870. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.C.; Bihuniak, J.D.; MacIntyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Ogando, J.; Sáez, M.E.; Santos, J.; Nuevo-Tapioles, C.; Gut, M.; Esteve-Codina, A.; Heath, S.; González-Pérez, A.; Cuezva, J.M.; Lacalle, R.A.; et al. PD-1 signaling affects cristae morphology and leads to mitochondrial dysfunction in human CD8+ T lymphocytes. J. Immunother. Cancer 2019, 7, 151. [Google Scholar] [CrossRef]
- Kumar Vodnala, S.; Eil, R.; Kishton, R.J.; Sukumar, M.; Yamamoto, T.N.; Ha, N.-H.; Lee, P.-H.; Shin, M.; Patel, S.J.; Yu, Z.; et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 2019, 363, 6434. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Rosen, D.B.; Grein, J.; Tedesco, D.; Joyce-Shaikh, B.; Ueda, R.; Semana, M.; Bauer, M.; Bang, K.; Stevenson, C.; et al. GITR agonism enhances cellular metabolism to support CD8+ T-cell proliferation and effector cytokine production in a mouse tumor model. Cancer Immunol. Res. 2018, 6, 1199–1211. [Google Scholar] [CrossRef]
- Walton, Z.E.; Patel, C.H.; Brooks, R.C.; Yu, Y.; Ibrahim-Hashim, A.; Riddle, M.; Porcu, A.; Jiang, T.; Ecker, B.L.; Tameire, F.; et al. Acid Suspends the Circadian Clock in Hypoxia through Inhibition of mTOR. Cell 2018, 174, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Delgoffe, G.M.; Meyer, C.F.; Chan, W.; Powell, J.D. Anergic T Cells Are Metabolically Anergic. J. Immunol. 2009, 183, 6095–6101. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, M.; Stabilini, A.; Roncarolo, M.G. Rapamycin selectively expands CD4+CD25+FoxP3 + regulatory T cells. Blood 2005, 105, 4743–4748. [Google Scholar] [CrossRef]
- Wang, H.; Franco, F.; Tsui, Y.-C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.-H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef]
- Mougiakakos, D.; Johansson, C.C.; Kiessling, R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood 2009, 113, 3542–3545. [Google Scholar] [CrossRef]
- Marijt, K.A.; Sluijter, M.; Blijleven, L.; Tolmeijer, S.H.; Scheeren, F.A.; Van Der Burg, S.H.; Van Hall, T. Metabolic stress in cancer cells induces immune escape through a PI3K-dependent blockade of IFNγreceptor signaling. J. Immunother. Cancer 2019, 7. [Google Scholar] [CrossRef]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef]
- Batista-Gonzalez, A.; Vidal, R.; Criollo, A.; Carreño, L.J. New Insights on the Role of Lipid Metabolism in the Metabolic Reprogramming of Macrophages. Front. Immunol. 2020, 10, 2993. [Google Scholar] [CrossRef]
- Ryan, D.G.; O’Neill, L.A.J. Krebs Cycle Reborn in Macrophage Immunometabolism. Annu. Rev. Immunol. 2020, 38, 289–313. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Artyomov, M.N. Itaconate: The poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 2019, 19, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Hooftman, A.; O’Neill, L.A.J. The Immunomodulatory Potential of the Metabolite Itaconate. Trends Immunol. 2019, 40, 687–698. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-Mcdermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Hayes, C.S.; Shicora, A.C.; Keough, M.P.; Snook, A.E.; Burns, M.R.; Gilmour, S.K. Polyamine-blocking therapy reverses immunosuppression in the tumor microenvironment. Cancer Immunol. Res. 2014, 2, 274–285. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Dowling, J.K.; Afzal, R.; Gearing, L.J.; Cervantes-Silva, M.P.; Annett, S.; Davis, G.M.; De Santi, C.; Assmann, N.; Dettmer, K.; Gough, D.J.; et al. Mitochondrial arginase-2 is essential for IL-10 metabolic reprogramming of inflammatory macrophages. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Liu, P.S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; DI Conza, G.; Cheng, W.C.; Chou, C.H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Aksoylar, H.I.; Yu, J.; Snyder, N.W.; Worth, A.J.; Iyer, S.S.; Wang, J.; Ben-Sahra, I.; Byles, V.; Polynne-Stapornkul, T.; et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife 2016, 5, e11612. [Google Scholar] [CrossRef]
- Dominguez, M.; Brüne, B.; Namgaladze, D. Exploring the Role of ATP-Citrate Lyase in the Immune System. Front. Immunol. 2021, 12, 14. [Google Scholar] [CrossRef]
- Csóka, B.; Selmeczy, Z.; Koscsó, B.; Németh, Z.H.; Pacher, P.; Murray, P.J.; Kepka-Lenhart, D.; Morris, S.M., Jr.; Gause, W.C.; Leibovich, S.J.; et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012, 26, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Henze, A.T.; Mazzone, M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Investig. 2016, 126, 3672–3679. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, C.; Tewari, B.N.; Kanchan, R.K.; Baghel, K.S.; Nautiyal, N.; Shrivastava, R.; Kaur, H.; Bramha Bhatt, M.L.; Bhadauria, S. Macrophages are recruited to hypoxic tumor areas and acquire a Pro-Angiogenic M2-Polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014, 5, 5350–5368. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Nasi, A.; Fekete, T.; Krishnamurthy, A.; Snowden, S.; Rajnavölgyi, E.; Catrina, A.I.; Wheelock, C.E.; Vivar, N.; Rethi, B. Dendritic Cell Reprogramming by Endogenously Produced Lactic Acid. J. Immunol. 2013, 191, 3090–3099. [Google Scholar] [CrossRef]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting tumor microenvironment for cancer therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Chen, Y.S.; Lin, E.Y.; Chiou, T.W.; Harn, H.J. Exosomes in clinical trial and their production in compliance with good manufacturing practice. Tzu Chi Med. J. 2020, 32, 113–120. [Google Scholar]
- Rosato, P.C.; Wijeyesinghe, S.; Stolley, J.M.; Nelson, C.E.; Davis, R.L.; Manlove, L.S.; Pennell, C.A.; Blazar, B.R.; Chen, C.C.; Geller, M.A.; et al. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Farrell, P.J. Epstein-Barr Virus and Cancer. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 29–53. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Zhao, Y.Y.; Shen, J.; Sun, X.; Liu, Y.; Liu, H.; Wang, Y.; Wang, J. Nanoenabled Modulation of Acidic Tumor Microenvironment Reverses Anergy of Infiltrating T Cells and Potentiates Anti-PD-1 Therapy. Nano Lett. 2019, 19, 2774–2783. [Google Scholar] [CrossRef]
- Daneshmandi, S.; Wegiel, B.; Seth, P. Blockade of lactate dehydrogenase-A (LDH-A) improves efficacy of anti-programmed cell death-1 (PD-1) therapy in melanoma. Cancers 2019, 11, 450. [Google Scholar] [CrossRef]
- Seth, P.; Csizmadia, E.; Hedblom, A.; Vuerich, M.; Xie, H.; Li, M.; Longhi, M.S.; Wegiel, B. Deletion of lactate dehydrogenase-A in myeloid cells triggers antitumor immunity. Cancer Res. 2017, 77, 3632–3643. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.A.; Geeganage, S.; Amaladas, N.; Lu, Z.H.; Rasmussen, E.R.; Sonyi, A.; Chin, D.; Capen, A.; Li, Y.; Meyer, C.M.; et al. The folate pathway inhibitor pemetrexed pleiotropically enhances effects of cancer immunotherapy. Clin. Cancer Res. 2019, 25, 7175–7188. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Stewart, K.M.; Wang, X.; Liu, K.; Xie, M.; Kyu Ryu, J.; Li, K.; Ma, T.; Wang, H.; Ni, L.; et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature 2017, 548, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Villa, M.; Sanin, D.E.; Buck, M.D.; O’Sullivan, D.; Ching, R.; Matsushita, M.; Grzes, K.M.; Winkler, F.; Chang, C.H.; et al. Acetate Promotes T Cell Effector Function during Glucose Restriction. Cell Rep. 2019, 27, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Kilgour, M.K.; MacPherson, S.; Zacharias, L.G.; Ellis, A.E.; Sheldon, R.D.; Liu, E.Y.; Keyes, S.; Pauly, B.; Carleton, G.; Allard, B.; et al. 1-Methylnicotinamide is an immune regulatory metabolite in human ovarian cancer. Sci. Adv. 2021, 7, eabe1174. [Google Scholar] [CrossRef] [PubMed]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor–Redirected T Cells as well as Bystander Cells from Oxidative Stress–Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef]
- Jian, S.L.; Chen, W.W.; Su, Y.C.; Su, Y.W.; Chuang, T.H.; Hsu, S.C.; Huang, L.R. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ROS-mediated apoptosis. Cell Death Dis. 2017, 8. [Google Scholar] [CrossRef]
- Aricò, E.; Castiello, L.; Capone, I.; Gabriele, L.; Belardelli, F. Type i interferons and cancer: An evolving story demanding novel clinical applications. Cancers 2019, 11, 1943. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Zhang, D.W.; Zheng, X.L.; Tang, C.K. Itaconate: An emerging determinant of inflammation in activated macrophages. Immunol. Cell Biol. 2019, 97, 134–141. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).