
Estrogen-Like Effect of Mitotane Explained by Its Agonist Activity on Estrogen Receptor-α

 , , , and
, , , and
Abstract
1. Introduction
2. Material and Methods
2.1. Ligand Docking Experiments
2.2. Molecular Dynamics
2.3. Cell Treatment
2.4. Cell Viability and Cell Proliferation Experiments
2.5. Transient Silencing of ER-α
2.6. Western Blot Analysis for ER-α Expression
2.7. Statistical Analysis
3. Results
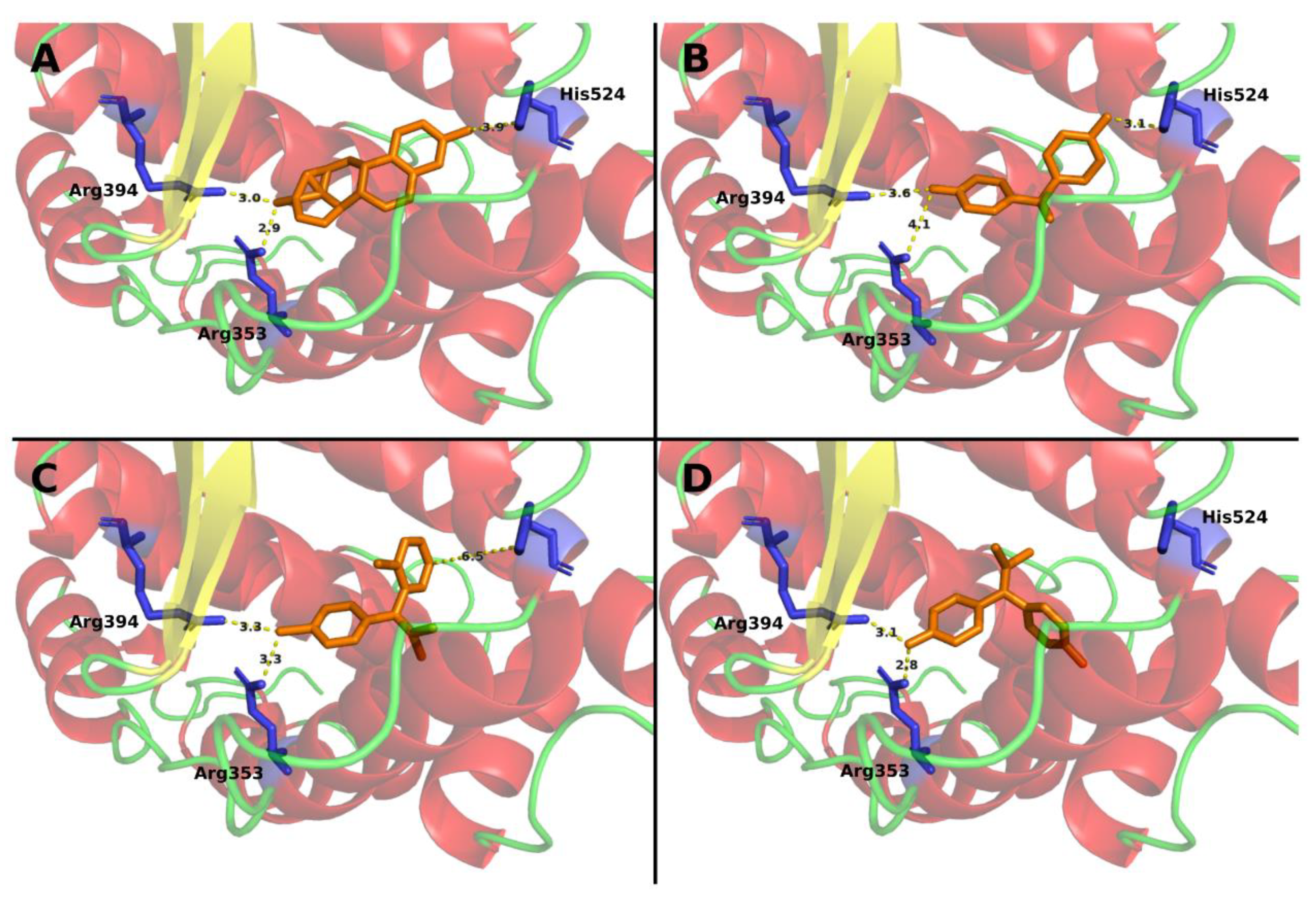
3.1. Mitotane Docking on ER-α
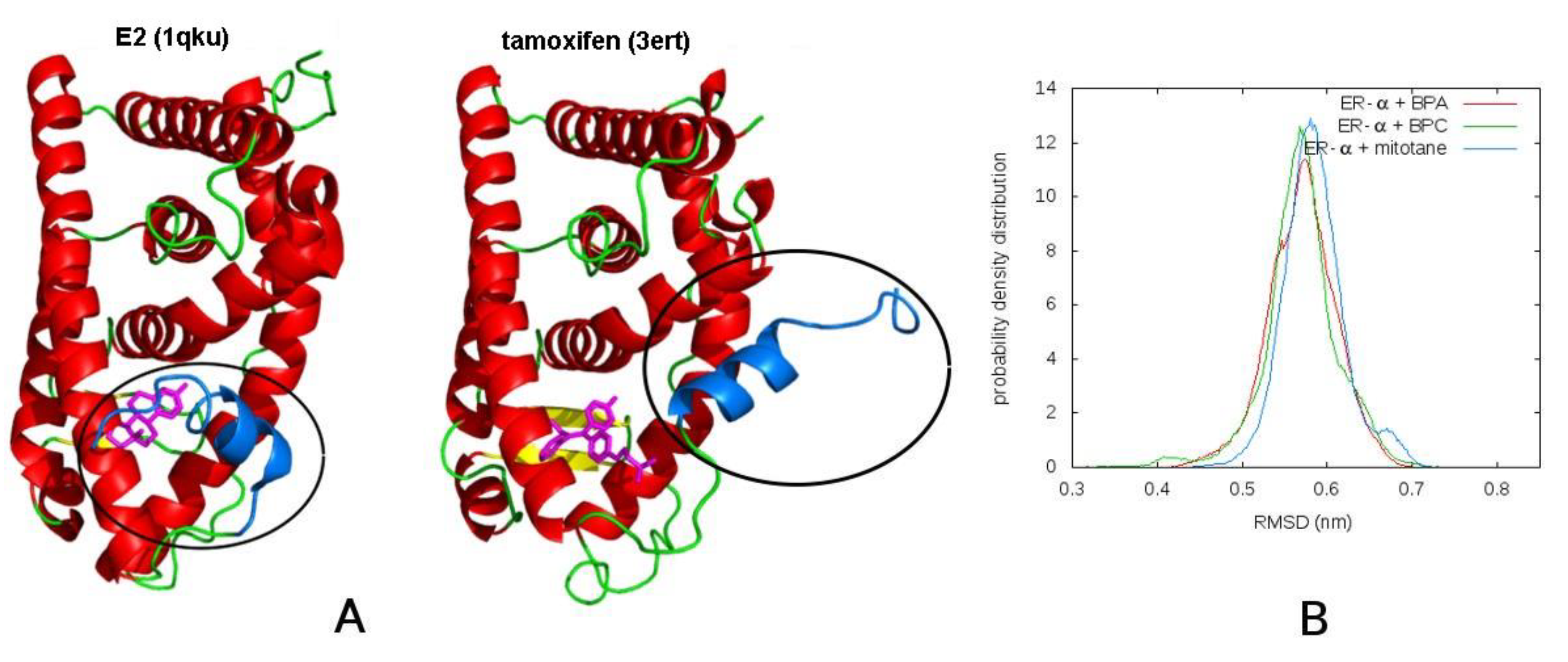
3.2. Molecular Dynamics
3.2.1. Mitotane in Molecular Dynamics Simulations Acts as an Agonist
3.2.2. Binding Free Energies from MD Calculations
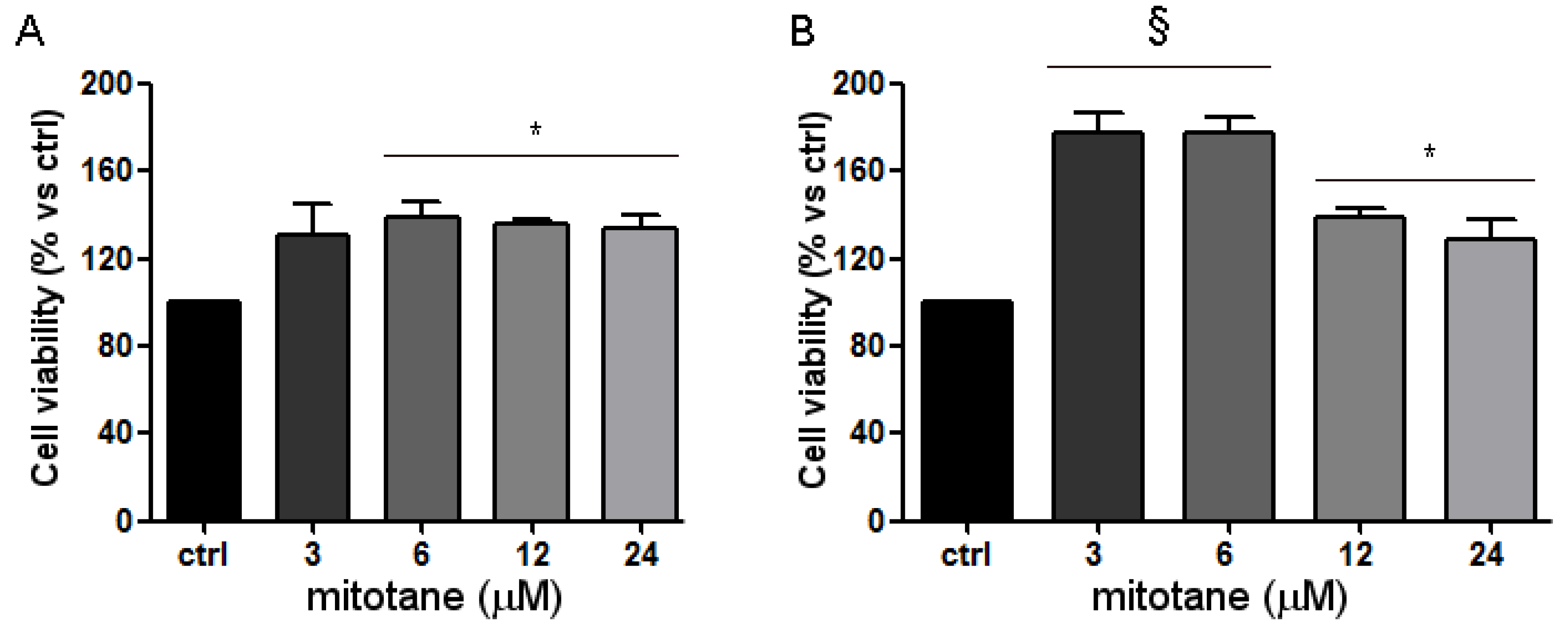
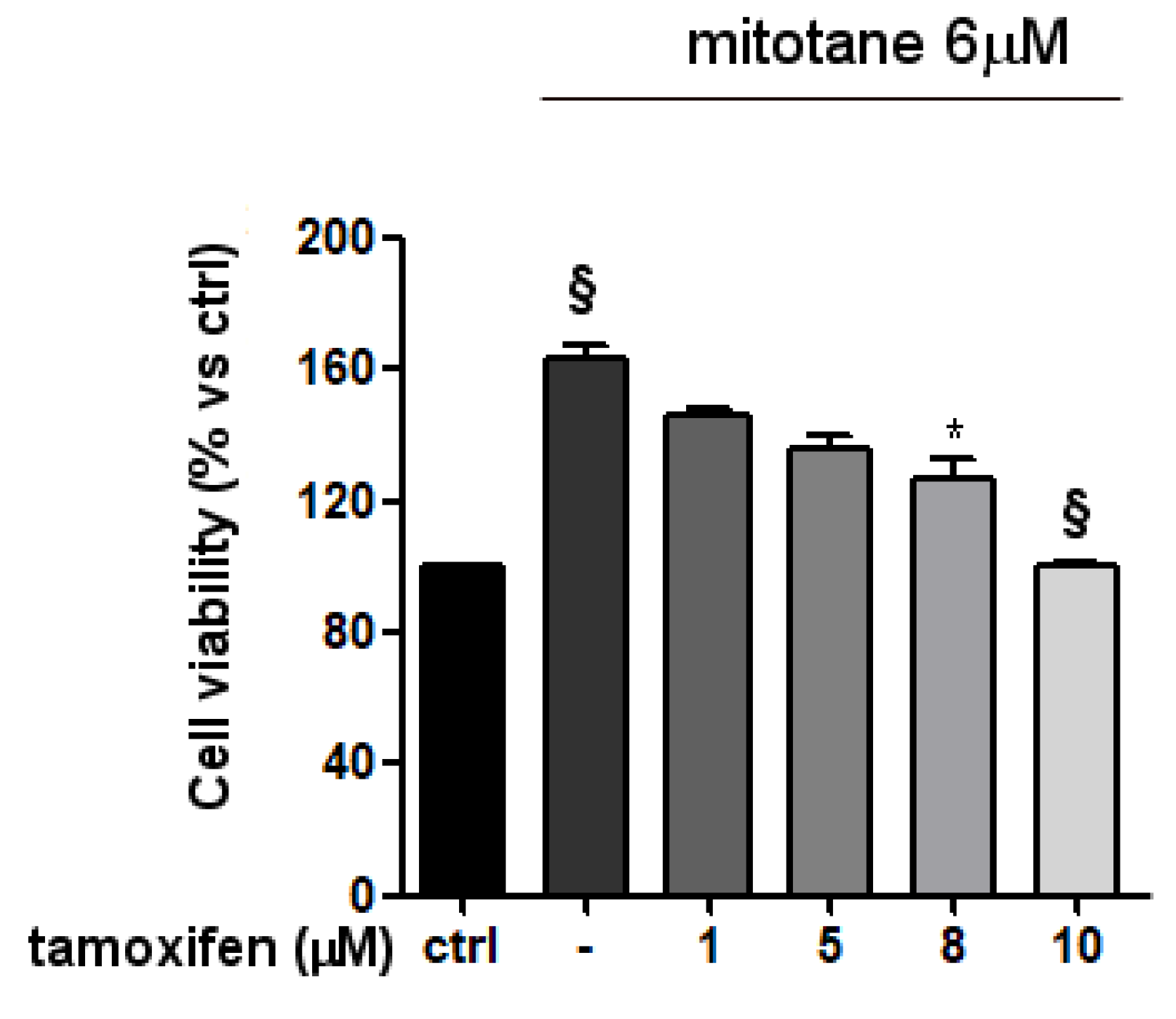
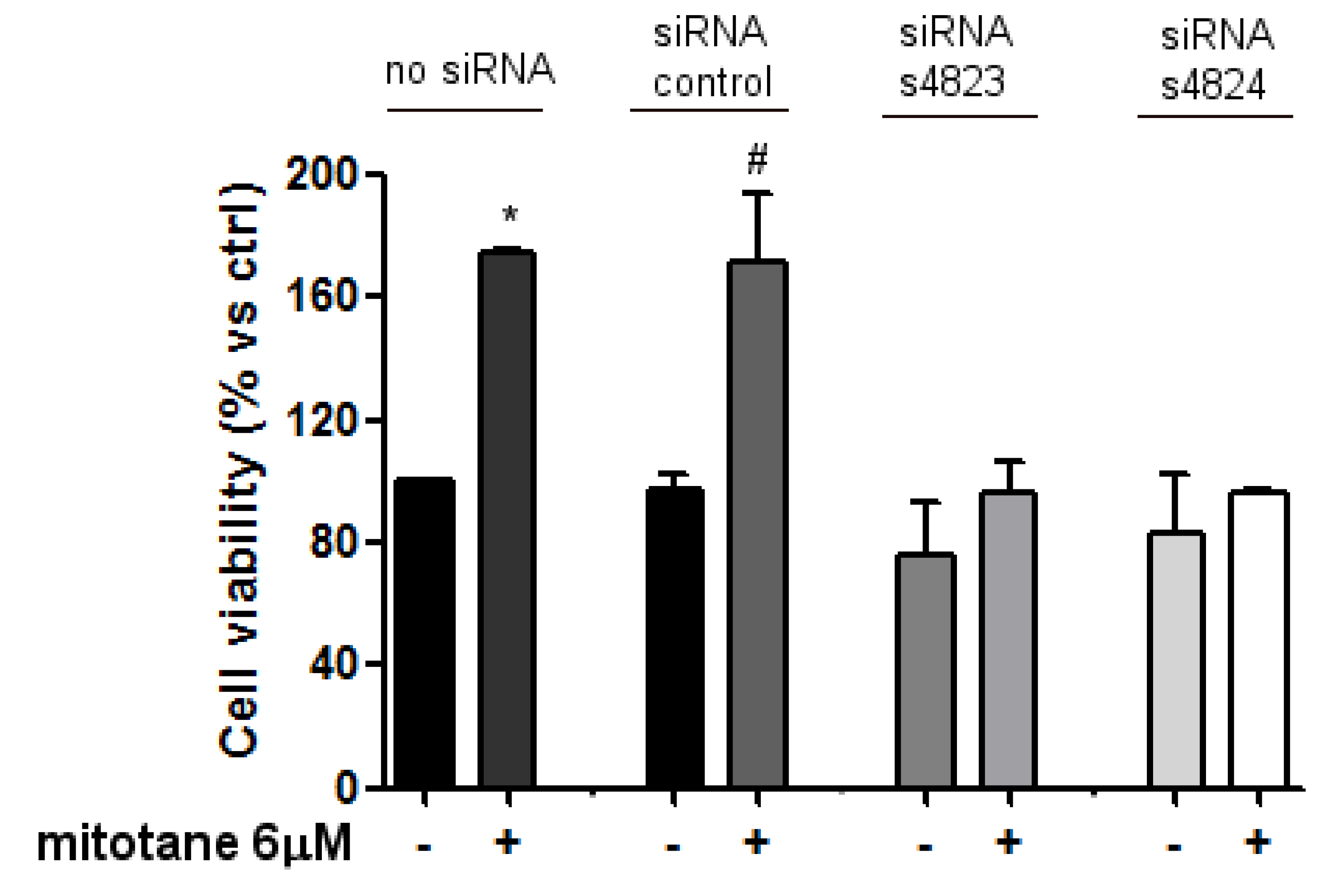
3.3. Mitotane Induced an Increase of MCF-7 Cell Viability and Proliferation via ER-α Stimulation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Notes
References
- Fassnacht, M.; Assie, G.; Baudin, E.; Eisenhofer, G.; de la Fouchardiere, C.; Haak, H.; de Krijger, R.; Porpiglia, F.; Terzolo, M.; Berruti, A. Adrenocortical carcinomas and malignant phaeochromocytomas: ESMO–EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1476–1490. [Google Scholar] [CrossRef]
- Fassnacht, M.; Dekkers, O.M.; Else, T.; Baudin, E.; Berruti, A.; de Krijger, R.R.; Haak, H.R.; Mihai, R.; Assie, G.; Terzolo, M. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur. J. Endocrinol. 2018, 179, G1–G46. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Berruti, A.; Fassnacht, M.; Haak, H.; Else, T.; Baudin, E.; Sperone, P.; Kroiss, M.; Kerkhofs, T.; Williams, A.R.; Ardito, A.; et al. Prognostic Role of Overt Hypercortisolism in Completely Operated Patients with Adrenocortical Cancer. Eur. Urol. 2014, 65, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Berruti, A.; Grisanti, S.; Pulzer, A.; Claps, M.; Daffara, F.; Loli, P.; Mannelli, M.; Boscaro, M.; Arvat, E.; Tiberio, G.; et al. Long-Term Outcomes of Adjuvant Mitotane Therapy in Patients with Radically Resected Adrenocortical Carcinoma. J. Clin. Endocrinol. Metab. 2017, 102, 1358–1365. [Google Scholar] [CrossRef]
- Terzolo, M.; Angeli, A.; Fassnacht, M.; Daffara, F.; Tauchmanova, L.; Conton, P.A.; Rossetto, R.; Buci, L.; Sperone, P.; Grossrubatscher, E.; et al. Adjuvant Mitotane Treatment for Adrenocortical Carcinoma. N. Engl. J. Med. 2007, 356, 2372–2380. [Google Scholar] [CrossRef] [PubMed]
- Laganà, M.; Grisanti, S.; Cosentini, D.; Ferrari, V.D.; Lazzari, B.; Ambrosini, R.; Sardini, C.; Volta, A.D.; Palumbo, C.; Poliani, P.L.; et al. Efficacy of the EDP-M Scheme Plus Adjunctive Surgery in the Management of Patients with Advanced Adrenocortical Carcinoma: The Brescia Experience. Cancers 2020, 12, 941. [Google Scholar] [CrossRef]
- Veytsman, I.; Nieman, L.; Fojo, T. Management of Endocrine Manifestations and the Use of Mitotane as a Chemotherapeutic Agent for Adrenocortical Carcinoma. J. Clin. Oncol. 2009, 27, 4619–4629. [Google Scholar] [CrossRef]
- Luton, J.-P.; Cerdas, S.; Billaud, L.; Thomas, G.; Guilhaume, B.; Bertagna, X.; Laudat, M.-H.; Louvel, A.; Chapuis, Y.; Blondeau, P.; et al. Clinical Features of Adrenocortical Carcinoma, Prognostic Factors, and the Effect of Mitotane Therapy. N. Engl. J. Med. 1990, 322, 1195–1201. [Google Scholar] [CrossRef]
- Basile, V.; Puglisi, S.; Calabrese, A.; Pia, A.; Perotti, P.; Berruti, A.; Reimondo, G.; Terzolo, M. Unwanted Hormonal and Metabolic Effects of Postoperative Adjuvant Mitotane Treatment for Adrenocortical Cancer. Cancers 2020, 12, 2615. [Google Scholar] [CrossRef]
- Salenave, S.; Bernard, V.; Cao, C.D.; Guignat, L.; Bachelot, A.; Leboulleux, S.; Droumaguet, C.; Bry-Gauillard, H.; Pierre, P.; Crinière, L.; et al. Ovarian macrocysts and gonadotrope–ovarian axis disruption in premenopausal women receiving mitotane for adrenocortical carcinoma or Cushing’s disease. Eur. J. Endocrinol. 2015, 172, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, G.; Ekerhovd, E.; Janson, P.O.; Jansson, S.; Ahlman, H.; Wängberg, B.; Norström, A. Ovarian cyst formation in women of reproductive age receiving mitotane as part of the treatment of adrenocortical carcinoma: Clinical and experimental observations. Acta Obstet. Gynecol. Scand. 2020, 99, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Oddie, P.D.; Albert, B.B.; Hofman, P.L.; Jefferies, C.; Laughton, S.; Carter, P.J. Mitotane in the treatment of childhood adrenocortical carcinoma: A potent endocrine disruptor. Endocrinol. Diabetes Metab. Case Rep. 2018, 2018, 18–0059. [Google Scholar] [CrossRef] [PubMed]
- Rosner, W. The Functions of Corticosteroid-Binding Globulin and Sex Hormone-Binding Globulin: Recent Advances. Endocr. Rev. 1990, 11, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Nader, N.; Raverot, G.; Emptoz-Bonneton, A.; Déchaud, H.; Bonnay, M.; Baudin, E.; Pugeat, M. Mitotane Has an Estrogenic Effect on Sex Hormone-Binding Globulin and Corticosteroid-Binding Globulin in Humans. J. Clin. Endocrinol. Metab. 2006, 91, 2165–2170. [Google Scholar] [CrossRef] [PubMed]
- Kaludjerovic, J.; Ward, W.E. The Interplay between Estrogen and Fetal Adrenal Cortex. J. Nutr. Metab. 2012, 2012, 1–12. [Google Scholar] [CrossRef]
- De Cremoux, P.; Rosenberg, D.; Goussard, J.; Bremont-Weil, C.; Tissier, F.; Tran-Perennou, C.; Groussin, L.; Bertagna, X.; Bertherat, J.; Raffin-Sanson, M.-L. Expression of progesterone and estradiol receptors in normal adrenal cortex, adrenocortical tumors, and primary pigmented nodular adrenocortical disease. Endocr. Relat. Cancer 2008, 15, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Baquedano, M.S.; Saraco, N.; Berensztein, E.; Pepe, C.; Bianchini, M.; Levy, E.; Goñi, J.; Rivarola, M.A.; Belgorosky, A. Identification and Developmental Changes of Aromatase and Estrogen Receptor Expression in Prepubertal and Pubertal Human Adrenal Tissues. J. Clin. Endocrinol. Metab. 2007, 92, 2215–2222. [Google Scholar] [CrossRef]
- Barzon, L.; Masi, G.; Pacenti, M.; Trevisan, M.; Fallo, F.; Remo, A.; Martignoni, G.; Montanaro, D.; Pezzi, V.; Palù, G. Expression of aromatase and estrogen receptors in human adrenocortical tumors. Virchows Archiv 2007, 452, 181–191. [Google Scholar] [CrossRef]
- Rossini, E.; Tamburello, M.; Abate, A.; Beretta, S.; Fragni, M.; Cominelli, M.; Cosentini, D.; Hantel, C.; Bono, F.; Grisanti, S.; et al. Cytotoxic Effect of Progesterone, Tamoxifen and Their Combination in Experimental Cell Models of Human Adrenocortical Cancer. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef]
- Klotz, D.M.; Beckman, B.S.; Hill, S.M.; A McLachlan, J.; Walters, M.R.; Arnold, S.F. Identification of environmental chemicals with estrogenic activity using a combination of in vitro assays. Environ. Health Perspect. 1996, 104, 1084–1089. [Google Scholar] [CrossRef]
- Chen, C.W.; Hurd, C.; Vorojeikina, D.P.; Arnold, S.F.; Notides, A.C. Transcriptional activation of the human estrogen receptor by DDT isomers and metabolites in yeast and MCF-7 cells. Biochem. Pharmacol 1997, 53, 1161–1172. [Google Scholar]
- Scippo, M.-L.; Argiris, C.; Muller, M.; Willemsen, P.; Martial, J.; Maghuin-Rogister, G. Recombinant human estrogen, androgen and progesterone receptors for detection of potential endocrine disruptors. Anal. Bioanal. Chem. 2004, 378, 664–669. [Google Scholar] [CrossRef]
- Kumar, R.; Zakharov, M.N.; Khan, S.H.; Miki, R.; Jang, H.; Toraldo, G.; Singh, R.; Bhasin, S.; Jasuja, R. The Dynamic Structure of the Estrogen Receptor. J. Amino Acids 2011, 2011, 1–7. [Google Scholar] [CrossRef]
- Gangloff, M.; Ruff, M.; Eiler, S.; Duclaud, S.; Wurtz, J.M.; Moras, D. Crystal Structure of a Mutant hERα Ligand-binding Domain Reveals Key Structural Features for the Mechanism of Partial Agonism. J. Biol. Chem. 2001, 276, 15059–15065. [Google Scholar] [CrossRef]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Schwabe, J.; Chapman, L.; Finch, J.T.; Rhodes, D.; Neuhaus, D. DNA recognition by the oestrogen receptor: From solution to the crystal. Structure 1993, 1, 187–204. [Google Scholar] [CrossRef]
- Farzaneh, S.; Zarghi, A. Estrogen Receptor Ligands: A Review (2013–2015). Sci. Pharm. 2016, 84, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Delfosse, V.; Grimaldi, M.; Pons, J.-L.; Boulahtouf, A.; le Maire, A.; Cavailles, V.; Labesse, G.; Bourguet, W.; Balaguer, P. Structural and mechanistic insights into bisphenols action provide guidelines for risk assessment and discovery of bisphenol A substitutes. Proc. Natl. Acad. Sci. USA 2012, 109, 14930–14935. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 3, W270–W277. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. EADock: Docking of small molecules into protein active sites with a multiobjective evolutionary optimization. Proteins: Struct. Funct. Bioinform. 2007, 67, 1010–1025. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Fragni, M.; Bonini, S.A.; Bettinsoli, P.; Bodei, S.; Generali, D.; Bottini, A.; Spano, P.F.; Memo, M.; Sigala, S. The miR-21/PTEN/Akt signaling pathway is involved in the anti-tumoral effects of zoledronic acid in human breast cancer cell lines. Naunyn-Schmiedeberg Arch. Pharmacol. 2016, 389, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Fragni, M.; Perego, P.; Vezzoli, S.; Bonini, S.A.; Tortoreto, M.; Galli, D.; Claps, M.; Tiberio, G.A.; Terzolo, M.; et al. Antisecretive and Antitumor Activity of Abiraterone Acetate in Human Adrenocortical Cancer: A Preclinical Study. J. Clin. Endocrinol. Metab. 2016, 101, 4594–4602. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, A.M.; Pike, A.C.W.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engström, O.; Öhman, L.; Greene, G.L.; Gustafsson, J.Å.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nat. Cell Biol. 1997, 389, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Rich, R.L.; Hoth, L.R.; Geoghegan, K.F.; Brown, T.A.; LeMotte, P.K.; Simons, S.P.; Hensley, P.; Myszka, D.G. Kinetic analysis of estrogen receptor/ligand interactions. Proc. Natl. Acad. Sci. USA 2002, 99, 8562–8567. [Google Scholar] [CrossRef] [PubMed]
- Corso, C.R.; Acco, A.; Bach, C.; Bonatto, S.J.R.; De Figueiredo, B.C.; De Souza, L.M. Pharmacological profile and effects of mitotane in adrenocortical carcinoma. Br. J. Clin. Pharmacol. 2020, 1–13. [Google Scholar] [CrossRef]
- Pavlin, M.; Spinello, A.; Pennati, M.; Zaffaroni, N.; Gobbi, S.; Bisi, A.; Colombo, G.; Magistrato, A. A Computational Assay of Estrogen Receptor α Antagonists Reveals the Key Common Structural Traits of Drugs Effectively Fighting Refractory Breast Cancers. Sci. Rep. 2018, 8, 649. [Google Scholar] [CrossRef]
- Fulvestrant, IBM Micromedex® DRUGDEX® (Electronic Version). IBM Watson Health, Greenwood Village, Colorado, USA. Available online: https://www.micromedexsolutions.com/ (accessed on 6 May 2021).
- Tamoxifen, IBM Micromedex® DRUGDEX® (Electronic Version). IBM Watson Health, Greenwood Village, Colorado, USA. Available online: https://www.micromedexsolutions.com/ (accessed on 6 May 2021).
- Puszkiel, A.; Arellano, C.; Vachoux, C.; Evrard, A.; Le Morvan, V.; Boyer, J.; Robert, J.; Delmas, C.; Dalenc, F.; Debled, M.; et al. Model-Based Quantification of Impact of Genetic Polymorphisms and Co-Medications on Pharmacokinetics of Tamoxifen and Six Metabolites in Breast Cancer. Clin. Pharmacol. Ther. 2021, 109, 1244–1255. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | BPA | BPC | Mitotane | Tamoxifen |
|---|---|---|---|---|
| MMGBSA | 12.8 | 10.1 | 9.9 | −4.6 |
| FEP | 11.0 | 10.4 | 8.4 | 2.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossini, E.; Giacopuzzi, E.; Gangemi, F.; Tamburello, M.; Cosentini, D.; Abate, A.; Laganà, M.; Berruti, A.; Grisanti, S.; Sigala, S. Estrogen-Like Effect of Mitotane Explained by Its Agonist Activity on Estrogen Receptor-α. Biomedicines 2021, 9, 681. https://doi.org/10.3390/biomedicines9060681
Rossini E, Giacopuzzi E, Gangemi F, Tamburello M, Cosentini D, Abate A, Laganà M, Berruti A, Grisanti S, Sigala S. Estrogen-Like Effect of Mitotane Explained by Its Agonist Activity on Estrogen Receptor-α. Biomedicines. 2021; 9(6):681. https://doi.org/10.3390/biomedicines9060681
Chicago/Turabian StyleRossini, Elisa, Edoardo Giacopuzzi, Fabrizio Gangemi, Mariangela Tamburello, Deborah Cosentini, Andrea Abate, Marta Laganà, Alfredo Berruti, Salvatore Grisanti, and Sandra Sigala. 2021. "Estrogen-Like Effect of Mitotane Explained by Its Agonist Activity on Estrogen Receptor-α" Biomedicines 9, no. 6: 681. https://doi.org/10.3390/biomedicines9060681
APA StyleRossini, E., Giacopuzzi, E., Gangemi, F., Tamburello, M., Cosentini, D., Abate, A., Laganà, M., Berruti, A., Grisanti, S., & Sigala, S. (2021). Estrogen-Like Effect of Mitotane Explained by Its Agonist Activity on Estrogen Receptor-α. Biomedicines, 9(6), 681. https://doi.org/10.3390/biomedicines9060681