
Exploiting Knowledge on Structure–Activity Relationships for Designing Peptidomimetics of Endogenous Peptides


Abstract
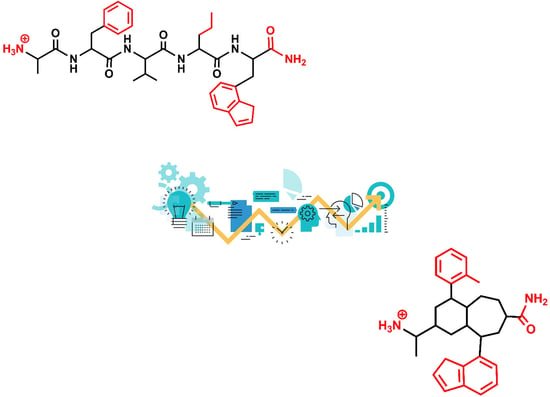
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
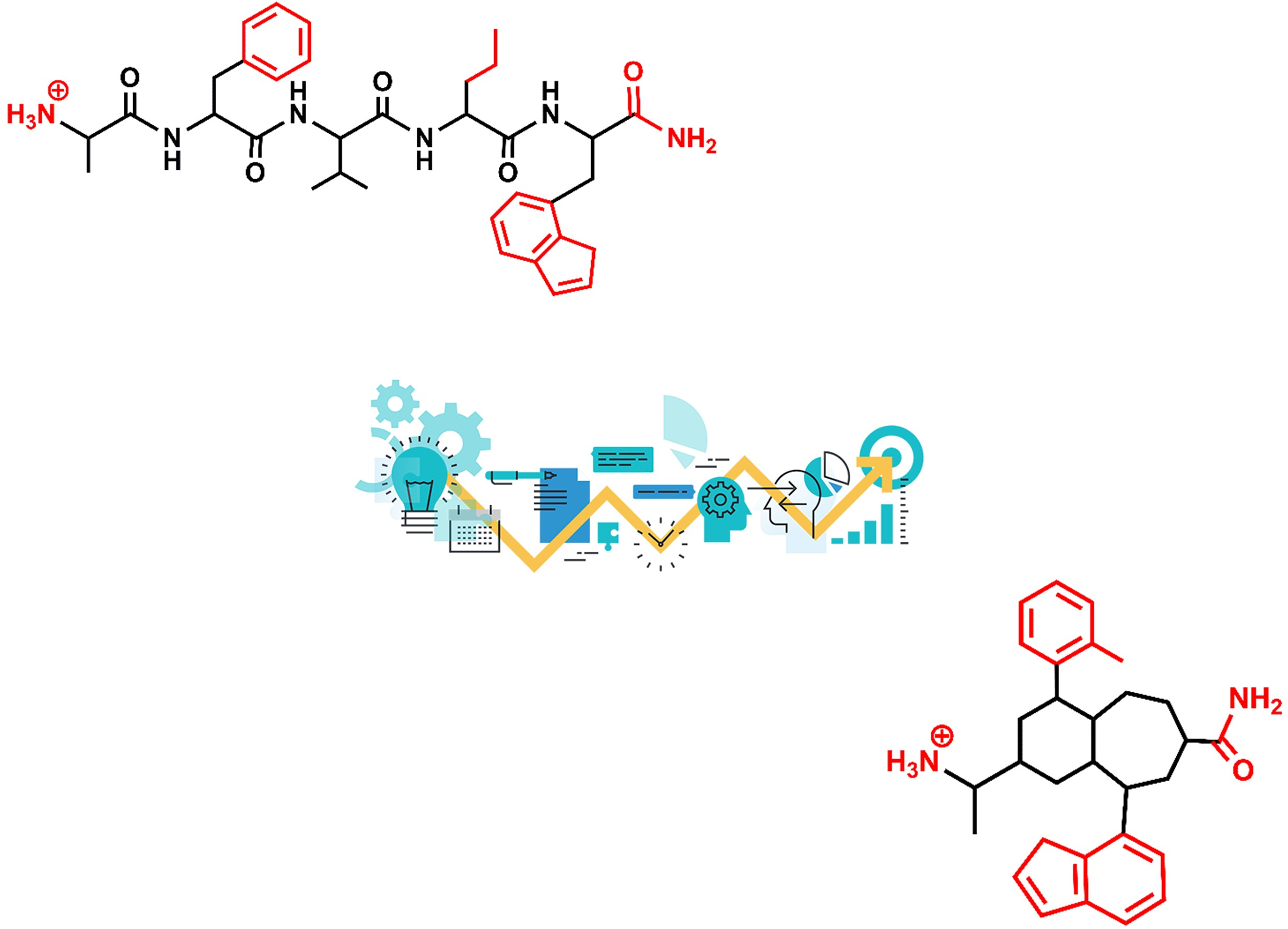
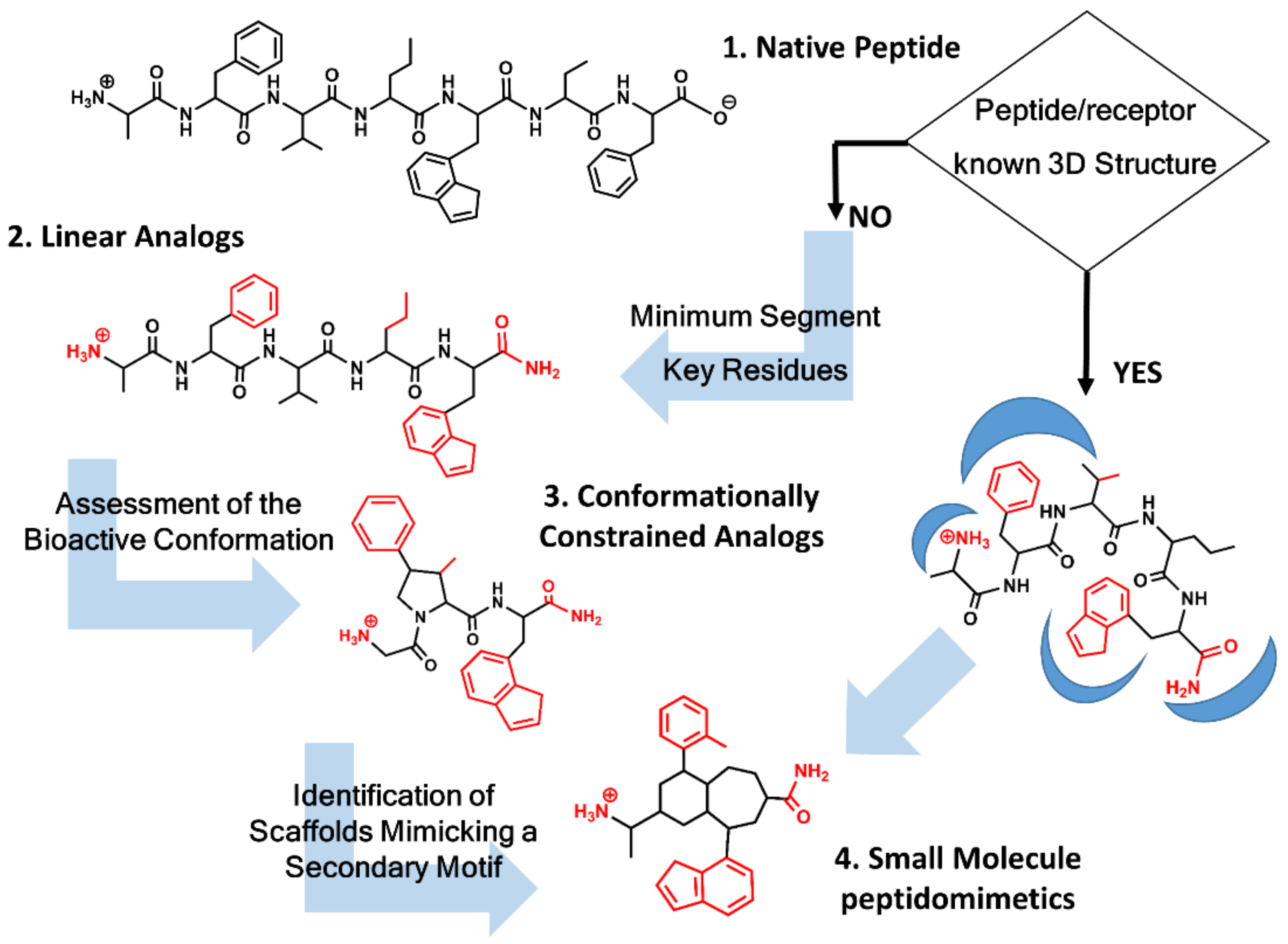
2. Roadmap to Design Peptidomimetics
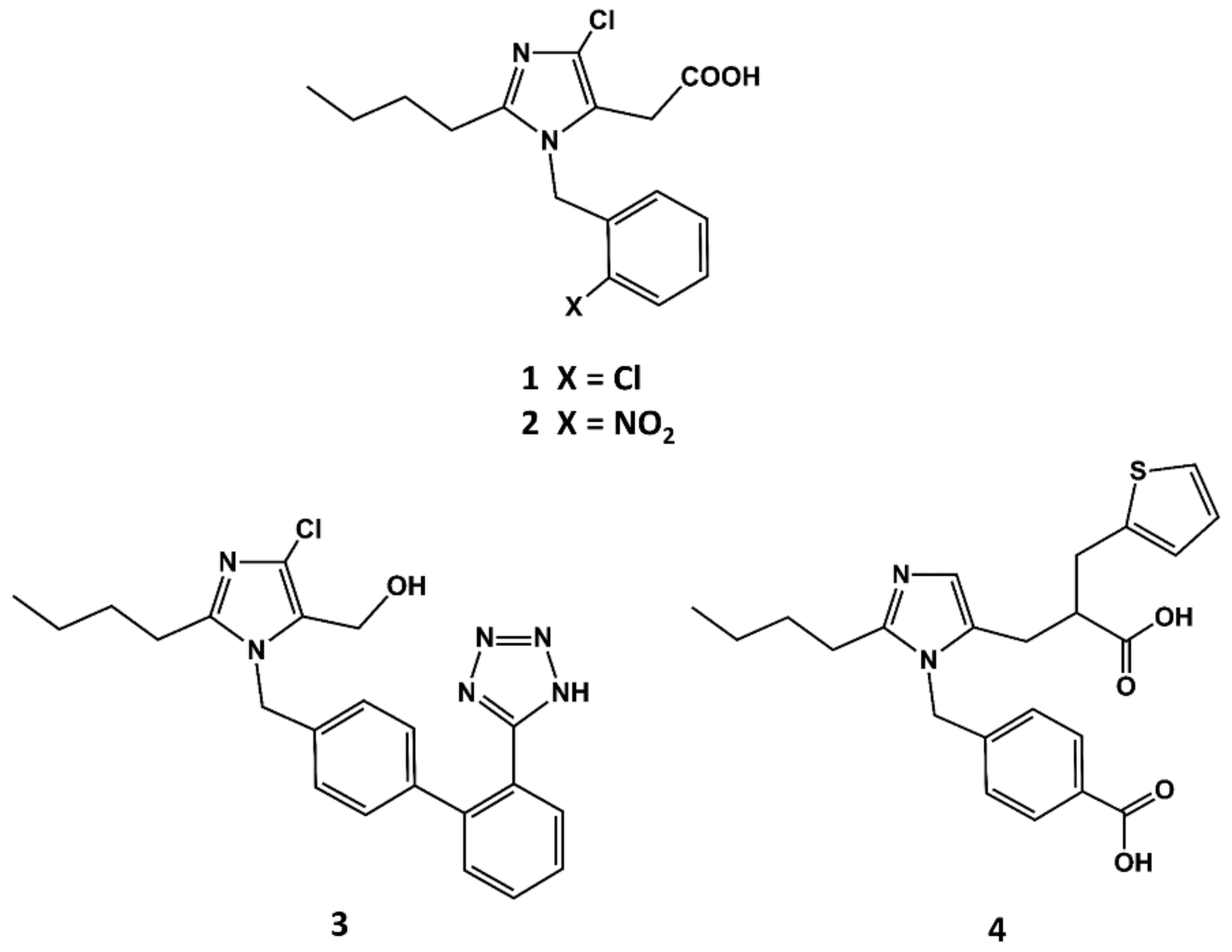
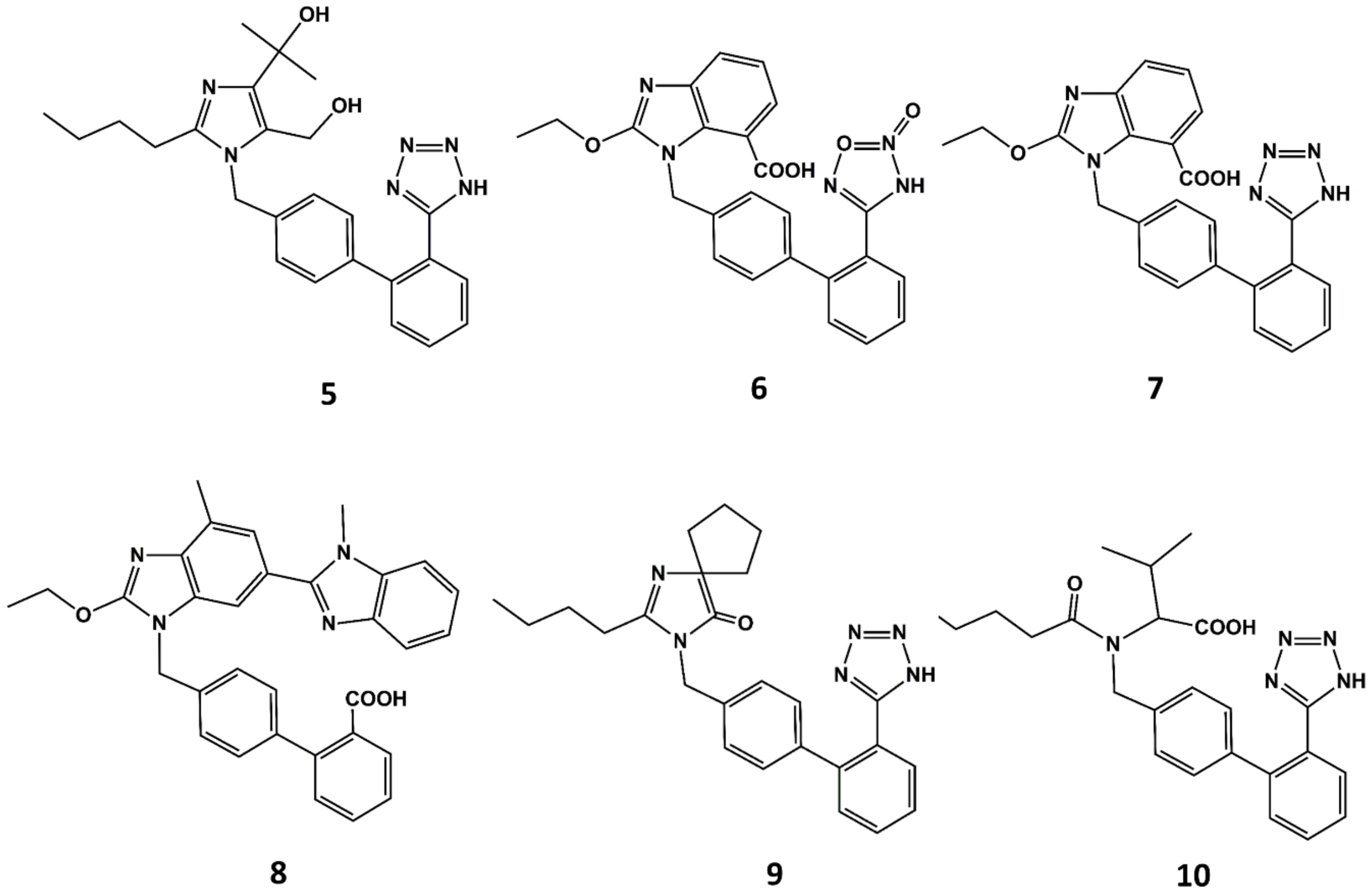
3. Example 1. Design of Angiotensin II AT1 Antagonists
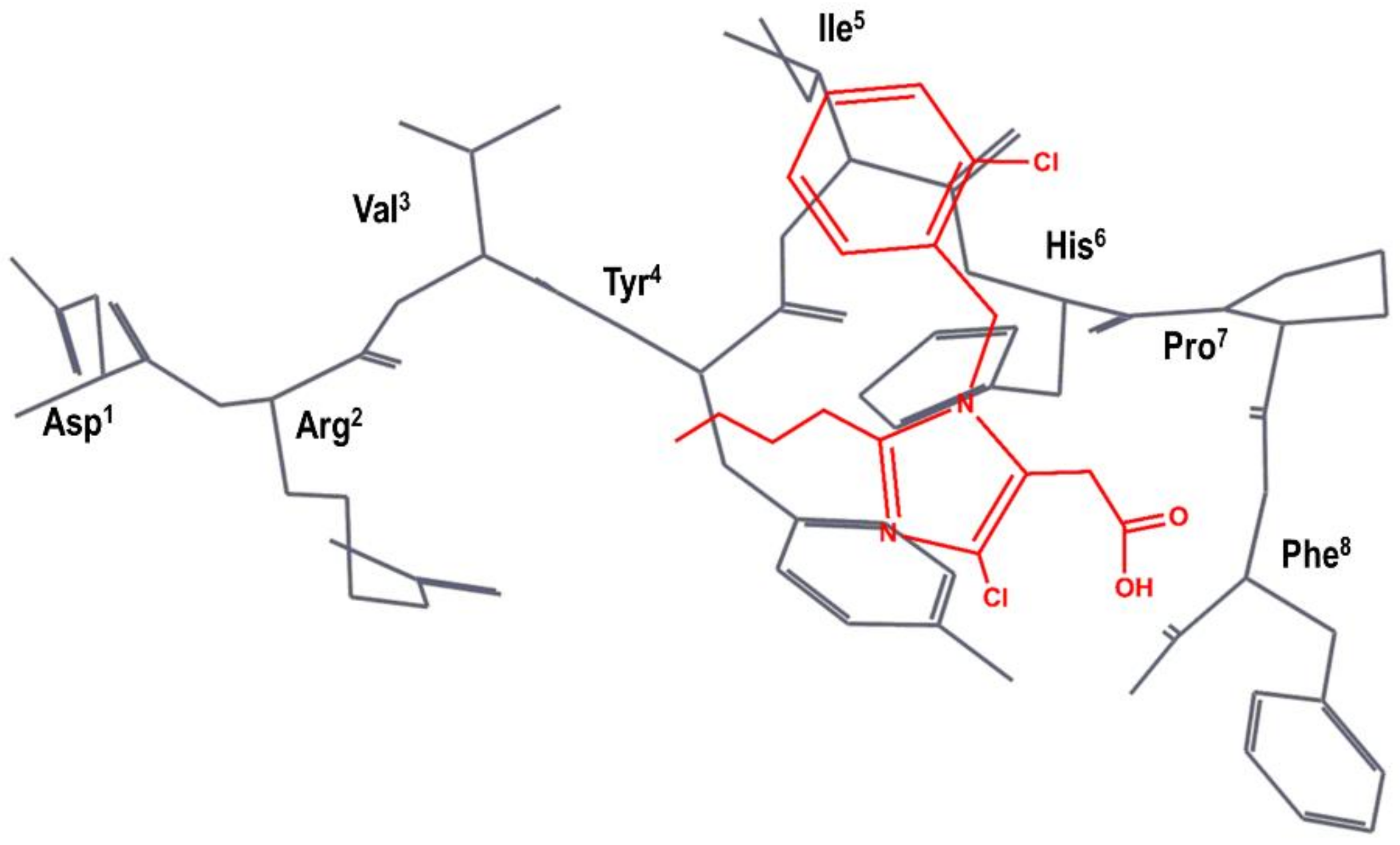
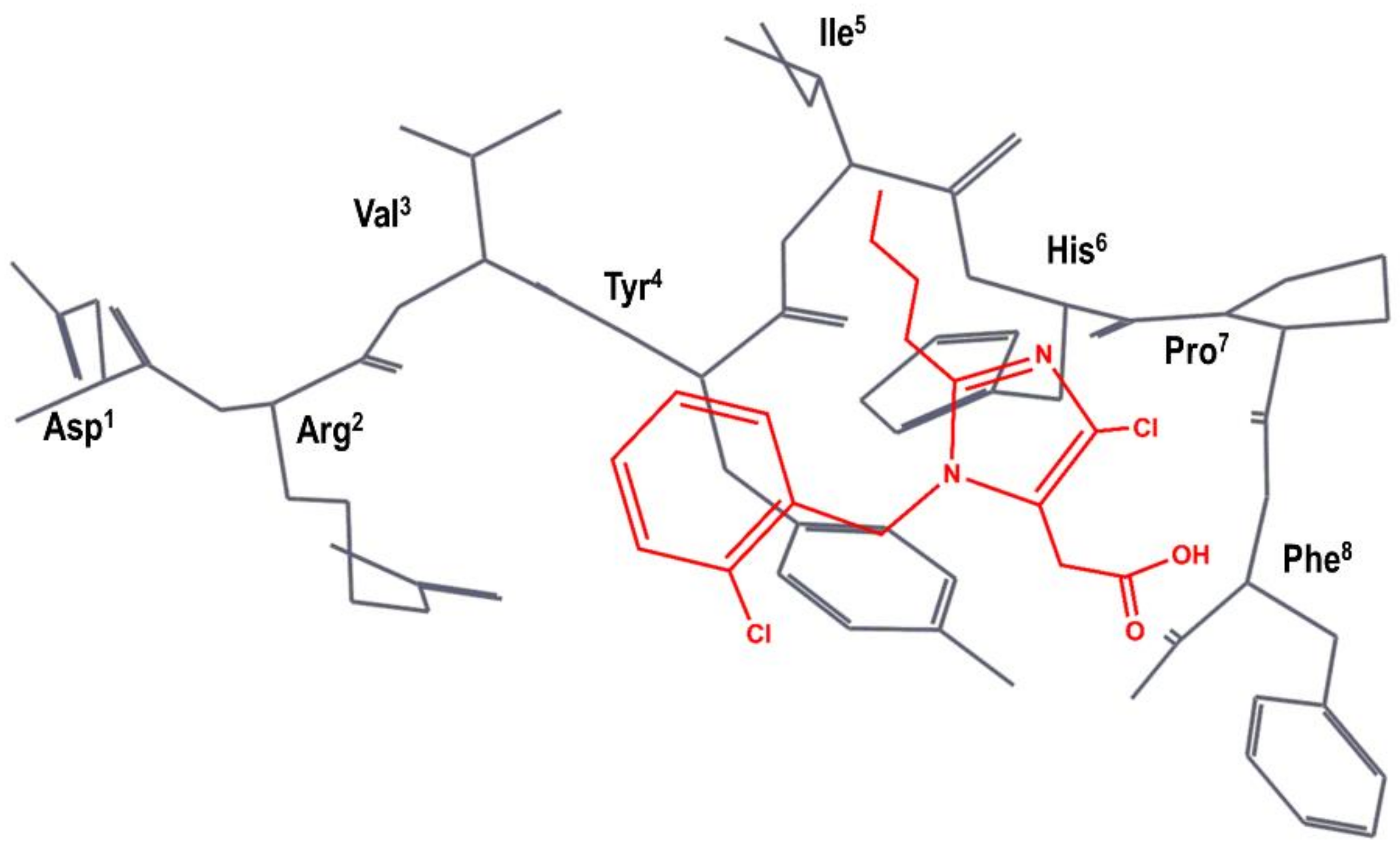
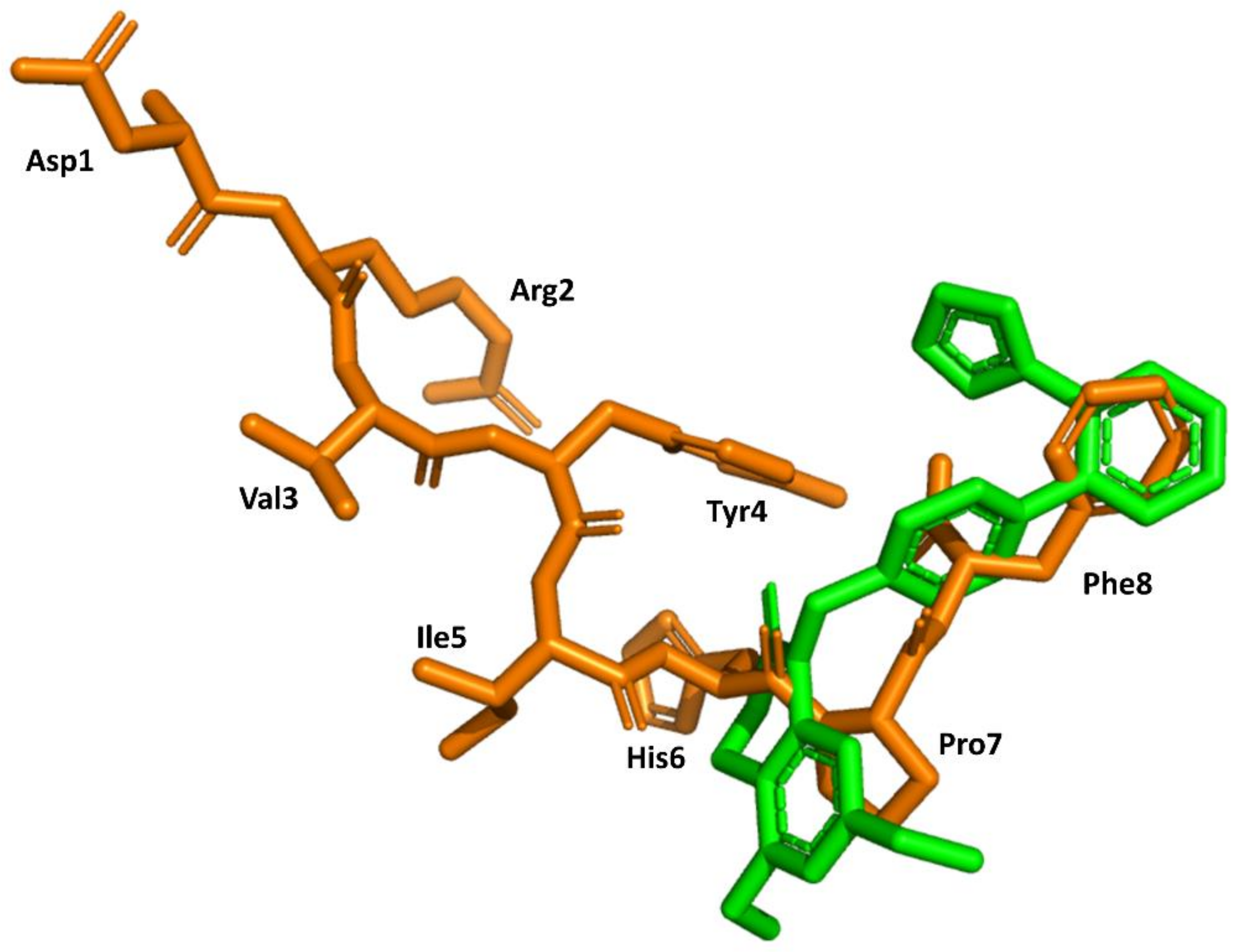
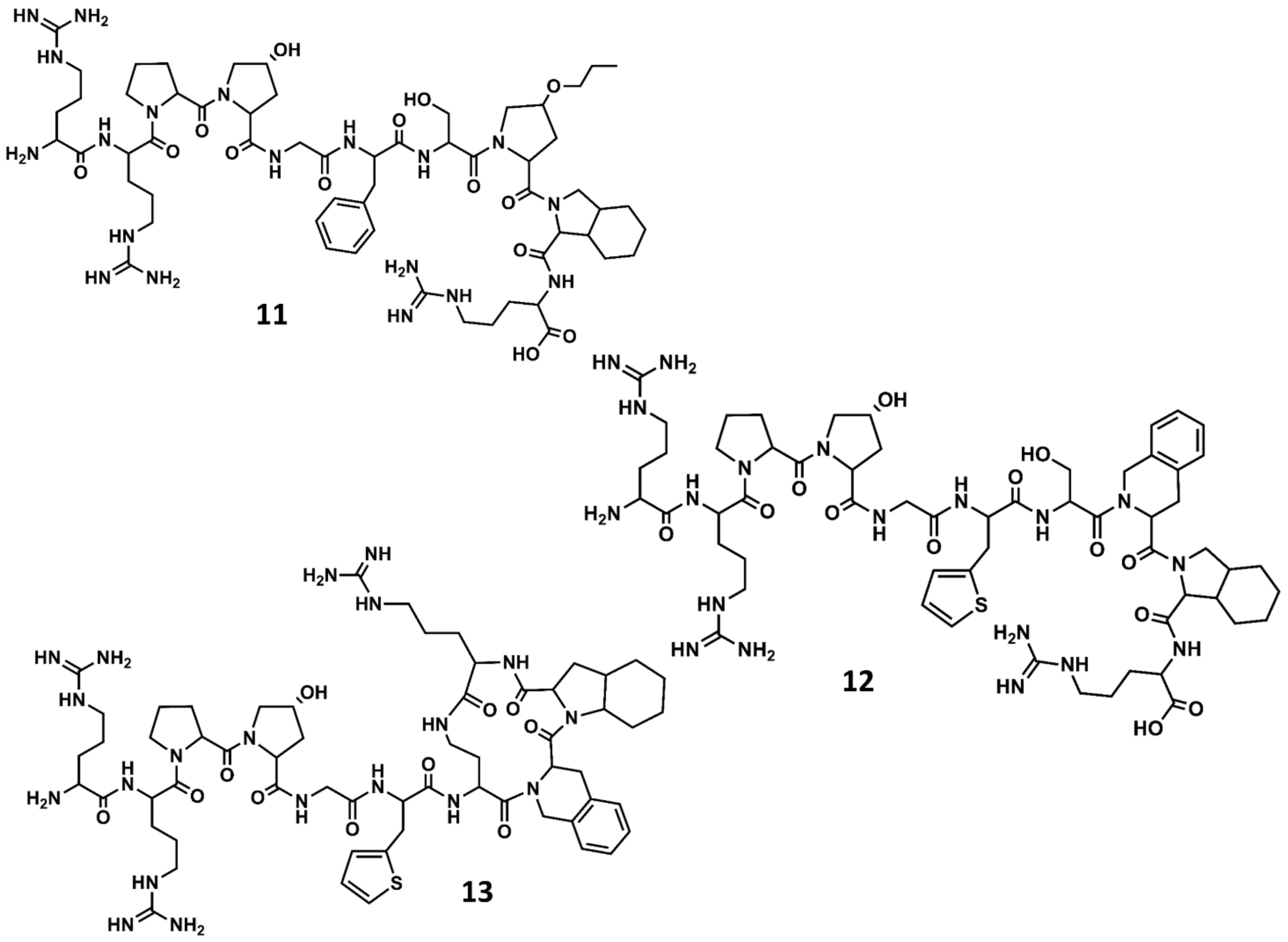
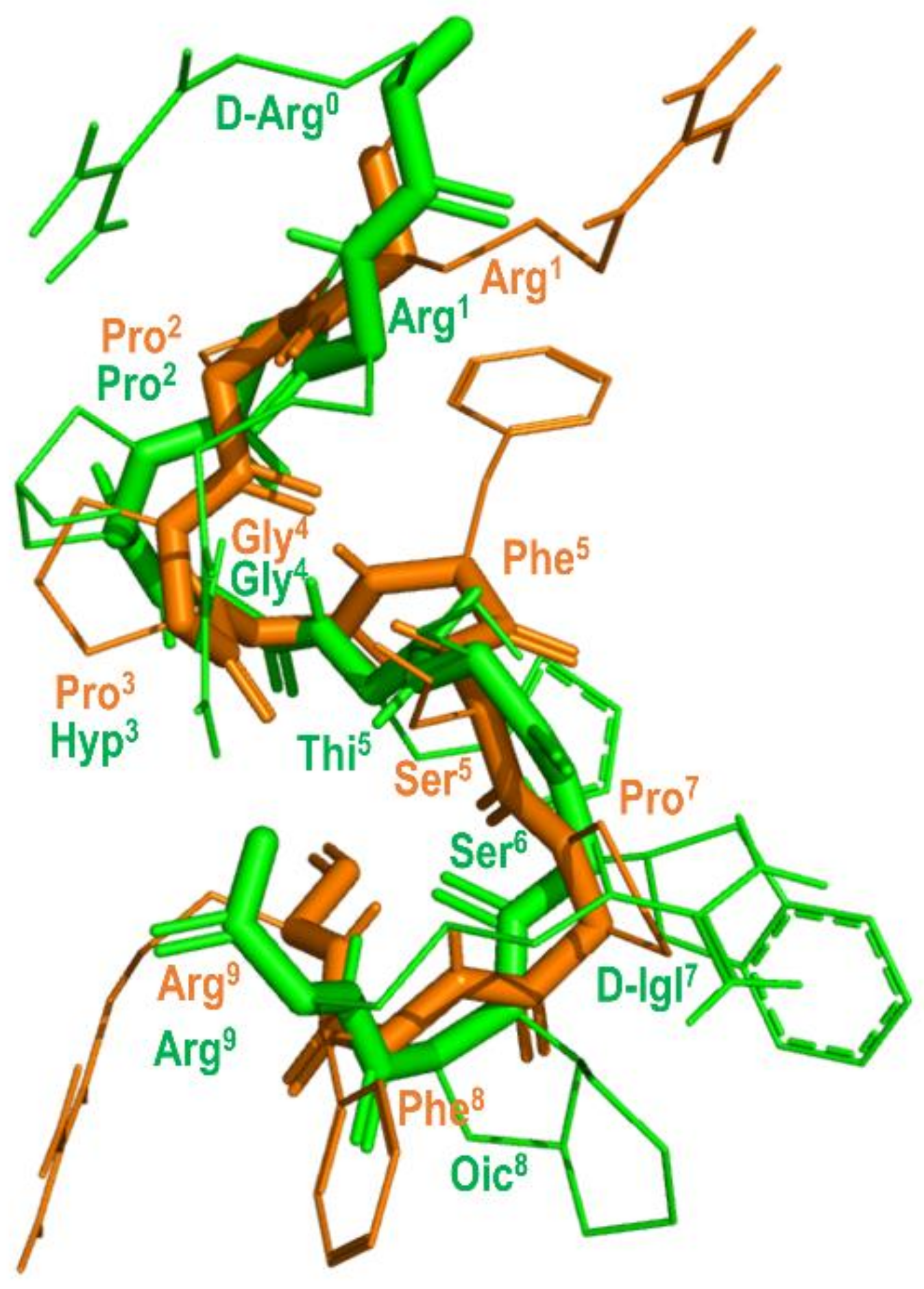
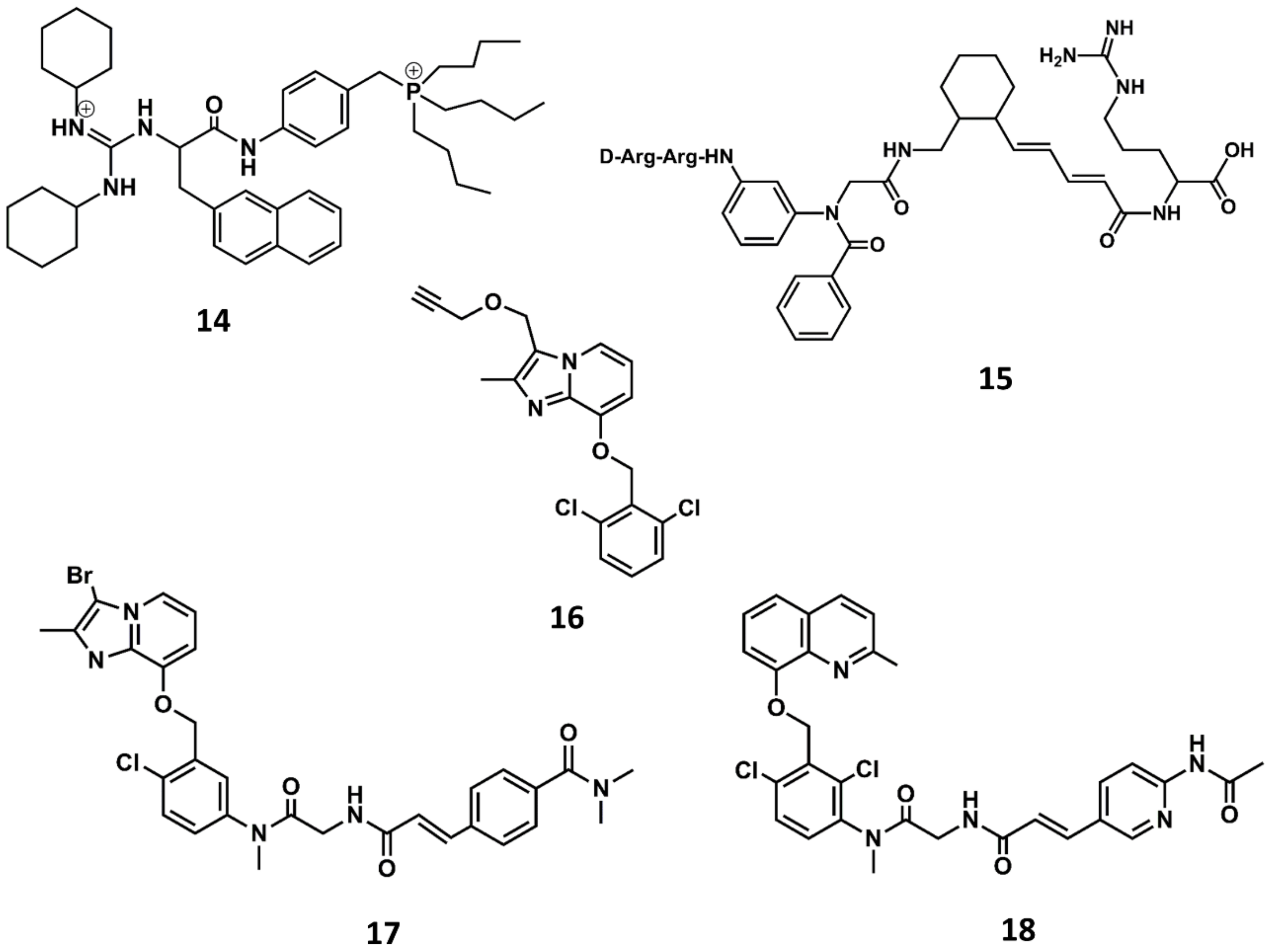
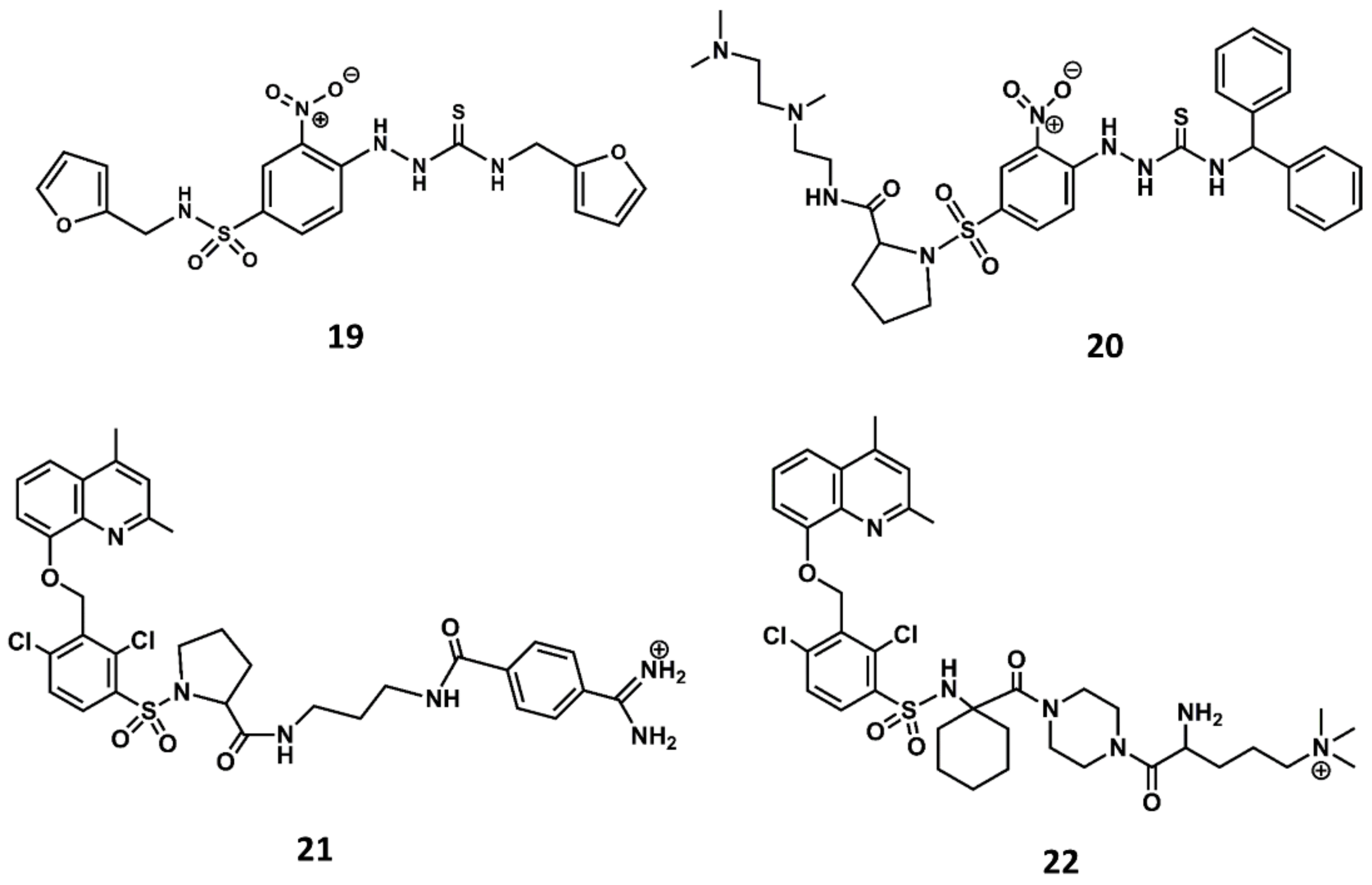
4. Example 2. Design of Bradykinin B2 Antagonists
5. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kastin, A.J. (Ed.) Handbook of Biological Active Peptides; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Hook, V.; Funkelstein, L.; Lu, D.; Bark, S.; Wegrzyn, J.; Hwang, S.R. Proteases for processing proneuropeptides into peptide neurotransmitters and hormones. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 393–423. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, A.D.; Saghatelian, A. Investigating endogenous peptides and peptidases using peptidomics. Biochemistry 2011, 50, 7447–7461. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Mordalski, S.; Pándy-Szekeres, G.; Esgerra, M.; Mamyrbekov, A.; Munk, C.; Keserű, G.M.; Gloriam, D.E. GPCRdb in 2021: Integrating GPCR sequence, structure and function. Nucl. Acids Res. 2020, 49, D335–D343. [Google Scholar] [CrossRef]
- Hubbard, S.R.; Till, J.H. Protein tyrosine kinase structure and function. Ann. Rev. Biochem. 2000, 69, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, M.J.; Ringrose, P.S. Peptides and Related Drugs: A Review of Their Absorption, Metabolism, and Excretion. Drug Metabol. Rev. 1986, 17, 283–310. [Google Scholar] [CrossRef]
- Malonis, R.J.; Lai, J.R.; Vergnolle, O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem. Rev. 2020, 120, 3210–3229. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Perez, J.J. Designing Peptidomimetics. Curr. Top. Med. Chem. 2018, 18, 566–590. [Google Scholar] [CrossRef]
- Perez, J.J.; Corcho, F.; Llorens, O. Molecular modeling in the design of peptidomimetics and peptide surrogates. Curr. Med. Chem. 2002, 9, 2209–2229. [Google Scholar] [CrossRef]
- Kosterlitz, H.W.; Waterfield, A.A. In vitro models in study of structure-activity-relationships of narcotic analgesics. Annu. Rev. Pharmacol. Toxicol. 1975, 15, 29–47. [Google Scholar] [CrossRef]
- Snyder, S.H.; Pasternak, G.W. Historical review: Opioid receptors. Trends Pharmacol. Sci. 2003, 24, 198–205. [Google Scholar] [CrossRef]
- Wu, Z.; Hruby, V.J. Backbone alignment modeling of the structure-activity relationships of opioid ligands. J. Chem. Inf. Model. 2011, 51, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, P.K.; Ala, P.; Woerner, F.J.; Chang, C.H.; Garber, S.S.; Anton, E.D.; Bacheler, L.T. Cyclic urea amides: HIV-1 protease inhibitors with low nanomolar potency against both wild type and protease inhibitor resistant mutants of HIV. J. Med. Chem. 1997, 40, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Hanske, J.; Sadian, Y.; Muller, C.W. The cryo-EM resolution revolution and transcription complexes. Curr. Opin. Struct. Biol. 2018, 52, 8–15. [Google Scholar] [CrossRef]
- Billeter, M.; Wagner, G.; Wuthrich, K. Solution NMR structure determination of proteins revisited. J. Biomol. NMR 2008, 42, 155–158. [Google Scholar] [CrossRef]
- Kang, C. Applications of In-Cell NMR in Structural Biology and Drug Discovery. Int. J. Mol. Sci. 2019, 20, 139. [Google Scholar] [CrossRef]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wuthrich, K. GPCR drug discovery: Integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 2019, 18, 59–82. [Google Scholar] [CrossRef]
- Hennig, J.; Sattler, M. The dynamic duo: Combining NMR and small angle scattering in structural biology. Protein Sci. 2014, 23, 669–682. [Google Scholar] [CrossRef]
- Iacobucci, C.; Gotze, M.; Ihling, C.H.; Piotrowski, C.; Arlt, C.; Schafer, M.; Hage, C.; Schmidt, R.; Sinz, A. A cross-linking/mass spectrometry workflow based on MS-cleavable cross-linkers and the MeroX software for studying protein structures and protein-protein interactions. Nat. Protoc. 2018, 13, 2864. [Google Scholar] [CrossRef]
- Zheng, J.; Yong, H.Y.; Panutdaporn, N.; Liu, C.; Tang, K.; Luo, D. High-resolution HDX-MS reveals distinct mechanisms of RNA recognition and activation by RIG-I and MDA5. Nucleic Acids Res. 2015, 43, 1216–1230. [Google Scholar] [CrossRef]
- Cox, B.M. A Concise Review of Concepts in Opioid Pharmacology up to the Discovery of Endogenous Opioids. Mol. Pharmacol. 2020, 98, 392–400. [Google Scholar] [CrossRef]
- Kaminski, D.L.; Ruwart, M.J.; Jellinek, M. Structure-function relationships of peptide fragments of gastrin and cholecystokinin. Am. J. Physiol. 1977, 233, E286–E292. [Google Scholar] [CrossRef] [PubMed]
- Morrison, K.L.; Weiss, G.A. Combinatorial Alanine-Scanning. Curr. Opin. Chem. Biol. 2001, 5, 302–307. [Google Scholar] [CrossRef]
- Manning, M.; Misicka, A.; Olma, A.; Bankowski, K.; Stoev, S.; Chini, B.; Durroux, T.; Mouillac, B.; Corbani, M.; Guillon, G. Oxytocin and vasopressin agonists and antagonists as research tools and potential therapeutics. J. Neuroendocrinol. 2012, 24, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.J.; Perez, R.A.; Perez, A. The Potential of Computer Modeling as a Tool to Investigate PPI: From Drug Design to Tissue Engineering. Front. Biomol. Sci. 2021, 8, 681617. [Google Scholar]
- Venkatraman, J.; Shankaramma, S.C.; Balaram, P. Design of folded peptides. Chem. Rev. 2001, 101, 3131–3152. [Google Scholar] [CrossRef]
- Balaram, P. Non-standard amino acids in peptide design and protein engineering. Curr. Opin. Struct. Biol. 1992, 2, 845–851. [Google Scholar] [CrossRef]
- Perdih, A.; Kikelj, D. The application of Freidinger lactams and their analogs in the design of conformationally constrained peptidomimetics. Curr. Med. Chem. 2006, 13, 1525–1556. [Google Scholar] [CrossRef]
- Yocum, R.R.; Rasmussen, J.R.; Strominger, J.L. The mechanism of action of penicillin. Penicillin acylates the active site of Bacillus stearothermophilus D-alanine carboxypeptidase. J. Biol. Chem. 1980, 25, 3977–3986. [Google Scholar] [CrossRef]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef]
- Müller, G.; Gurrath, M.; Kessler, H. Pharmacophore refinement of gpIIb/IIIa antagonists based on comparative studies of antiadhesive cyclic and acyclic RGD peptides. J. Comp. Aided Mol. Design. 1994, 8, 709–730. [Google Scholar] [CrossRef]
- Goodman, S.L.; Hölzemann, G.; Sulyok, G.A.G.; Kessler, H. Nanomolar Small Molecule Inhibitors for ανβ6, ανβ5, and ανβ3 Integrins. J. Med. Chem. 2002, 45, 1045–1051. [Google Scholar] [CrossRef]
- Dina, R.; Jafari, M. Angiotensin II-receptor antagonists: An overview. Am. J. Health Syst. Pharm. 2000, 57, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- de Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International Union of Pharmacology. XXIII. The Angiotensin II Receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar]
- Goodfriend, T.L. Angiotensin receptors: History and mysteries. Am. J. Hypertens. 2000, 13, 442–449. [Google Scholar] [CrossRef]
- Türker, R.K.; Hall, M.M.; Yamamoto, M.; Sweet, C.S.; Bumpus, F.M. A new, long-lasting competitive inhibitor of angiotensin. Science 1972, 177, 1203–1205. [Google Scholar] [CrossRef]
- Johnson, J.A.; Davis, J.O. Angiotensin II: Important role in the maintenance of arterial blood pressure. Science 1973, 179, 906–907. [Google Scholar] [CrossRef]
- Anderson, G.H.; Streeten, D.H.; Dalakos, T.G. Pressor response to 1-Sar-8-Ala-angiotensin II (saralasin) in hypertensive subjects. Circ. Res. 1977, 40, 243–250. [Google Scholar] [CrossRef]
- Printz, M.P.; Nemethy, G.; Bleich, H. Proposed Models for Angiotensin II in Aqueous Solution and Conclusions about Receptor Topography. Nat. New Biol. 1972, 237, 135–140. [Google Scholar] [CrossRef]
- Matsoukas, J.M.; Bigam, G.; Zhou, N.; Moore, G.J.I. 1H-NMR studies of [Sar1]angiotensin II conformation by nuclear Overhauser effect spectroscopy in the rotating frame (ROESY): Clustering of the aromatic rings in dimethylsulfoxide. Peptides 1990, 11, 359–366. [Google Scholar] [CrossRef]
- Spear, K.L.; Brown, M.S.; Reinhard, E.J.; McMahon, E.G.; Olins, G.M.; Palomo, M.A.; Patton, D.R. Conformational Restriction of Angiotensin II: Cyclic Analogues Having High Potency. J. Med. Chem. 1990, 33, 1935–1940. [Google Scholar] [CrossRef]
- Plucinska, K.; Kataoka, T.; Yodo, M.; Cody, W.L.; He, J.X.; Humblet, C.; Lu, G.H.; Lunney, E.; Major, T.C.; Panek, R.L.; et al. Multiple Binding Modes for the Receptor-Bound Conformations of Cyclic AII Agonists. J. Med. Chem. 1998, 36, 1902–1913. [Google Scholar] [CrossRef]
- Nikiforovich, G.V.; Marshall, G.R. 3-Dimensional recognition requirements for angiotensin agonists—A novel solution for an old problem. Biochem. Biophys. Res. Commun. 1993, 195, 222–228. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kishimoto, S.; Nishikawa, K. Hypotensive Imidazole-5-Acetic Acid Derivatives. U.S. Patent 4,355,040, 20 October 1982. [Google Scholar]
- Smeby, R.R.; Fermandjian, S. Conformation of Angiotensin II. In Chemistry and Biochemistry of Amino Acids, Peptides and Proteins, 5th ed.; Weinstein, B., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1978; p. 177. [Google Scholar]
- Duncia, J.V.; Chiu, A.T.; Carini, D.J.; Gregory, G.B.; Johnson, A.L.; Price, W.A.; Wells, G.J.; Wong, P.C.; Calabrese, J.C.; Timmermans, P.B.M.W. The Discovery of Potent Nonpeptide Angiotensin II Receptor Antagonists: A New Class of Potent Antihypertensives. J. Med. Chem. 1990, 33, 1312–1329. [Google Scholar] [CrossRef]
- Mais, D.E.; Bowling, N.L.; True, T.A.; Naka, M.; Morinelli, T.A.; Oatis, J.E., Jr.; Hamanaka, N.; Halushka, P.V. 1-(Carboxybenzyl)imidazole-S-acrylic Acids: Potent and Selective Angiotensin II Receptor Antagonists. J. Med. Chem. 1991, 34, 1514–1517. [Google Scholar]
- Aulakh, G.K.; Sodhi, R.K.; Singh, M. An update on non-peptide angiotensin receptor antagonists and related RAAS modulators. Life Sci. 2007, 81, 615–639. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.C.; Foster, C.; Brunner, H.R.; Liu, L. A Systematic Comparison of the Properties of Clinically Used Angiotensin II Type 1 Receptor Antagonists. Pharmacol. Rev. 2013, 65, 809–848. [Google Scholar] [CrossRef]
- Naik, P.; Murumkar, P.; Giridhar, R.; Yadav, M.R. Angiotensin II receptor type 1 (AT1) selective nonpeptidic antagonists-A perspective. Bioorg. Med. Chem. 2010, 18, 8418–8456. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of The Angiotensin Receptor Revealed by Serial Femtosecond Crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Desnoyer, R.; Han, G.W.; Patel, N.; Katritch, V.; Karnik, S.S.; Cherezov, V.; Stevens, R.C. Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J. Biol. Chem. 2015, 290, 29127–29139. [Google Scholar] [CrossRef]
- Wingler, L.M.; Skiba, M.A.; McMahon, C.; Staus, D.P.; Kleinhenz, A.L.W.; Suomivuori, C.M.; Latorraca, N.R.; Dror, R.O.; Lefkowitz, R.J.; Kruse, A.C. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 2020, 367, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.D.; Karnik, S.S. Angiotensin Type 1 Receptor Blockers in Heart Failure. Curr. Drug Targets 2020, 21, 125–131. [Google Scholar] [CrossRef]
- Violin, J.D.; DeWire, S.M.; Yamashita, D.; Rominger, D.H.; Nguyen, L.; Schiller, K.; Whalen, E.J.; Gowen, M.; Lark, M.W. Selectively Engaging β-Arrestins at the Angiotensin II Type 1 Receptor Reduces Blood Pressure and Increases Cardiac Performance. J. Pharmacol. Exp. Ther. 2010, 335, 572–579. [Google Scholar] [CrossRef]
- Wingler, L.M.; Elgeti, M.; Hilger, D.; Latorraca, N.R.; Lerch, M.T.; Staus, D.P.; Dror, R.O.; Kobilka, B.K.; Hubbell, W.L.; Lefkowitz, R.J. Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell 2019, 176, 468–478. [Google Scholar] [CrossRef]
- Zhang, H.; Luginina, A.; Mishin, A.; Baidya, M.; Shukla, A.K.; Cherezov, V. Structural insights into ligand recognition and activation of angiotensin receptors. Trends Pharmacol. Sci. 2021, 10. [Google Scholar] [CrossRef]
- Linz, W.; Wiemer, G.; Gohlke, P.; Unger, T.E.; Scholkens, B.A. Contribution of kinins to the cardiovascular actions of angiotensin converting enzyme inhibitors. Pharmacol. Rev. 1995, 47, 25–49. [Google Scholar]
- Leeb-Lundberg, L.M.F.; Marceau, F.; Muller-Esterl, W.; Pettibone, D.J.; Zuraw, B.L. International Union of Pharmacology. XLV. Classification of the Kinin Receptor Family: From Molecular Mechanisms to Pathophysiological Consequences. Pharmacol. Rev. 2005, 57, 27–77. [Google Scholar] [CrossRef]
- Marceau, F.; Bachelard, H.; Bouthillier, J.; Fortin, J.-P.; Morissette, J.-P.G.; Bawolak, M.-T.; Charest-Morin, X.; Gera, L. Bradykinin receptors: Agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. Int. Immunopharmacol. 2020, 82, 106305. [Google Scholar] [CrossRef]
- McCarthy, C.G.; Wilczynski, S.; Wenceslau, C.F.; Webb, R.C. A new storm on the horizon in COVID-19: Bradykinin-induced vascular complications. Vascul. Pharmacol. 2021, 137, 106826. [Google Scholar] [CrossRef]
- Blaes, N.; Girolami, J.-P. Targeting the ‘Janus face’ of the B2-bradykinin receptor. Expert Opin. Ther. Targets 2013, 17, 1145–1166. [Google Scholar] [CrossRef]
- Vavrek, R.J.; Stewart, J.M. Competitive Antagonists of Bradykinin. Peptides 1985, 6, 161–164. [Google Scholar] [CrossRef]
- Stewart, J.M. Bradykinin antagonists: Discovery and development. Peptides 2004, 25, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Kyle, D.J.; Blake, P.R.; Smithwick, D.; Green, L.M.; Summers, M.F.; Martin, J.A.; Sinsko, J.A. NMR and computational evidence that high-affinity bradykinin receptor antagonists adopt C-terminal β-turns. J. Med. Chem. 1993, 36, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Kyle, D.J.; Burch, R.M. Recent advances toward novel bradykinin antagonists. Drugs Future 1992, 17, 305–312. [Google Scholar]
- Hock, F.J.; Wirth, K.; Albus, U.; Linz, W.; Gerhards, G.H.; Wiemer, G.; Henke, S.; Breipohl, G.; Konig, W.; Knolle, J.; et al. HOE 140 a New Potent and Long-Acting Bradykinin-Antagonist: In vitro Studies. Br. J. Pharmacol. 1991, 102, 769–773. [Google Scholar] [CrossRef]
- Sawutz, D.G.; Salvino, J.M.; Seoane, P.R.; Douty, B.D.; Houck, W.T.; Bobko, M.A.; Doleman, M.S.; Dolle, R.E.; Wolfe, H.R. Synthesis, characterization, and conformational analysis of the D/L-Tic7 stereoisomers of the bradykinin receptor antagonist D-Arg0[Hyp3,Thi5,D-Tic7,Oic8]bradykinin. Biochemistry 1994, 33, 2373–2379. [Google Scholar] [CrossRef] [PubMed]
- Guba, W.; Haessner, R.; Breipohl, G.; Henke, S.; Knolle, J.; Santagada, V.; Kessler, H. Combined Approach of NMR and Molecular Dynamics within a Biphasic Membrane Mimetic: Conformation and Orientation of the Bradykinin Antagonist Hoe 140. J. Am. Chem. Soc. 1994, 116, 7532–7540. [Google Scholar] [CrossRef]
- Filizola, M.; Centeno, N.B.; Carteni-Farina, M.; Perez, J.J. Conformational analysis of the highly potent bradykinin antagonist HOE-140 by means of two different computational methods. J. Biomol. Struct. Dyn. 1998, 4, 639–652. [Google Scholar] [CrossRef]
- Lopez, J.J.; Shukla, A.K.; Reinhart, C.; Schwalbe, H.; Michel, H.; Glaubitz, C. The Structure of the Neuropeptide Bradykinin Bound to the Human G-Protein Coupled Receptor Bradykinin B2 as Determined by Solid-State NMR Spectroscopy. Angew. Chem. Int. Ed. 2008, 47, 1668–1671. [Google Scholar] [CrossRef]
- Sejbal, J.; Cann, J.R.; Stewart, J.M.; Gera, L.; Kotovych, G. An NMR, CD, molecular dynamics, and fluorometric study of the conformation of the bradykinin antagonist B-9340 in water and in aqueous micellar solutions. J. Med. Chem. 1996, 39, 1281–1292. [Google Scholar] [CrossRef]
- Thurieau, C.; Feleton, M.; Hennig, P.; Raimbaud, E.; Canet, E.; Fauchere, J.-L. Design and Synthesis of New Linear and Cyclic Bradykinin Antagonists. J. Med. Chem. 1996, 39, 2095–2101. [Google Scholar] [CrossRef]
- Monteagudo, E.S.; Calvani, F.; Catrambone, F.; Fincham, C.I.; Madami, A.; Meini, S.; Terracciano, R. New conformationally homogeneous beta-turn antagonists of the human B-2 kinin receptor. J. Pept. Sci. 2001, 7, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Kyle, D.J.; Chakravarty, S.; Sinsko, J.A.; Stormann, T.M. A Proposed Model of Bradykinin Bound to the Rat B2 Receptor and its Utility for Drug Design. J. Med. Chem. 1994, 37, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Meini, S.; Quartara, L.; Rizzi, A.; Patacchini, R.; Cucchi, P.; Giolitti, A.; Calo, G.; Regoli, D.; Criscuoli, M.; Maggi, C.A. MEN 11270 a novel selective constrained peptide antagonist with high affinity at the human B2 kinin receptor. J. Pharmacol. Exp. Ther. 1999, 289, 1250–1256. [Google Scholar] [PubMed]
- Bork, K.; Yasothan, U.; Kirkpatrick, P. Icatibant. Nat. Rev. Drug Discov. 2008, 7, 801–802. [Google Scholar] [CrossRef]
- Dziadulewicz, E.D. Non-peptide ligands for bradykinin receptors 1995–2004. Expert Opin. Ther. Patents 2005, 15, 829–859. [Google Scholar] [CrossRef]
- Heitsch, H. Non-peptide antagonists and agonists of the bradykinin B2 receptor. Curr. Med. Chem. 2002, 9, 913–928. [Google Scholar] [CrossRef]
- Sawutz, D.G.; Salvino, J.M.; Dolle, R.E.; Casiano, F.; Ward, S.J.; Houck, W.T.; Faunce, D.M.; Douty, B.D.; Baizman, E.; Awad, M.M.A.; et al. The Nonpeptide WIN 64338 is a Bradykinin B2 Receptor Antagonist. Proc. Natl. Acad. Sci. USA 1994, 91, 4693–4697. [Google Scholar] [CrossRef] [PubMed]
- Charkravarty, S.; Mavunkel, B.J.; Gochung, R.R.; Kyle, D.J. Novel bradykinin receptor antagonists from a structurally directed non-peptide combinatorial library. Immunopharmacology 1996, 33, 61–67. [Google Scholar] [CrossRef]
- Asano, M.; Inamura, N.; Hatori, C.; Sawai, H.; Fujiwara, T.; Katayama, A.; Kayakiri, H.; Satoh, S.; Abe, Y.; Inoue, T.; et al. The identification of an orally active nonpeptide bradykinin B2 receptor antagonist FR173657. Br. J. Pharmacol. 1997, 120, 617–624. [Google Scholar] [CrossRef]
- Abe, Y.; Kayakiri, H.; Satoh, S.; Inoue, T.; Sawada, Y.; Imai, K.; Inamura, N.; Asano, M.; Hatori, C.; Katayama, A.; et al. A novel class of orally active non-peptide bradykinin B2 receptor antagonists. 1. Construction of the basic Framework. J. Med. Chem. 1998, 41, 564–578. [Google Scholar] [CrossRef]
- Dziadulewicz, E.K.; Ritchie, T.J.; Hallett, A.; Snell, C.R.; Ko, S.Y.; Wrigglesworth, R.; Hughes, G.A.; Dunstan, A.R.; Bloomfield, G.C.; Drake, G.S.; et al. 1-(2-Nitrophenyl)thiosemicarbazides: A Novel Class of Potent, Orally Active Non-Peptide Antagonist for the Bradykinin B2 Receptor. J. Med. Chem. 2000, 43, 769–771. [Google Scholar] [CrossRef]
- Burgess, G.M.; Perkins, M.N.; Rang, H.P.; Campbell, E.A.; Brown, M.C.; McIntyre, P.; Urban, L.; Dziadulewicz, E.K.; Ritchie, T.J.; Hallett, A.; et al. Bradyzide a potent non-peptide B2 bradykinin receptor antagonist with long-lasting oral activity in animal models of inflammatory hyperalgesia. Br. J. Pharmacol. 2000, 129, 77–86. [Google Scholar] [CrossRef]
- Pruneau, D.; Paquet, J.L.; Luccarini, J.M.; Defrene, E.; Fouchet, C.; Franck, R.M.; Loillier, B.; Robert, C.; Belichard, P.; Duclos, H.; et al. Pharmacological profile of LF 16-0687 a new potent non-peptide bradykinin B2 receptor antagonist. Immunopharmacology 1999, 43, 187–194. [Google Scholar] [CrossRef]
- Cucchi, P.; Meini, S.; Bressan, A.; Catalani, C.; Bellucci, F.; Santicioli, P.; Lecci, A.; Faiella, A.; Rotondaro, L.; Giuliani, S.; et al. MEN16132 a novel potent and selective nonpeptide antagonist for the human bradykinin B2 receptor. In vitro pharmacology and molecular characterization. Eur. J. Pharmacol. 2005, 528, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Shakur, H.; Andrews, P.; Asser, T.; Balica, L.; Boeriu, C.; Quintero, J.D.; Dewan, Y.; Druwé, P.; Fletcher, O.; Frost, C.; et al. The BRAIN TRIAL: A randomised, placebo controlled trial of a Bradykinin B2 receptor antagonist (Anatibant) in patients with traumatic brain injury. Trials 2009, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Tenti, S.; Pascarelli, N.A.; Cheleschi, S.; Guidelli, G.M.; Fioravanti, A. The emerging role of bradykinin in the pathogenesis of osteoarthritis and its possible clinical implications. Curr. Rheumatol. Rev. 2016, 12, 177–184. [Google Scholar] [CrossRef]
- Lupala, C.S.; Gomez-Gutierrez, P.; Perez, J.J. New insights into the stereochemical requirements of the bradykinin B2 receptor antagonist binding. J. Comp. Aided Mol. Design 2016, 30, 85–101. [Google Scholar] [CrossRef]
- Lupala, C.S.; Gomez-Gutierrez, P.; Perez, J.J. New insights into the stereochemical requirements of the bradykinin B1 receptor antagonist binding. J. Mol. Graphics Model. 2016, 68, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Rasaeifar, B.; Lupala, C.S.; Gomez-Gutierrez, P.; Perez, J.J. Molecular Features Characterizing Non-peptide B1 and B2 Bradykinin Receptor Selectivity. Bioorg. Med. Chem. Lett. 2019, 29, 11–14. [Google Scholar] [CrossRef]
- Gibson, C.; Schnatbaum, K.; Pfeifer, J.R.; Locardi, E.; Paschke, M.; Reimer, U.; Richter, U.; Scharn, D.; Faussner, A.; Tradler, T. Novel Small Molecule Bradykinin B2 Receptor Antagonists. J. Med. Chem. 2009, 52, 4370–4379. [Google Scholar] [CrossRef] [PubMed]
- Lesage, A.; Gibson, C.; Marceau, F.; Ambrosi, H.D.; Saupe, J.; Katzer, W.; Loenders, B.; Charest-Morin, X.; Knolle, J. In Vitro Pharmacological Profile of a New Small Molecule Bradykinin B2 Receptor Antagonist. Front. Pharmacol. 2020, 11, 916. [Google Scholar] [CrossRef]
- Rasaeifar, B.; Gomez-Gutierrez, P.; Perez, J.J. Molecular features of non-selective small molecule antagonists of the Bradykinin Receptors. Pharmaceuticals 2020, 13, 259. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gutierrez, P.; Perez, J.J. Discovery of a Bradykinin B2 Partial Agonist Profile of Raloxifene in a Drug Repurposing Campaign. Int. J. Mol. Sci. 2021, 22, 257. [Google Scholar] [CrossRef] [PubMed]
- I-SPY COVID TRIAL: An Adaptive Platform Trial to Reduce Mortality and Ventilator Requirements for Critically Ill Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT04488081?term=icatibant&draw=3&rank=19 (accessed on 30 April 2021).
- Multicenter, Adaptive, Randomized, Placebo-Controlled, Double Blind, Parallel-Group Phase 2/3 Trial, to Study Efficacy and Safety of Two Doses of Raloxifene in Adult Paucisymptomatic COVID-19 Patients. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2020-003936-25 (accessed on 30 April 2021).
- Cushman, D.W.; Cheung, H.S.; Sabo, E.F.; Ondetti, M.A. Design of potent competitive inhibitors of angiotensin-converting enzyme. Carboxyalkanoyl and mercaptoalkanoyl amino acids. Biochemistry 1977, 16, 5484–5491. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.A.; Martin, J.A.; Kinchington, D.; Broadhurst, A.V.; Craig, J.C.; Duncan, I.B.; Galpin, S.A.; Handa, B.K.; Kay, J.; Kröhn, A.; et al. Rational design of peptide-based HIV proteinase inhibitors. Science 1990, 248, 358–361. [Google Scholar] [CrossRef]
- Ciancetta, A.; Jacobson, K.A. Breakthrough in GPCR Crystallography and Its Impact on Computer-Aided Drug Design. Methods Mol. Biol. 2018, 1705, 45–72. [Google Scholar] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez, J.J. Exploiting Knowledge on Structure–Activity Relationships for Designing Peptidomimetics of Endogenous Peptides. Biomedicines 2021, 9, 651. https://doi.org/10.3390/biomedicines9060651
Perez JJ. Exploiting Knowledge on Structure–Activity Relationships for Designing Peptidomimetics of Endogenous Peptides. Biomedicines. 2021; 9(6):651. https://doi.org/10.3390/biomedicines9060651
Chicago/Turabian StylePerez, Juan J. 2021. "Exploiting Knowledge on Structure–Activity Relationships for Designing Peptidomimetics of Endogenous Peptides" Biomedicines 9, no. 6: 651. https://doi.org/10.3390/biomedicines9060651
APA StylePerez, J. J. (2021). Exploiting Knowledge on Structure–Activity Relationships for Designing Peptidomimetics of Endogenous Peptides. Biomedicines, 9(6), 651. https://doi.org/10.3390/biomedicines9060651