
Multifunctional Enkephalin Analogs with a New Biological Profile: MOR/DOR Agonism and KOR Antagonism

 ,
,  , ,
, ,  ,
, 
Abstract
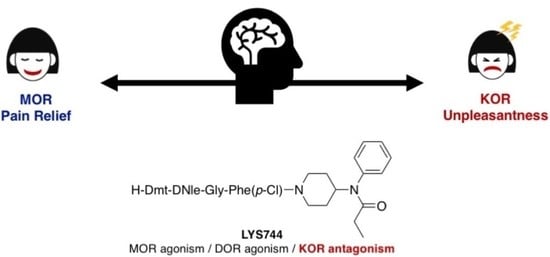
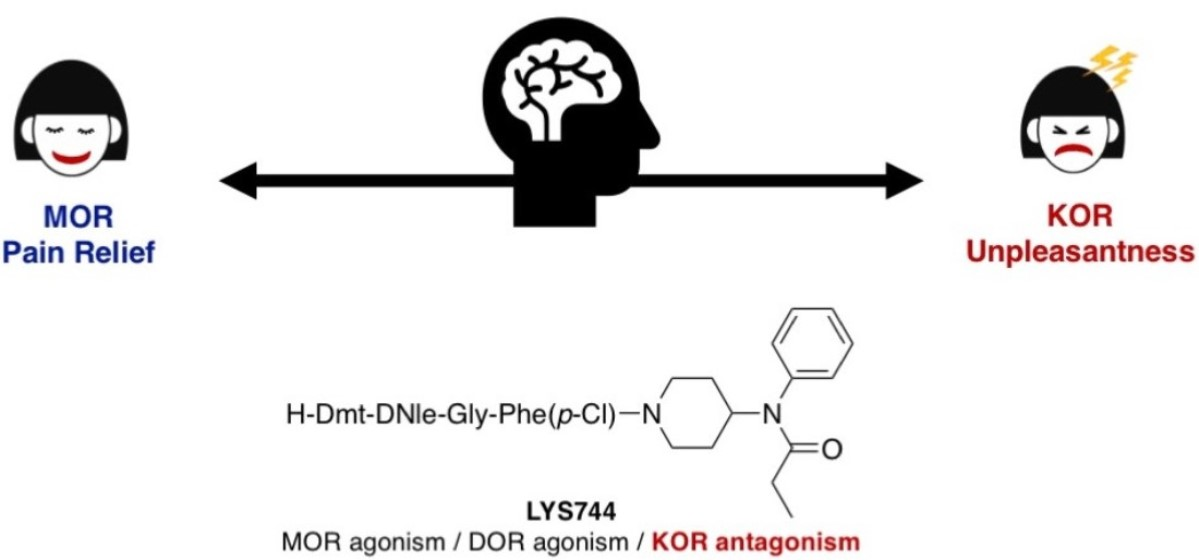
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Radioligand Labeled Binding Assays at the KOR
2.3. [35S]GTPγS Assays
2.4. TAM Experiments
2.5. Plasma Stability Tests
3. Results and Discussion
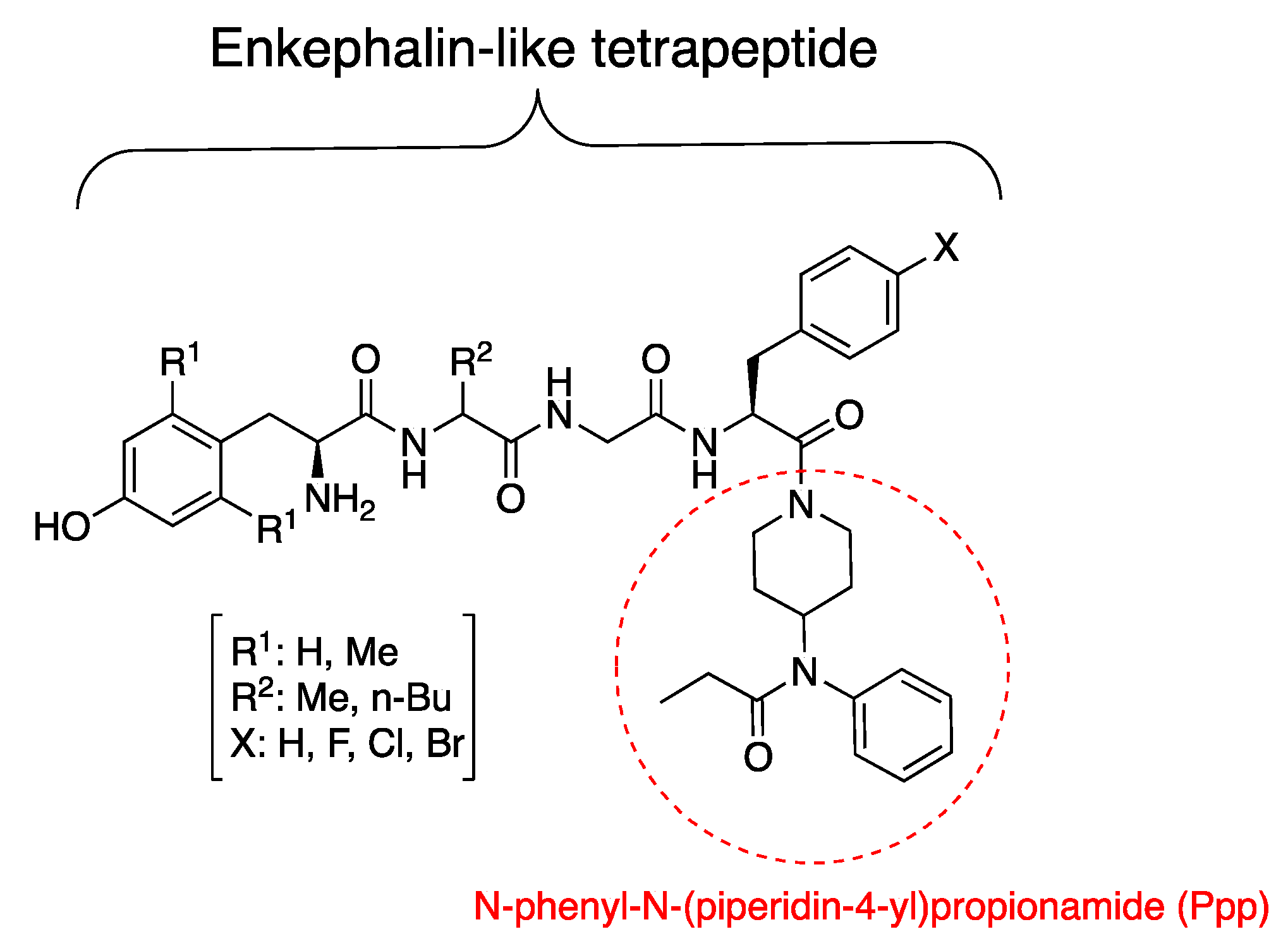
3.1. Synthesis
3.2. Binding Affinities
3.3. Functional Activity: [35S]GTPγS Assay
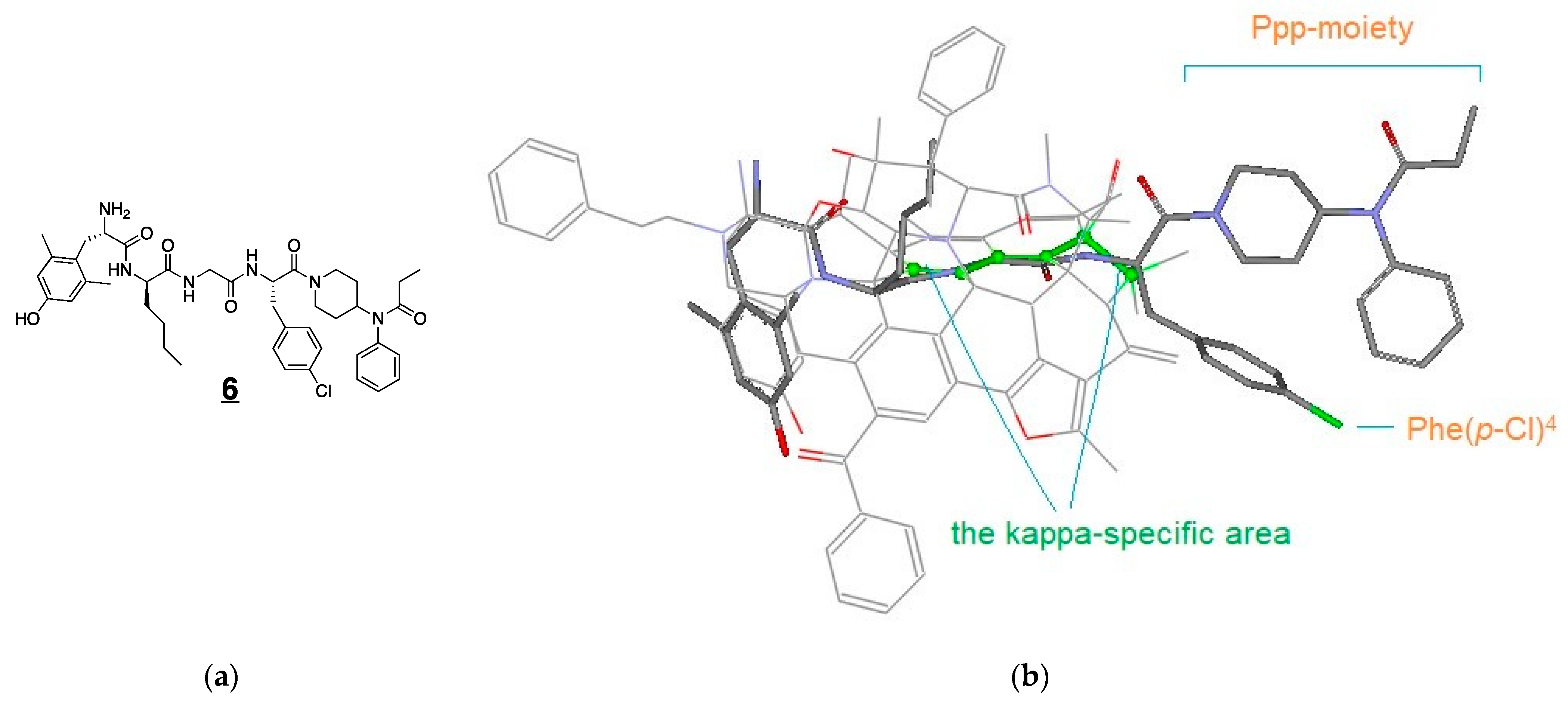
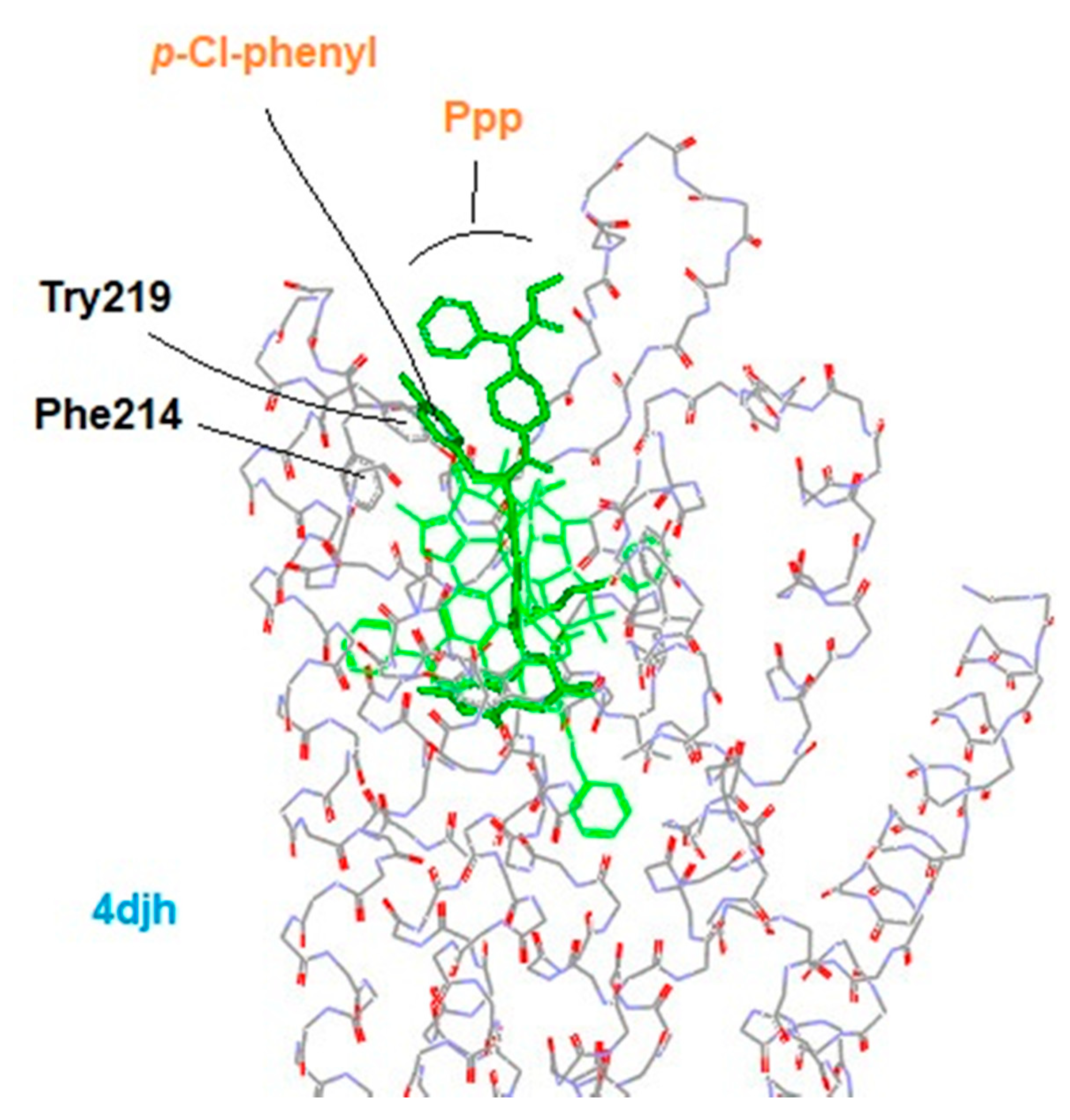
3.4. Modeling Experiments: TAM Approach
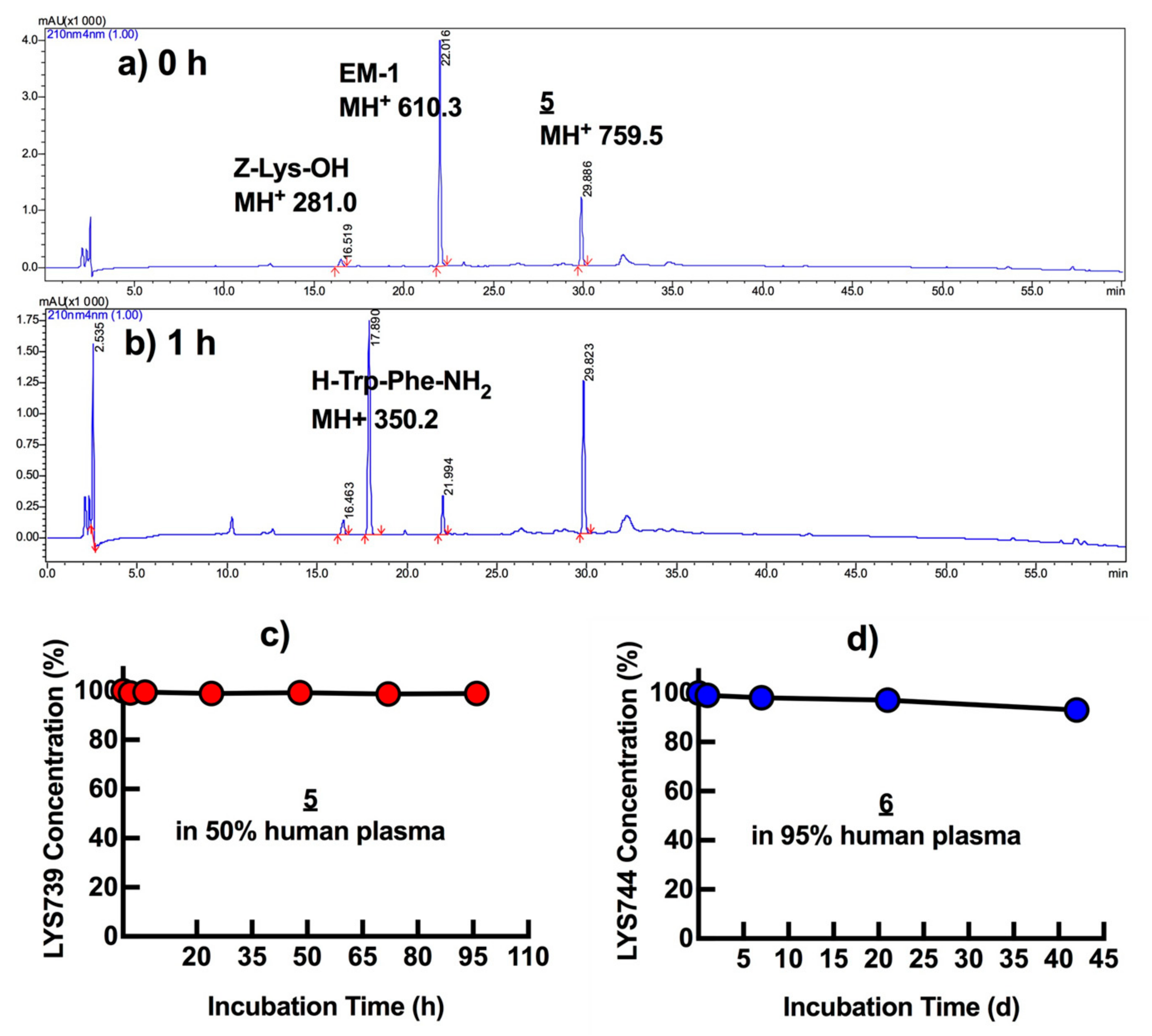
3.5. In Vitro Plasma Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anand, J.P.; Montgomery, D. Multifunctional opioid ligands. Handb. Exp. Pharmacol. 2018, 247, 21–51. [Google Scholar]
- Morphy, R.; Kay, C.; Rankovic, Z. From magic bullets to designed multiple ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designing multiple ligands-medicinal chemistry strategies and challenges. Curr. Pharm. Des. 2009, 15, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Dietis, N.; Guerrini, R.; Calo, G.; Salvadori, S.; Rowbotham, D.J.; Lambert, D.G. Simultaneous targeting of multiple opioid receptors: A strategy to improve side-effect profile. Br. J. Anaesth. 2009, 103, 38–49. [Google Scholar] [CrossRef]
- Balboni, G.; Onnis, V.; Congiu, C.; Zotti, M.; Sasaki, Y.; Ambo, A.; Bryant, S.D.; Jinsmaa, Y.; Lazarus, L.H.; Lazzari, I.; et al. Further studies on the effect of lysine at the C-terminus of the Dmt-Tic opioid pharmacophore. Bioorg. Med. Chem. 2007, 15, 3143–3151. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Balboni, G.; Guerrini, R.; Salvadori, S.; Bianchi, C.; Rizzi, D.; Bryant, S.D.; Lazarus, L.H. Evaluation of the Dmt-Tic pharmacophore: Conversion of a potent delta-opioid receptor antagonist into a potent delta agonist and ligands with mixed properties. J. Med. Chem. 2002, 45, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cook, C.; Thai, E.; Pickens, S.; Taylor, A.M.; Tea, V.D.; Carroll, I.; Leslie, F.M.; Evans, C.J.; Cahill, C.M. Neuropathic pain alters reward and affect via kappa opioid receptor (KOR) upregulation. FASEB J. 2016, 30 Supplement 928.5. [Google Scholar]
- Bruchas, M.R.; Land, B.B.; Chavkin, C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010, 1314, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Van’t Veer, A.; Carlezon, W.A. Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology 2013, 229, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Kishimoto, Y.; Ise, Y.; Yajima, Y.; Misawa, K.; Suzuki, T. Direct evidence for the involvement of the mesolimbic kappa-opioid system in the morphine-induced rewarding effect under an inflammatory pain-like state. Neuropsychopharmacology 2005, 30, 111–118. [Google Scholar] [CrossRef]
- Rasmussen, K.; White, D.A.; Acri, J.B. NIDA’s medication development priorities in response to the opioid crisis: Ten most wanted. Neuropsychopharmacology 2019, 44, 657–659. [Google Scholar] [CrossRef]
- Emrich, H.M.; Vogt, P.; Herz, A. Possible antidepressive effects of opioids: Action of buprenorphine. Ann. N. Y. Acad. Sci. 1982, 398, 108–112. [Google Scholar] [CrossRef]
- Falcon, E.; Maier, K.; Robinson, S.A.; Hill-Smith, T.E.; Lucki, I.J.P. Effects of buprenorphine on behavioral tests for antidepressant and anxiolytic drugs in mice. Psychopharmacology 2015, 232, 907–915. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kulkarani, V.; Cowell, S.M.; Ma, S.W.; Davis, P.; Hanlon, K.E.; Vanderah, T.W.; Lai, J.; Porreca, F.; Vardanyan, R.; et al. Development of potent mu and delta opioid agonists with high lipophilicity. J. Med. Chem. 2011, 54, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Petrov, R.; Park, C.K.; Ma, S.W.; Davis, P.; Lai, J.; Porreca, F.; Vardanyan, R.; Hruby, V.J. Development of novel enkephalin analogues that have enhanced opioid activities at both mu and delta opioid receptors. J. Med. Chem. 2007, 50, 5528–5532. [Google Scholar] [CrossRef] [PubMed]
- Brownson, E.A.; Abbruscato, T.J.; Gillespie, T.J.; Hruby, V.J.; Davis, T.P. Effect of peptidases at the blood brain barrier on the permeability of enkephalin. J. Pharmacol. Exp. Ther. 1994, 270, 675–680. [Google Scholar]
- Altman, R.A.; Sharma, K.K.; Rajewski, L.G.; Toren, P.C.; Baltezor, M.J.; Pal, M.; Karad, S.N. Tyr1-ψ [(Z) CF═ CH]-Gly2 fluorinated peptidomimetic improves distribution and metabolism properties of leu-enkephalin. ACS Chem. Neurosci. 2018, 9, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Habgood, M.D.; Begley, D.J.; Abbott, N.J. Determinants of passive drug entry into the central nervous system. Cell Mol. Neurobiol. 2000, 20, 231–253. [Google Scholar] [CrossRef]
- Gentry, C.L.; Egleton, R.D.; Gillespie, T.; Abbruscato, T.J.; Bechowski, H.B.; Hruby, V.J.; Davis, T.P. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides 1999, 20, 1229–1238. [Google Scholar] [CrossRef]
- Schiller, P.W.; Nguyen, T.M.; Berezowska, I.; Dupuis, S.; Weltrowska, G.; Chung, N.N.; Lemieux, C. Synthesis and in vitro opioid activity profiles of DALDA analogues. Eur. J. Med. Chem. 2000, 35, 895–901. [Google Scholar] [CrossRef]
- Wu, Z.; Hruby, V.J. Toward a universal μ-agonist template for template-based alignment modeling of opioid ligands. ACS Omega 2019, 4, 17457–17476. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S. Gram-scale preparation of c-terminal-modified enkephalin analogues by typical liquid-phase peptide synthesis. Curr. Protoc. Protein Sci. 2019, 98, e97. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Colon, C.N.; Lee, Y.S.; Remesic, M.; Hall, S.M.; LaVigne, J.; Davis, P.; Sandweiss, A.J.; McIntosh, M.I.; Hanson, J.; Largent-Milnes, T.M.; et al. Structure-activity relationships of [des-Arg(7)]Dynorphin a analogues at the kappa opioid receptor. J. Med. Chem. 2016, 59, 10291–10298. [Google Scholar] [CrossRef]
- Lai, J.; Ma, S.W.; Zhu, R.H.; Rothman, R.B.; Lentes, K.U.; Porreca, F. Pharmacological characterization of the cloned kappa opioid receptor as a kappa 1b subtype. Neuroreport 1994, 5, 2161–2164. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.L.; Streicher, J.M.; Groer, C.E.; Munro, T.A.; Zhou, L.; Bohn, L.M. Functional selectivity of 6’-guanidinonaltrindole (6’-GNTI) at kappa-opioid receptors in striatal neurons. J. Biol. Chem. 2013, 288, 22387–22398. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA Discovery Studio Visualizer. Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 20 April 2020).
- National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2018-00023-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA. Available online: https://pdsp.unc.edu/pdspweb/ (accessed on 25 May 2020).
- Kroeze, W.K.; Sassano, M.F.; Huang, X.P.; Lansu, K.; McCorvy, J.D.; Giguere, P.M.; Sciaky, N.; Roth, B.L. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 2015, 22, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Besnard, J.; Ruda, G.F.; Setola, V.; Abecassis, K.; Rodriguiz, R.M.; Huang, X.P.; Norval, S.; Sassano, M.F.; Shin, A.I.; Webster, L.A.; et al. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Rosa, M.; Caltabiano, G.; Barreto-Valer, K.; Gonzalez-Nunez, V.; Gomez-Tamayo, J.C.; Arda, A.; Jimenez-Barbero, J.; Pardo, L.; Rodriguez, R.E.; Arsequell, G.; et al. Modulation of the interaction between a peptide ligand and a G protein-coupled receptor by halogen atoms. ACS Med. Chem. Lett. 2015, 6, 872–876. [Google Scholar] [CrossRef]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The sigma-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Spahn, V.; Del Vecchio, G.; Labuz, D.; Rodriguez-Gaztelumendi, A.; Massaly, N.; Temp, J.; Durmaz, V.; Sabri, P.; Reidelbach, M.; Machelska, H. A nontoxic pain killer designed by modeling of pathological receptor conformations. Science 2017, 355, 966–969. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Knapp, S.; Hruby, V. Template-based alignment modeling: An innovative ligand-based approach for medicinal chemists. Med. Chem. Res. 2020, 29, 1160–1167. [Google Scholar] [CrossRef]
- Wu, Z.; Hruby, V.J. Backbone alignment modeling of the structure-activity relationships of opioid ligands. J. Chem. Inf. Model. 2011, 51, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.C.; Chao, C.C.; Takemori, A.E.; Gekker, G.; Hu, S.; Peterson, P.K.; Portoghese, P.S. Arylacetamide-derived fluorescent probes: Synthesis, biological evaluation, and direct fluorescent labeling of kappa opioid receptors in mouse microglial cells. J. Med. Chem. 1996, 39, 1729–1735. [Google Scholar] [CrossRef]
- Makwana, K.M.; Mahalakshmi, R. Implications of aromatic–aromatic interactions: From protein structures to peptide models. Prot. Sci. 2015, 24, 1920–1933. [Google Scholar] [CrossRef]
- Wheeler, S.E. Understanding substituent effects in noncovalent interactions involving aromatic rings. Acc. Chem. Res. 2013, 46, 1029–1038. [Google Scholar] [CrossRef]
- Aldrich, J.V.; McLaughlin, J.P. Peptide kappa opioid receptor ligands: Potential for drug development. AAPS J. 2009, 11, 312–322. [Google Scholar] [CrossRef]
- Orosz, G.; Ronai, A.Z.; Bajusz, S.; Medzihradszky, K. N-terminally protected penta- and tetrapeptide opioid antagonists based on a pentapeptide sequence found in the venom of Philippine cobra. Biochem. Biophys. Res. Commun. 1994, 202, 1285–1290. [Google Scholar] [CrossRef]
- Lemaire, S.; Turcotte, A. Synthesis and biological activity of analogs of dynorphin-A(1-13) substituted in positions 2 and 4: Design of [Ala2,Trp4]-Dyn-A(1-13) as a putative selective opioid antagonist. Can. J. Physiol. Pharmacol. 1986, 64, 673–678. [Google Scholar] [CrossRef]
- Werle, M.; Bernkop-Schnurch, A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. [Google Scholar] [CrossRef]
- Chung, T.D.Y.; Terry, D.B.; Smith, L.H. In vitro and in vivo assessment of ADME and PK properties during lead selection and lead optimization-guidelines, benchmarks and rules of thumb. In Assay Guidance Manual; Sittampalam, G.S., Coussens, N.P., Brimacombe, K., Grossman, A., Arkin, M., Auld, D., Austin, C., Baell, J., Bejcek, B., Caaveiro, J.M.M., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Janecka, A.; Staniszewska, R.; Gach, K.; Fichna, J. Enzymatic degradation of endomorphins. Peptides 2008, 29, 2066–2073. [Google Scholar] [CrossRef]
- Rashedul Islam, M.; Yang, L.; Lee, Y.S.; Hruby, V.J.; Karamyan, V.J.; Abbruscato, T.J. Enkephalin-fentanyl multifunctional opioids as potential neuroprotectants for ischemic stroke treatment. Curr. Pharm. Des. 2016, 22, 6459–6468. [Google Scholar] [CrossRef] [PubMed]
- Creative Bioarray. Available online: https://www.creative-bioarray.com/Services/mdck-permeability-assay.htm (accessed on 25 May 2021).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analog | Structure | hKOR 1 | rMOR 5 | hDOR 5 | |
|---|---|---|---|---|---|
| [3H]U69,593 2 | [3H]DAMGO | [3H]DPDPE | |||
| Ki (nM) 3 | −logIC50 4 | Ki (nM) | Ki (nM) | ||
| LYS729 (1) | Tyr-DAla-Gly-Phe-NH2 | 230 | 6.65 ± 0.13 | 2.8 | 300 |
| LYS436 (2) | Tyr-DAla-Gly-Phe-Ppp | 220 | 6.66 ± 0.10 | 23 | 0.69 |
| LYS540 (3) | Dmt-DAla-Gly-Phe-Ppp | 21 | 7.68 ± 0.24 | 0.38 | 0.36 |
| LYS644 (4) | Dmt-DNle-Gly-Phe-Ppp | 4.8 | 8.32 ± 0.12 | 0.39 | 0.18 |
| LYS739 (5) | Dmt-DNle-Gly-Phe(p-F)-Ppp | 0.89 | 10.5 ± 0.10 | 0.02 | 0.40 |
| LYS744 (6) | Dmt-DNle-Gly-Phe(p-Cl)-Ppp | 1.3 | 8.89 ± 0.15 | 0.10 | 0.08 |
| MR119 (7) | Dmt-DNle-Gly-Phe(p-Br)-Ppp | 7.4 | 8.13 ± 0.12 | 1.5 | 1.1 |
| LYS707 (8) | Dmt-DAla-Gly-Phe(p-Cl)-Ppp | 2.4 | 8.62 ± 0.07 | 0.14 | 0.14 |
| LYS745 (9) | Dmt-DTic-Gly-Phe(p-Cl)-Ppp | 3.8 | 8.42 ± 0.05 | 0.15 | 0.11 |
| Salvinorin A | 2.4 | 8.62 ± 0.10 | - | - | |
| Analog | KOR 1 | |||
|---|---|---|---|---|
| Agonist Mode | Antagonist Mode 2 | |||
| EC50 (nM) 3 | Emax (%) 4 | IC50 (nM) 5 | Imax (%) 6 | |
| 1 | - | <30 7 | - | <10 8 |
| 2 | - | <10 7 | - | <10 8 |
| 3 | 538 ± 75 | 39 | 521 ± 71 | 64 |
| 4 | 210 ± 51 | 41 | 386 ± 35 | 57 |
| 5 | 21.1 ± 7.2 | 39 | 59.8 ± 10.0 | 65 |
| 6 | - | <10 7 | 52.4 ± 1.2 | 122 |
| naloxone | - | - | 58.6 ± 4.4 | 100 |
| U50,488 | 13.8 ± 2.5 | 100 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.S.; Remesic, M.; Ramos-Colon, C.; Wu, Z.; LaVigne, J.; Molnar, G.; Tymecka, D.; Misicka, A.; Streicher, J.M.; Hruby, V.J.; et al. Multifunctional Enkephalin Analogs with a New Biological Profile: MOR/DOR Agonism and KOR Antagonism. Biomedicines 2021, 9, 625. https://doi.org/10.3390/biomedicines9060625
Lee YS, Remesic M, Ramos-Colon C, Wu Z, LaVigne J, Molnar G, Tymecka D, Misicka A, Streicher JM, Hruby VJ, et al. Multifunctional Enkephalin Analogs with a New Biological Profile: MOR/DOR Agonism and KOR Antagonism. Biomedicines. 2021; 9(6):625. https://doi.org/10.3390/biomedicines9060625
Chicago/Turabian StyleLee, Yeon Sun, Michael Remesic, Cyf Ramos-Colon, Zhijun Wu, Justin LaVigne, Gabriella Molnar, Dagmara Tymecka, Aleksandra Misicka, John M. Streicher, Victor J. Hruby, and et al. 2021. "Multifunctional Enkephalin Analogs with a New Biological Profile: MOR/DOR Agonism and KOR Antagonism" Biomedicines 9, no. 6: 625. https://doi.org/10.3390/biomedicines9060625
APA StyleLee, Y. S., Remesic, M., Ramos-Colon, C., Wu, Z., LaVigne, J., Molnar, G., Tymecka, D., Misicka, A., Streicher, J. M., Hruby, V. J., & Porreca, F. (2021). Multifunctional Enkephalin Analogs with a New Biological Profile: MOR/DOR Agonism and KOR Antagonism. Biomedicines, 9(6), 625. https://doi.org/10.3390/biomedicines9060625