
The Emerging Roles of Autophagy in Human Diseases


Abstract
:1. Introduction to Autophagy
2. Autophagy and Cancer
2.1. Autophagy and Tumor Suppression
2.2. Autophagy and Tumor Promotion
2.3. Autophagy and Tumor Metastasis
2.3.1. Autophagy and Cancer Cell Motility
2.3.2. Autophagy and Resistance to Anoikis
2.4. Autophagy and Cancer Stem Cells
2.5. Autophagy and Dormant Cancer Cells
2.6. Autophagy and Cancer Therapy
3. Autophagy and Neurodegenerative Diseases
3.1. Parkinson Disease
3.2. Alzheimer Disease
3.3. Huntington Disease
3.4. Autophagy and Therapy of Neurodegenerative Diseases
4. Autophagy and Infectious Diseases
4.1. Autophagy and Bacterial Infection
4.2. Autophagy and Viral Infection
4.3. Autophagy and COVID-19
5. Autophagy and Metabolic Disorders
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer disease |
| ATG | autophagy related |
| BAT | brown adipose tissue |
| CQ | chloroquine |
| CoV | coronavirus |
| CSCs | cancer stem cells |
| ECM | extracellular matrix |
| FA | focal adhesion |
| GWAS | genome-wide association studies |
| HCQ | hydroxychloroquine |
| HD | Huntington disease |
| LAMs | LIR-motif associated mutations |
| LIR | LC3-interacting region |
| MHC-I | major histocompability complex class Ι |
| mHTT | mutant HTT (huntingtin) |
| MSI | microsatellite instability |
| MTORC1 | MTOR complex 1 |
| OMM | outer mitochondrial membrane |
| PD | Parkinson disease |
| polyQ | polyglutamine |
| PtdIns3K | phosphatidylinositol 3-kinase |
| PtdIns3P | phosphatidylinositol-3-phosphate |
| ROS | reactive oxygen species |
| SCV | Salmonella-containing vacuole |
| T2DM | type 2 diabetes mellitus |
| WAT | white adipose tissue |
References
- Wong, P.M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef] [Green Version]
- Nascimbeni, A.C.; Codogno, P.; Morel, E. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. FEBS J. 2017, 284, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Graef, M.; Friedman, J.R.; Graham, C.; Babu, M.; Nunnari, J. ER exit sites are physical and functional core autophagosome biogenesis components. Mol. Biol. Cell. 2013, 24, 2918–2931. [Google Scholar] [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell. Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Puri, C.; Renna, M.; Bento, C.F.; Moreau, K.; Rubinsztein, D.C. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 2013, 154, 1285–1299. [Google Scholar] [CrossRef] [Green Version]
- Puri, C.; Vicinanza, M.; Ashkenazi, A.; Gratian, M.J.; Zhang, Q.; Bento, C.F.; Renna, M.; Menzies, F.M.; Rubinsztein, D.C. The RAB11A-Positive Compartment Is a Primary Platform for Autophagosome Assembly Mediated by WIPI2 Recognition of PI3P-RAB11A. Dev. Cell. 2018, 45, 114–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Popelka, H.; Lei, Y.; Yang, Y.; Klionsky, D.J. The Roles of Ubiquitin in Mediating Autophagy. Cells 2020, 9, 2025. [Google Scholar] [CrossRef]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell. 2014, 55, 238–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [Green Version]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell. Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Bakula, D.; Scheibye-Knudsen, M. MitophAging: Mitophagy in Aging and Disease. Front. Cell. Dev. Biol. 2020, 8, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, K.; Kondo-Okamoto, N.; Ohsumi, Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell. 2009, 17, 87–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanki, T.; Wang, K.; Cao, Y.; Baba, M.; Klionsky, D.J. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell. 2009, 17, 98–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo-Okamoto, N.; Noda, N.N.; Suzuki, S.W.; Nakatogawa, H.; Takahashi, I.; Matsunami, M.; Hashimoto, A.; Inagaki, F.; Ohsumi, Y.; Okamoto, K. Autophagy-related protein 32 acts as autophagic degron and directly initiates mitophagy. J. Biol. Chem. 2012, 287, 10631–10638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.J.; Klionsky, D.J. Yeast mitophagy: Unanswered questions. Biochim. Biophys. Acta. Gen. Subj. 2021, 1865, 129932. [Google Scholar] [CrossRef]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell. Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trempe, J.F.; Sauvé, V.; Grenier, K.; Seirafi, M.; Tang, M.Y.; Ménade, M.; Al-Abdul-Wahid, S.; Krett, J.; Wong, K.; Kozlov, G.; et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 2013, 340, 1451–1455. [Google Scholar] [CrossRef]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [Green Version]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell. Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, M.; Kujuro, Y.; Okatsu, K.; Koyano, F.; Kosako, H.; Kimura, M.; Suzuki, N.; Uchiyama, S.; Tanaka, K.; Matsuda, N. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J. Biol. Chem. 2013, 288, 22019–22032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell. 2015, 60, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. Bioessays 2018, 40, e1800008. [Google Scholar] [CrossRef] [PubMed]
- Schuck, S. Microautophagy - distinct molecular mechanisms handle cargoes of many sizes. J. Cell. Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Alfaro, I.E.; Albornoz, A.; Molina, A.; Moreno, J.; Cordero, K.; Criollo, A.; Budini, M. Chaperone Mediated Autophagy in the Crosstalk of Neurodegenerative Diseases and Metabolic Disorders. Front. Endocrinol. 2018, 9, 778. [Google Scholar] [CrossRef]
- Allen, E.A.; Baehrecke, E.H. Autophagy in animal development. Cell. Death. Differ. 2020, 27, 903–918. [Google Scholar] [CrossRef]
- Chen, X.; He, Y.; Lu, F. Autophagy in Stem Cell Biology: A Perspective on Stem Cell Self-Renewal and Differentiation. Stem Cells Int. 2018, 2018, 9131397. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Wang, Q.; Kao, Y.R.; Diaz, A.; Tasset, I.; Kaushik, S.; Thiruthuvanathan, V.; Zintiridou, A.; Nieves, E.; Dzieciatkowska, M.; et al. Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 2021, 591, 117–123. [Google Scholar] [CrossRef]
- Germic, N.; Frangez, Z.; Yousefi, S.; Simon, H.U. Regulation of the innate immune system by autophagy: Monocytes, macrophages, dendritic cells and antigen presentation. Cell. Death. Differ. 2019, 26, 715–727. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Ding, Z.B.; Shi, Y.H.; Zhou, J.; Qiu, S.J.; Xu, Y.; Dai, Z.; Shi, G.M.; Wang, X.Y.; Ke, A.W.; Wu, B.; et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008, 68, 9167–9175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.H.; Tang, F.; Xu, J.; Wu, X.; Yang, S.B.; Feng, Z.Y.; Ding, Y.G.; Wan, X.B.; Guan, Z.; Li, H.G.; et al. Low expression of Beclin 1, associated with high Bcl-xL, predicts a malignant phenotype and poor prognosis of gastric cancer. Autophagy 2012, 8, 389–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicotra, G.; Mercalli, F.; Peracchio, C.; Castino, R.; Follo, C.; Valente, G.; Isidoro, C. Autophagy-active beclin-1 correlates with favourable clinical outcome in non-Hodgkin lymphomas. Mod. Pathol. 2010, 23, 937–950. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef]
- Kim, M.S.; Jeong, E.G.; Ahn, C.H.; Kim, S.S.; Lee, S.H.; Yoo, N.J. Frameshift mutation of UVRAG, an autophagy-related gene, in gastric carcinomas with microsatellite instability. Hum. Pathol. 2008, 39, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zhao, Z.; Yang, Y.; O’Connell, D.; Zhang, X.; Oh, S.; Ma, B.; Lee, J.H.; Zhang, T.; Varghese, B.; et al. Truncating mutation in the autophagy gene UVRAG confers oncogenic properties and chemosensitivity in colorectal cancers. Nat. Commun. 2015, 6, 7839. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Fimia, G.M.; Di Bartolomeo, S.; Piacentini, M.; Cecconi, F. Unleashing the Ambra1-Beclin 1 complex from dynein chains: Ulk1 sets Ambra1 free to induce autophagy. Autophagy 2011, 7, 115–117. [Google Scholar] [CrossRef] [Green Version]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 706. [Google Scholar] [CrossRef] [Green Version]
- Maiani, E.; Milletti, G.; Nazio, F.; Holdgaard, S.G.; Bartkova, J.; Rizza, S.; Cianfanelli, V.; Lorente, M.; Simoneschi, D.; Di Marco, M.; et al. AMBRA1 regulates cyclin D to guard S-phase entry and genomic integrity. Nature 2021, 592, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Velho, S.; Fernandes, M.S.; Leite, M.; Figueiredo, C.; Seruca, R. Causes and consequences of microsatellite instability in gastric carcinogenesis. World J. Gastroenterol. 2014, 20, 16433–16442. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Kim, S.S.; Ahn, C.H.; Yoo, N.J.; Lee, S.H. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J. Pathol. 2009, 217, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Nishimura, T.; Sakamaki, Y.; Itakura, E.; Hatta, T.; Natsume, T.; Mizushima, N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell. 2014, 25, 1327–1337. [Google Scholar] [CrossRef]
- An, C.H.; Kim, Y.R.; Kim, H.S.; Kim, S.S.; Yoo, N.J.; Lee, S.H. Frameshift mutations of vacuolar protein sorting genes in gastric and colorectal cancers with microsatellite instability. Hum. Pathol. 2012, 43, 40–47. [Google Scholar] [CrossRef]
- Wible, D.J.; Chao, H.P.; Tang, D.G.; Bratton, S.B. Cancer mutations and alternative mRNA splicing reveal a conjugation switch that regulates ATG12-ATG5-ATG16L1 complex assembly and autophagy. Cell Discov. 2019, 5, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebovitz, C.B.; Robertson, A.G.; Goya, R.; Jones, S.J.; Morin, R.D.; Marra, M.A.; Gorski, S.M. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 2015, 11, 1668–1687. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014, 4, 914–927. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Zhang, W.; Ning, W.; Wang, C.; Deng, W.; Li, Z.; Shang, Z.; Shen, X.; Liu, X.; Baba, O.; et al. Model-based analysis uncovers mutations altering autophagy selectivity in human cancer. Nat. Commun. 2021, 12, 3258. [Google Scholar] [CrossRef] [PubMed]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, A.; Miyata, H.; Doki, Y.; Yamasaki, M.; Sohma, I.; Gotoh, K.; Takiguchi, S.; Fujiwara, Y.; Uchiyama, Y.; Monden, M. LC3, an autophagosome marker, is highly expressed in gastrointestinal cancers. Int. J. Oncol. 2008, 33, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Wang, C.; Croce, C.M.; Guan, J.L. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014, 28, 1204–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammoh, N.; Fraser, J.; Puente, C.; Syred, H.M.; Kang, H.; Ozawa, T.; Lam, D.; Acosta, J.C.; Finch, A.J.; Holland, E.; et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 2016, 12, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Lévy, J.; Cacheux, W.; Bara, M.A.; L’Hermitte, A.; Lepage, P.; Fraudeau, M.; Trentesaux, C.; Lemarchand, J.; Durand, A.; Crain, A.M.; et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat. Cell Biol. 2015, 17, 1062–1073. [Google Scholar] [CrossRef]
- Santanam, U.; Banach-Petrosky, W.; Abate-Shen, C.; Shen, M.M.; White, E.; DiPaola, R.S. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 2016, 30, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poillet-Perez, L.; White, E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019, 33, 610–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, V.; Khayati, K.; Hu, Z.S.; Lee, A.; Kamran, W.; Su, X.; Guo, J.Y. Autophagy modulates lipid metabolism to maintain metabolic flexibility for. Genes Dev. 2019, 33, 150–165. [Google Scholar] [CrossRef] [Green Version]
- Huo, Y.; Cai, H.; Teplova, I.; Bowman-Colin, C.; Chen, G.; Price, S.; Barnard, N.; Ganesan, S.; Karantza, V.; White, E.; et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov. 2013, 3, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.; Tortola, L.; Perlot, T.; Wirnsberger, G.; Novatchkova, M.; Nitsch, R.; Sykacek, P.; Frank, L.; Schramek, D.; Komnenovic, V.; et al. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 2014, 5, 3056. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Lazova, R.; Camp, R.L.; Klump, V.; Siddiqui, S.F.; Amaravadi, R.K.; Pawelek, J.M. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin. Cancer Res. 2012, 18, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yang, M.; Zhao, J.; Wang, J.; Zhang, Y.; Zhang, Q. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med. Oncol. 2013, 30, 475. [Google Scholar] [CrossRef]
- Nagano, M.; Hoshino, D.; Koshikawa, N.; Akizawa, T.; Seiki, M. Turnover of focal adhesions and cancer cell migration. Int. J. Cell. Biol. 2012, 2012, 310616. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Bijian, K.; Qiu, D.; Su, J.; Saad, A.; Dahabieh, M.S.; Miller, W.H.; Alaoui-Jamali, M.A. Endosomal sorting and c-Cbl targeting of paxillin to autophagosomes regulate cell-matrix adhesion turnover in human breast cancer cells. Oncotarget 2017, 8, 31199–31214. [Google Scholar] [CrossRef]
- Sharifi, M.N.; Mowers, E.E.; Drake, L.E.; Collier, C.; Chen, H.; Zamora, M.; Mui, S.; Macleod, K.F. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell. Rep. 2016, 15, 1660–1672. [Google Scholar] [CrossRef] [Green Version]
- Assar, E.A.; Tumbarello, D.A. Loss of the Essential Autophagy Regulators FIP200 or Atg5 Leads to Distinct Effects on Focal Adhesion Composition and Organization. Front. Cell. Dev. Biol. 2020, 8, 733. [Google Scholar] [CrossRef] [PubMed]
- Caino, M.C.; Chae, Y.C.; Vaira, V.; Ferrero, S.; Nosotti, M.; Martin, N.M.; Weeraratna, A.; O’Connell, M.; Jernigan, D.; Fatatis, A.; et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J. Clin. Invest. 2013, 123, 2907–2920. [Google Scholar] [CrossRef] [Green Version]
- Kenific, C.M.; Stehbens, S.J.; Goldsmith, J.; Leidal, A.M.; Faure, N.; Ye, J.; Wittmann, T.; Debnath, J. NBR1 enables autophagy-dependent focal adhesion turnover. J. Cell. Biol. 2016, 212, 577–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, T.; Kenific, C.M.; Suresh, D.; Gonzalez, H.; Shamir, E.R.; Mei, W.; Tankka, A.; Leidal, A.M.; Kalavacherla, S.; Woo, K.; et al. Autophagic Degradation of NBR1 Restricts Metastatic Outgrowth during Mammary Tumor Progression. Dev. Cell. 2020, 52, 591–604. [Google Scholar] [CrossRef]
- Peng, Y.F.; Shi, Y.H.; Shen, Y.H.; Ding, Z.B.; Ke, A.W.; Zhou, J.; Qiu, S.J.; Fan, J. Promoting colonization in metastatic HCC cells by modulation of autophagy. PLoS ONE 2013, 8, e74407. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, P.; Sou, Y.S.; Kageyama, S.; Koike, M.; Waguri, S.; Komatsu, M. NBR1-mediated p62-liquid droplets enhance the Keap1-Nrf2 system. EMBO Rep. 2020, 21, e48902. [Google Scholar] [CrossRef]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yang, B.; Zhou, Q.; Wu, Y.; Shang, D.; Guo, Y.; Song, Z.; Zheng, Q.; Xiong, J. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 2013, 34, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.H.; Baek, S.H.; Kim, E.K.; Ha, J.M.; Jin, S.Y.; Lee, H.S.; Ha, H.K.; Song, S.H.; Kim, S.J.; Shin, H.K.; et al. Uncoordinated 51-like kinase 2 signaling pathway regulates epithelial-mesenchymal transition in A549 lung cancer cells. FEBS Lett. 2016, 590, 1365–1374. [Google Scholar] [CrossRef]
- Zhang, W.; Yuan, W.; Song, J.; Wang, S.; Gu, X. LncRNA CPS1-IT1 suppresses EMT and metastasis of colorectal cancer by inhibiting hypoxia-induced autophagy through inactivation of HIF-1α. Biochimie 2018, 144, 21–27. [Google Scholar] [CrossRef]
- Ren, T.; Zheng, B.; Huang, Y.; Wang, S.; Bao, X.; Liu, K.; Guo, W. Osteosarcoma cell intrinsic PD-L2 signals promote invasion and metastasis via the RhoA-ROCK-LIMK2 and autophagy pathways. Cell. Death Dis. 2019, 10, 261. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, J.; Glogowska, A.; Thliveris, J.; Kalantari, F.; Shojaei, S.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 749–768. [Google Scholar] [CrossRef]
- Kiyono, K.; Suzuki, H.I.; Matsuyama, H.; Morishita, Y.; Komuro, A.; Kano, M.R.; Sugimoto, K.; Miyazono, K. Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 2009, 69, 8844–8852. [Google Scholar] [CrossRef] [Green Version]
- Lv, Q.; Wang, W.; Xue, J.; Hua, F.; Mu, R.; Lin, H.; Yan, J.; Lv, X.; Chen, X.; Hu, Z.W. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res. 2012, 72, 3238–3250. [Google Scholar] [CrossRef] [Green Version]
- Grassi, G.; Di Caprio, G.; Santangelo, L.; Fimia, G.M.; Cozzolino, A.M.; Komatsu, M.; Ippolito, G.; Tripodi, M.; Alonzi, T. Autophagy regulates hepatocyte identity and epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions promoting Snail degradation. Cell Death Dis. 2015, 6, e1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiang, L.; Zhao, B.; Ming, M.; Wang, N.; He, T.C.; Hwang, S.; Thorburn, A.; He, Y.Y. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc. Natl. Acad. Sci. USA 2014, 111, 9241–9246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi Roshan, M.; Soltani, A.; Soleimani, A.; Rezaie Kahkhaie, K.; Afshari, A.R.; Soukhtanloo, M. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie 2019, 165, 229–234. [Google Scholar] [CrossRef]
- Dower, C.M.; Wills, C.A.; Frisch, S.M.; Wang, H.G. Mechanisms and context underlying the role of autophagy in cancer metastasis. Autophagy 2018, 14, 1110–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmore, A.P. Anoikis. Cell Death Differ. 2005, 12 (Suppl. 2), 1473–1477. [Google Scholar] [CrossRef]
- Avivar-Valderas, A.; Salas, E.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Nagi, C.; Debnath, J.; Aguirre-Ghiso, J.A. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol. Cell. Biol. 2011, 31, 3616–3629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avivar-Valderas, A.; Bobrovnikova-Marjon, E.; Alan Diehl, J.; Bardeesy, N.; Debnath, J.; Aguirre-Ghiso, J.A. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene 2013, 32, 4932–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell. 2008, 19, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Debnath, J. IκB kinase complex (IKK) triggers detachment-induced autophagy in mammary epithelial cells independently of the PI3K-AKT-MTORC1 pathway. Autophagy 2013, 9, 1214–1227. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.D.; Liu, L.; Deng, H.; Li, Z.B.; Sheng, J.Q.; He, X.X.; Tian, D.A.; Li, P.Y. Astrocyte elevated gene 1 (AEG-1) promotes anoikis resistance and metastasis by inducing autophagy in hepatocellular carcinoma. J. Cell. Physiol. 2020, 235, 5084–5095. [Google Scholar] [CrossRef]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Yang, Y.; Xing, D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011, 278, 403–413. [Google Scholar] [CrossRef]
- Buchheit, C.L.; Angarola, B.L.; Steiner, A.; Weigel, K.J.; Schafer, Z.T. Anoikis evasion in inflammatory breast cancer cells is mediated by Bim-EL sequestration. Cell Death Differ. 2015, 22, 1275–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reginato, M.J.; Mills, K.R.; Paulus, J.K.; Lynch, D.K.; Sgroi, D.C.; Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 2003, 5, 733–740. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Athonvarangkul, D.; Mishall, P.; Sahu, S.; Singh, R. Autophagy proteins regulate ERK phosphorylation. Nat. Commun. 2013, 4, 2799. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Deloménie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell. Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhao, Q.; Sun, H.; Yin, L.; Wu, J.; Xu, J.; He, T.; Yang, C.; Liang, C. Defective autophagy leads to the suppression of stem-like features of CD271. J. Biomed. Sci. 2016, 23, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.K.; Yang, C.Y.; Jeng, Y.M.; Chen, C.L.; Wu, H.H.; Chang, Y.C.; Ma, C.; Kuo, W.H.; Chang, K.J.; Shew, J.Y.; et al. Autocrine/paracrine mechanism of interleukin-17B receptor promotes breast tumorigenesis through NF-κB-mediated antiapoptotic pathway. Oncogene 2014, 33, 2968–2977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bie, Q.; Song, H.; Chen, X.; Yang, X.; Shi, S.; Zhang, L.; Zhao, R.; Wei, L.; Zhang, B.; Xiong, H. IL-17B/IL-17RB signaling cascade contributes to self-renewal and tumorigenesis of cancer stem cells by regulating Beclin-1 ubiquitination. Oncogene 2021, 40, 2200–2216. [Google Scholar] [CrossRef] [PubMed]
- Bortnik, S.; Tessier-Cloutier, B.; Leung, S.; Xu, J.; Asleh, K.; Burugu, S.; Magrill, J.; Greening, K.; Derakhshan, F.; Yip, S.; et al. Differential expression and prognostic relevance of autophagy-related markers ATG4B, GABARAP, and LC3B in breast cancer. Breast Cancer Res. Treat. 2020, 183, 525–547. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, D.; Sun, Z.; Ye, T.; Li, J.; Zeng, B.; Zhao, Q.; Rosie Xing, H. Autophagy augments the self-renewal of lung cancer stem cells by the degradation of ubiquitinated p53. Cell. Death Dis. 2021, 12, 98. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 2015, 13, 651–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, S.K.; Wen, J.; Chen, S.; Guan, J.L. Autophagy Differentially Regulates Distinct Breast Cancer Stem-like Cells in Murine Models via EGFR/Stat3 and Tgfβ/Smad Signaling. Cancer Res. 2016, 76, 3397–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Q.; Qin, J.; Zhang, Y.; Cheng, X.; Wang, X.; Lu, W.; Xie, X.; Zhang, S. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J. Exp. Clin. Cancer Res. 2017, 36, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Balaguer, A.; Ortiz-Martínez, F.; García-Martínez, A.; Pomares-Navarro, C.; Lerma, E.; Peiró, G. FOXA2 mRNA expression is associated with relapse in patients with Triple-Negative/Basal-like breast carcinoma. Breast Cancer Res. Treat. 2015, 153, 465–474. [Google Scholar] [CrossRef]
- Ghatak, D.; Das Ghosh, D.; Roychoudhury, S. Cancer Stemness: p53 at the Wheel. Front Oncol. 2020, 10, 604124. [Google Scholar] [CrossRef]
- Sharif, T.; Martell, E.; Dai, C.; Kennedy, B.E.; Murphy, P.; Clements, D.R.; Kim, Y.; Lee, P.W.; Gujar, S.A. Autophagic homeostasis is required for the pluripotency of cancer stem cells. Autophagy 2017, 13, 264–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Yeh, A.C.; Ramaswamy, S. Mechanisms of Cancer Cell Dormancy--Another Hallmark of Cancer? Cancer Res. 2015, 75, 5014–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef]
- Akkoc, Y.; Peker, N.; Akcay, A.; Gozuacik, D. Autophagy and Cancer Dormancy. Front. Oncol. 2021, 11, 627023. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Baquero, M.T.; Yang, H.; Yang, M.; Reger, A.S.; Kim, C.; Levine, D.A.; Clarke, C.H.; Liao, W.S.; Bast, R.C. DIRAS3 regulates the autophagosome initiation complex in dormant ovarian cancer cells. Autophagy 2014, 10, 1071–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig. 2008, 118, 3917–3929. [Google Scholar] [CrossRef] [Green Version]
- Correa, R.J.; Valdes, Y.R.; Peart, T.M.; Fazio, E.N.; Bertrand, M.; McGee, J.; Préfontaine, M.; Sugimoto, A.; DiMattia, G.E.; Shepherd, T.G. Combination of AKT inhibition with autophagy blockade effectively reduces ascites-derived ovarian cancer cell viability. Carcinogenesis 2014, 35, 1951–1961. [Google Scholar] [CrossRef] [Green Version]
- Washington, M.N.; Suh, G.; Orozco, A.F.; Sutton, M.N.; Yang, H.; Wang, Y.; Mao, W.; Millward, S.; Ornelas, A.; Atkinson, N.; et al. ARHI (DIRAS3)-mediated autophagy-associated cell death enhances chemosensitivity to cisplatin in ovarian cancer cell lines and xenografts. Cell. Death Dis. 2015, 6, e1836. [Google Scholar] [CrossRef] [Green Version]
- La Belle Flynn, A.; Calhoun, B.C.; Sharma, A.; Chang, J.C.; Almasan, A.; Schiemann, W.P. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat. Commun. 2019, 10, 3668. [Google Scholar] [CrossRef] [Green Version]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shang, Z.; Zhou, Y.; Hu, X.; Chen, Y.; Fan, Y.; Wei, X.; Wu, L.; Liang, Q.; Zhang, J.; et al. Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival. Cell. Death Dis. 2018, 9, 213. [Google Scholar] [CrossRef] [Green Version]
- Aqbi, H.F.; Tyutyunyk-Massey, L.; Keim, R.C.; Butler, S.E.; Thekkudan, T.; Joshi, S.; Smith, T.M.; Bandyopadhyay, D.; Idowu, M.O.; Bear, H.D.; et al. Autophagy-deficient breast cancer shows early tumor recurrence and escape from dormancy. Oncotarget 2018, 9, 22113–22122. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Pan, H.; Liu, Z.; Xie, J.; Han, W. Roles of PFKFB3 in cancer. Signal Transduct. Target. Ther. 2017, 2, 17044. [Google Scholar] [CrossRef]
- Ueda, S.; Takanashi, M.; Sudo, K.; Kanekura, K.; Kuroda, M. miR-27a ameliorates chemoresistance of breast cancer cells by disruption of reactive oxygen species homeostasis and impairment of autophagy. Lab. Investig. 2020, 100, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Blessing, A.M.; Santiago-O’Farrill, J.M.; Mao, W.; Pang, L.; Ning, J.; Pak, D.; Bollu, L.R.; Rask, P.; Iles, L.; Yang, H.; et al. Elimination of dormant, autophagic ovarian cancer cells and xenografts through enhanced sensitivity to anaplastic lymphoma kinase inhibition. Cancer 2020, 126, 3579–3592. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Hu, T.; Li, P.; Luo, Z.; Chen, X.; Zhang, J.; Wang, C.; Chen, P.; Dong, Z. Chloroquine inhibits hepatocellular carcinoma cell growth in vitro and in vivo. Oncol Rep 2016, 35, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Zhao, Y.L.; Deng, X.; Yang, S.; Mao, Y.; Li, Z.; Jiang, P.; Zhao, X.; Wei, Y. Chloroquine inhibits colon cancer cell growth in vitro and tumor growth in vivo via induction of apoptosis. Cancer Investig. 2009, 27, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell. Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [Green Version]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; Liotta, L.A.; et al. Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef]
- Maes, H.; Kuchnio, A.; Peric, A.; Moens, S.; Nys, K.; De Bock, K.; Quaegebeur, A.; Schoors, S.; Georgiadou, M.; Wouters, J.; et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 2014, 26, 190–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, P.; Strambi, A.; Zipoli, C.; Hägg-Olofsson, M.; Buoncervello, M.; Linder, S.; De Milito, A. Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: Implications for cancer therapies. Autophagy 2014, 10, 562–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maycotte, P.; Gearheart, C.M.; Barnard, R.; Aryal, S.; Mulcahy Levy, J.M.; Fosmire, S.P.; Hansen, R.J.; Morgan, M.J.; Porter, C.C.; Gustafson, D.L.; et al. STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res. 2014, 74, 2579–2590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Hu, P.; Yang, Z.; Xue, C.; Gong, J.; Sun, S.; Shi, L.; Zhang, S.; Li, Z.; Yang, C.; et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol. Rep. 2017, 37, 3449–3458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronan, B.; Flamand, O.; Vescovi, L.; Dureuil, C.; Durand, L.; Fassy, F.; Bachelot, M.F.; Lamberton, A.; Mathieu, M.; Bertrand, T.; et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 2014, 10, 1013–1019. [Google Scholar] [CrossRef]
- Dyczynski, M.; Yu, Y.; Otrocka, M.; Parpal, S.; Braga, T.; Henley, A.B.; Zazzi, H.; Lerner, M.; Wennerberg, K.; Viklund, J.; et al. Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett. 2018, 435, 32–43. [Google Scholar] [CrossRef]
- Kurdi, A.; Cleenewerck, M.; Vangestel, C.; Lyssens, S.; Declercq, W.; Timmermans, J.P.; Stroobants, S.; Augustyns, K.; De Meyer, G.R.Y.; Van Der Veken, P.; et al. ATG4B inhibitors with a benzotropolone core structure block autophagy and augment efficiency of chemotherapy in mice. Biochem. Pharmacol. 2017, 138, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Akin, D.; Wang, S.K.; Habibzadegah-Tari, P.; Law, B.; Ostrov, D.; Li, M.; Yin, X.M.; Kim, J.S.; Horenstein, N.; Dunn, W.A. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 2014, 10, 2021–2035. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Hong, L.; Xu, J.; Zhong, G.; Gu, Q.; Guan, Y.; Zheng, X.; Dai, Q.; Luo, X.; Liu, C.; et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 2019, 15, 295–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [Green Version]
- Law, B.K. Rapamycin: An anti-cancer immunosuppressant? Crit. Rev. Oncol. Hematol. 2005, 56, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, F.S.; Chen, Y.L.; Hung, M.H.; Chu, P.Y.; Tsai, M.H.; Chen, L.J.; Hsiao, Y.J.; Shih, C.T.; Chang, M.J.; Chao, T.I.; et al. Palbociclib induces activation of AMPK and inhibits hepatocellular carcinoma in a CDK4/6-independent manner. Mol. Oncol. 2017, 11, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Tremel, S.; Williams, R.L. VPS34 complexes from a structural perspective. J. Lipid. Res. 2019, 60, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Pavlinov, I.; Salkovski, M.; Aldrich, L.N. Beclin 1-ATG14L Protein-Protein Interaction Inhibitor Selectively Inhibits Autophagy through Disruption of VPS34 Complex I. J. Am. Chem. Soc. 2020, 142, 8174–8182. [Google Scholar] [CrossRef]
- Xia, H.; Green, D.R.; Zou, W. Autophagy in tumour immunity and therapy. Nat. Rev. Cancer 2021, 21, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Bustos, S.O.; Antunes, F.; Rangel, M.C.; Chammas, R. Emerging Autophagy Functions Shape the Tumor Microenvironment and Play a Role in Cancer Progression - Implications for Cancer Therapy. Front Oncol. 2020, 10, 606436. [Google Scholar] [CrossRef] [PubMed]
- Towers, C.G.; Fitzwalter, B.E.; Regan, D.; Goodspeed, A.; Morgan, M.J.; Liu, C.W.; Gustafson, D.L.; Thorburn, A. Cancer Cells Upregulate NRF2 Signaling to Adapt to Autophagy Inhibition. Dev. Cell. 2019, 50, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Kerins, M.J.; Liu, P.; Tian, W.; Mannheim, W.; Zhang, D.D.; Ooi, A. Genome-Wide CRISPR Screen Reveals Autophagy Disruption as the Convergence Mechanism That Regulates the NRF2 Transcription Factor. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease - lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain 2013, 136, 2130–2146. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 2009, 4, e5515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Wang, X.; Feng, X.; Zhang, A.; Li, J.; Gu, K.; Huang, J.; Pang, S.; Dong, H.; Gao, H.; et al. Altered expression of autophagic genes in the peripheral leukocytes of patients with sporadic Parkinson’s disease. Brain Res. 2011, 1394, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015, 30, 1639–1647. [Google Scholar] [CrossRef]
- Kabuta, T.; Furuta, A.; Aoki, S.; Furuta, K.; Wada, K. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J. Biol. Chem. 2008, 283, 23731–23738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell. Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.Q.; Yuan, Y.H.; Gao, Y.N.; Huang, J.Y.; Ma, K.L.; Gao, Y.; Zhang, W.Q.; Guo, X.F.; Chen, N.H. Overexpression of human E46K mutant α-synuclein impairs macroautophagy via inactivation of JNK1-Bcl-2 pathway. Mol. Neurobiol. 2014, 50, 685–701. [Google Scholar] [CrossRef]
- Sarkar, S.; Olsen, A.L.; Sygnecka, K.; Lohr, K.M.; Feany, M.B. α-synuclein impairs autophagosome maturation through abnormal actin stabilization. PLoS Genet. 2021, 17, e1009359. [Google Scholar] [CrossRef]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M. Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Fussi, N.; Höllerhage, M.; Chakroun, T.; Nykänen, N.P.; Rösler, T.W.; Koeglsperger, T.; Wurst, W.; Behrends, C.; Höglinger, G.U. Exosomal secretion of α-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell. Death Dis. 2018, 9, 757. [Google Scholar] [CrossRef] [Green Version]
- Minakaki, G.; Menges, S.; Kittel, A.; Emmanouilidou, E.; Schaeffner, I.; Barkovits, K.; Bergmann, A.; Rockenstein, E.; Adame, A.; Marxreiter, F.; et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 2018, 14, 98–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poehler, A.M.; Xiang, W.; Spitzer, P.; May, V.E.; Meixner, H.; Rockenstein, E.; Chutna, O.; Outeiro, T.F.; Winkler, J.; Masliah, E.; et al. Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014, 10, 2171–2192. [Google Scholar] [CrossRef] [Green Version]
- Angot, E.; Steiner, J.A.; Lema Tomé, C.M.; Ekström, P.; Mattsson, B.; Björklund, A.; Brundin, P. Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS ONE 2012, 7, e39465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, S.J.; Kuo, S.H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.W.; Leung, C.T.; Liu, H.; Pang, S.Y.; Lam, C.S.; Xian, J.; Li, L.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: Role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy 2020, 16, 347–370. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Mamais, A.; Roosen, D.A.; Dihanich, S.; Soutar, M.P.; Plun-Favreau, H.; Bandopadhyay, R.; Hardy, J.; Tooze, S.A.; Cookson, M.R.; et al. mTOR independent regulation of macroautophagy by Leucine Rich Repeat Kinase 2 via Beclin-1. Sci. Rep. 2016, 6, 35106. [Google Scholar] [CrossRef] [PubMed]
- Obergasteiger, J.; Frapporti, G.; Lamonaca, G.; Pizzi, S.; Picard, A.; Lavdas, A.A.; Pischedda, F.; Piccoli, G.; Hilfiker, S.; Lobbestael, E.; et al. Kinase inhibition of G2019S-LRRK2 enhances autolysosome formation and function to reduce endogenous alpha-synuclein intracellular inclusions. Cell. Death Discov. 2020, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Bravo-San Pedro, J.M.; Niso-Santano, M.; Gómez-Sánchez, R.; Pizarro-Estrella, E.; Aiastui-Pujana, A.; Gorostidi, A.; Climent, V.; López de Maturana, R.; Sanchez-Pernaute, R.; López de Munain, A.; et al. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell. Mol. Life Sci. 2013, 70, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Giaime, E.; Tong, Y.; Wagner, L.K.; Yuan, Y.; Huang, G.; Shen, J. Age-Dependent Dopaminergic Neurodegeneration and Impairment of the Autophagy-Lysosomal Pathway in LRRK-Deficient Mice. Neuron 2017, 96, 796–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, S.; Holmström, K.M.; Treis, A.; Skujat, D.; Weber, S.S.; Fiesel, F.C.; Kahle, P.J.; Springer, W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2010, 6, 871–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganjam, G.K.; Bolte, K.; Matschke, L.A.; Neitemeier, S.; Dolga, A.M.; Höllerhage, M.; Höglinger, G.U.; Adamczyk, A.; Decher, N.; Oertel, W.H.; et al. Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 2019, 10, 865. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Little, D.; Luft, C.; Mosaku, O.; Lorvellec, M.; Yao, Z.; Paillusson, S.; Kriston-Vizi, J.; Gandhi, S.; Abramov, A.Y.; Ketteler, R.; et al. A single cell high content assay detects mitochondrial dysfunction in iPSC-derived neurons with mutations in SNCA. Sci. Rep. 2018, 8, 9033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Burgess, J.D.; Faroqi, A.H.; DeMeo, N.N.; Fiesel, F.C.; Springer, W.; Delenclos, M.; McLean, P.J. Alpha-synuclein-induced mitochondrial dysfunction is mediated via a sirtuin 3-dependent pathway. Mol. Neurodegener. 2020, 15, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampaio-Marques, B.; Felgueiras, C.; Silva, A.; Rodrigues, M.; Tenreiro, S.; Franssens, V.; Reichert, A.S.; Outeiro, T.F.; Winderickx, J.; Ludovico, P. SNCA (α-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy. Autophagy 2012, 8, 1494–1509. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem. Cell. 2016, 19, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Bonello, F.; Hassoun, S.M.; Mouton-Liger, F.; Shin, Y.S.; Muscat, A.; Tesson, C.; Lesage, S.; Beart, P.M.; Brice, A.; Krupp, J.; et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: Pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 2019, 28, 1645–1660. [Google Scholar] [CrossRef] [PubMed]
- Wauters, F.; Cornelissen, T.; Imberechts, D.; Martin, S.; Koentjoro, B.; Sue, C.; Vangheluwe, P.; Vandenberghe, W. Mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 2020, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Du, T.T.; Wang, L.; Duan, C.L.; Lu, L.L.; Zhang, J.L.; Gao, G.; Qiu, X.B.; Wang, X.M.; Yang, H. GBA deficiency promotes SNCA/α-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015, 11, 1803–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Ham, A.; Ma, T.C.; Kuo, S.H.; Kanter, E.; Kim, D.; Ko, H.S.; Quan, Y.; Sardi, S.P.; Li, A.; et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 2019, 15, 113–130. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zavodszky, E.; Seaman, M.N.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.L.; Erion, J.R.; Tian, Y.; Liu, W.; Yin, D.M.; Ye, J.; Tang, B.; Mei, L.; Xiong, W.C. VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for α-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J. Neurosci. 2015, 35, 10613–10628. [Google Scholar] [CrossRef]
- Ma, K.Y.; Fokkens, M.R.; Reggiori, F.; Mari, M.; Verbeek, D.S. Parkinson’s disease-associated VPS35 mutant reduces mitochondrial membrane potential and impairs PINK1/Parkin-mediated mitophagy. Transl. Neurodegener. 2021, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Nash, Y.; Schmukler, E.; Trudler, D.; Pinkas-Kramarski, R.; Frenkel, D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J. Neurochem. 2017, 143, 584–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.Y.; Kang, W.Y.; Chen, Y.M.; Jiang, T.F.; Zhang, J.; Zhang, L.N.; Ding, J.Q.; Liu, J.; Chen, S.D. DJ-1 Inhibits α-Synuclein Aggregation by Regulating Chaperone-Mediated Autophagy. Front Aging Neurosci. 2017, 9, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, L.Y.; Giasson, B.I.; Bonini, N.M. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivatt, R.M.; Sanchez-Martinez, A.; Godena, V.K.; Brown, S.; Ziviani, E.; Whitworth, A.J. Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 8494–8499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.D.; Xie, S.P.; Sathiyamoorthy, S.; Saw, W.T.; Sing, T.Y.; Ng, S.H.; Chua, H.P.; Tang, A.M.; Shaffra, F.; Li, Z.; et al. F-box protein 7 mutations promote protein aggregation in mitochondria and inhibit mitophagy. Hum. Mol. Genet. 2015, 24, 6314–6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinn, S.; Drolet, R.E.; Cramer, P.E.; Wong, A.H.; Toolan, D.M.; Gretzula, C.A.; Voleti, B.; Vassileva, G.; Disa, J.; Tadin-Strapps, M.; et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 2389–2394. [Google Scholar] [CrossRef] [Green Version]
- Jinn, S.; Blauwendraat, C.; Toolan, D.; Gretzula, C.A.; Drolet, R.E.; Smith, S.; Nalls, M.A.; Marcus, J.; Singleton, A.B.; Stone, D.J. Functionalization of the TMEM175 p.M393T variant as a risk factor for Parkinson disease. Hum. Mol. Genet. 2019, 28, 3244–3254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, H.; Yamashita, C.; Shiba-Fukushima, K.; Inoshita, T.; Funayama, M.; Sato, S.; Hatta, T.; Natsume, T.; Umitsu, M.; Takagi, J.; et al. Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun. 2017, 8, 15500. [Google Scholar] [CrossRef] [Green Version]
- Grünewald, A.; Arns, B.; Seibler, P.; Rakovic, A.; Münchau, A.; Ramirez, A.; Sue, C.M.; Klein, C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol. Aging 2012, 33, 1841–1843. [Google Scholar] [CrossRef]
- Ramonet, D.; Podhajska, A.; Stafa, K.; Sonnay, S.; Trancikova, A.; Tsika, E.; Pletnikova, O.; Troncoso, J.C.; Glauser, L.; Moore, D.J. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum. Mol. Genet. 2012, 21, 1725–1743. [Google Scholar] [CrossRef]
- Hunn, B.H.M.; Vingill, S.; Threlfell, S.; Alegre-Abarrategui, J.; Magdelyns, M.; Deltheil, T.; Bengoa-Vergniory, N.; Oliver, P.L.; Cioroch, M.; Doig, N.M.; et al. Impairment of Macroautophagy in Dopamine Neurons Has Opposing Effects on Parkinsonian Pathology and Behavior. Cell Rep. 2019, 29, 920–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, R.N.; Lambracht-Washington, D.; Yu, G.; Xia, W. Genomics of Alzheimer Disease: A Review. JAMA Neurol. 2016, 73, 867–874. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Varo, R.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Torres, M.; Baglietto-Vargas, D.; Moreno-Gonzalez, I.; De Castro, V.; Jimenez, S.; Ruano, D.; Vizuete, M.; et al. Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol. 2012, 123, 53–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Coffey, E.E.; Beckel, J.M.; Laties, A.M.; Mitchell, C.H. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 2014, 263, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, K.; Fleming, A.; Imarisio, S.; Lopez Ramirez, A.; Mercer, J.L.; Jimenez-Sanchez, M.; Bento, C.F.; Puri, C.; Zavodszky, E.; Siddiqi, F.; et al. PICALM modulates autophagy activity and tau accumulation. Nat. Commun. 2014, 5, 4998. [Google Scholar] [CrossRef] [Green Version]
- Ando, K.; Tomimura, K.; Sazdovitch, V.; Suain, V.; Yilmaz, Z.; Authelet, M.; Ndjim, M.; Vergara, C.; Belkouch, M.; Potier, M.C.; et al. Level of PICALM, a key component of clathrin-mediated endocytosis, is correlated with levels of phosphotau and autophagy-related proteins and is associated with tau inclusions in AD, PSP and Pick disease. Neurobiol. Dis. 2016, 94, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Ando, K.; De Decker, R.; Vergara, C.; Yilmaz, Z.; Mansour, S.; Suain, V.; Sleegers, K.; de Fisenne, M.A.; Houben, S.; Potier, M.C.; et al. Picalm reduction exacerbates tau pathology in a murine tauopathy model. Acta Neuropathol. 2020, 139, 773–789. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Su, L.Y.; Li, G.; Yang, J.; Liu, Q.; Yang, L.X.; Zhang, D.F.; Zhou, H.; Xu, M.; Fan, Y.; et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 2020, 16, 52–69. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.C.; Wu, A.J.; Huang, A.S.; Dong, R.; Malampati, S.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Zhu, Z.; Su, C.; et al. Lysosomal TPCN (two pore segment channel) inhibition ameliorates beta-amyloid pathology and mitigates memory impairment in Alzheimer disease. Autophagy 2021, 1–19. [Google Scholar] [CrossRef]
- Zheng, X.; Lin, W.; Jiang, Y.; Lu, K.; Wei, W.; Huo, Q.; Cui, S.; Yang, X.; Li, M.; Xu, N.; et al. Electroacupuncture ameliorates beta-amyloid pathology and cognitive impairment in Alzheimer disease via a novel mechanism involving activation of TFEB (transcription factor EB). Autophagy 2021, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Hong, C.G.; Yue, T.; Li, H.M.; Duan, R.; Hu, W.B.; Cao, J.; Wang, Z.X.; Chen, C.Y.; Hu, X.K.; et al. Inhibition of miR-331-3p and miR-9-5p ameliorates Alzheimer’s disease by enhancing autophagy. Theranostics 2021, 11, 2395–2409. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benito-Cuesta, I.; Ordóñez-Gutiérrez, L.; Wandosell, F. AMPK activation does not enhance autophagy in neurons in contrast to MTORC1 inhibition: Different impact on β-amyloid clearance. Autophagy 2021, 17, 656–671. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, D.H.; Yoon, S.Y. Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep. 2016, 49, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Bhaskar, K. Degradation and Transmission of Tau by Autophagic-Endolysosomal Networks and Potential Therapeutic Targets for Tauopathy. Front Mol. Neurosci. 2020, 13, 586731. [Google Scholar] [CrossRef] [PubMed]
- Caballero, B.; Wang, Y.; Diaz, A.; Tasset, I.; Juste, Y.R.; Stiller, B.; Mandelkow, E.M.; Mandelkow, E.; Cuervo, A.M. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 2018, 17, e12692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.C.; Nandi, G.A.; Tentarelli, S.; Gurrell, I.K.; Jamier, T.; Lucente, D.; Dickerson, B.C.; Brown, D.G.; Brandon, N.J.; Haggarty, S.J. Prolonged tau clearance and stress vulnerability rescue by pharmacological activation of autophagy in tauopathy neurons. Nat. Commun. 2020, 11, 3258. [Google Scholar] [CrossRef]
- Bourdenx, M.; Martín-Segura, A.; Scrivo, A.; Rodriguez-Navarro, J.A.; Kaushik, S.; Tasset, I.; Diaz, A.; Storm, N.J.; Xin, Q.; Juste, Y.R.; et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 2021, 184, 2696–2714. [Google Scholar] [CrossRef]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951. [Google Scholar] [CrossRef]
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 792–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Hu, Y.; Li, X.C.; Wang, Z.H.; Luo, Y.; Zhang, X.; Liu, X.P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nopoulos, P.C. Huntington disease: a single-gene degenerative disorder of the striatum. Dialogues Clin. Neurosci. 2016, 18, 91–98. [Google Scholar]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.C.; Li, H.Y.; Chen, G.C.; Chern, Y.; Tu, P.H. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy 2015, 11, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Kurosawa, M.; Matsumoto, G.; Kino, Y.; Okuno, M.; Kurosawa-Yamada, M.; Washizu, C.; Taniguchi, H.; Nakaso, K.; Yanagawa, T.; Warabi, E.; et al. Depletion of p62 reduces nuclear inclusions and paradoxically ameliorates disease phenotypes in Huntington’s model mice. Hum. Mol. Genet. 2015, 24, 1092–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.D.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wold, M.S.; Lim, J.; Lachance, V.; Deng, Z.; Yue, Z. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington’s disease models. Mol. Neurodegener. 2016, 11, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Ravikumar, B.; Floto, R.A.; Rubinsztein, D.C. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009, 16, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Wei, W.; Gaertig, M.A.; Li, S.; Li, X.J. Therapeutic Effect of Berberine on Huntington’s Disease Transgenic Mouse Model. PLoS ONE 2015, 10, e0134142. [Google Scholar] [CrossRef]
- Feng, X.; Luo, S.; Lu, B. Conformation Polymorphism of Polyglutamine Proteins. Trends Biochem. Sci. 2018, 43, 424–435. [Google Scholar] [CrossRef]
- Fu, Y.; Wu, P.; Pan, Y.; Sun, X.; Yang, H.; Difiglia, M.; Lu, B. A toxic mutant huntingtin species is resistant to selective autophagy. Nat Chem Biol 2017, 13, 1152–1154. [Google Scholar] [CrossRef]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djajadikerta, A.; Keshri, S.; Pavel, M.; Prestil, R.; Ryan, L.; Rubinsztein, D.C. Autophagy Induction as a Therapeutic Strategy for Neurodegenerative Diseases. J. Mol. Biol. 2020, 432, 2799–2821. [Google Scholar] [CrossRef] [PubMed]
- Ozcelik, S.; Fraser, G.; Castets, P.; Schaeffer, V.; Skachokova, Z.; Breu, K.; Clavaguera, F.; Sinnreich, M.; Kappos, L.; Goedert, M.; et al. Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS ONE 2013, 8, e62459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Zhang, Q.Q.; Cao, L.; Wang, H.F.; Tan, M.S.; Gao, Q.; Qin, H.; Zhang, Y.D.; et al. Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology 2014, 85, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Wey, M.C.; Fernandez, E.; Hart, M.J.; Gelfond, J.; Bokov, A.F.; Rani, S.; Strong, R. Rapamycin improves motor function, reduces 4-hydroxynonenal adducted protein in brain, and attenuates synaptic injury in a mouse model of synucleinopathy. Pathobiol. Aging Age Relat. Dis. 2015, 5, 28743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masini, D.; Bonito-Oliva, A.; Bertho, M.; Fisone, G. Inhibition of mTORC1 Signaling Reverts Cognitive and Affective Deficits in a Mouse Model of Parkinson’s Disease. Front Neurol. 2018, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Koprich, J.B.; Wang, Y.; Yu, W.B.; Xiao, B.G.; Brotchie, J.M.; Wang, J. Treatment with Trehalose Prevents Behavioral and Neurochemical Deficits Produced in an AAV alpha-Synuclein Rat Model of Parkinson’s Disease. Mol. Neurobiol. 2016, 53, 2258–2268. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef]
- Pupyshev, A.B.; Tikhonova, M.A.; Akopyan, A.A.; Tenditnik, M.V.; Dubrovina, N.I.; Korolenko, T.A. Therapeutic activation of autophagy by combined treatment with rapamycin and trehalose in a mouse MPTP-induced model of Parkinson’s disease. Pharmacol. Biochem. Behav. 2019, 177, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, F.H.; Menzies, F.M.; Lopez, A.; Stamatakou, E.; Karabiyik, C.; Ureshino, R.; Ricketts, T.; Jimenez-Sanchez, M.; Esteban, M.A.; Lai, L.; et al. Felodipine induces autophagy in mouse brains with pharmacokinetics amenable to repurposing. Nat. Commun. 2019, 10, 1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.; Jia, N.; Zhong, Y.; Shang, X. S14G-humanin alleviates insulin resistance and increases autophagy in neurons of APP/PS1 transgenic mouse. J. Cell Biochem. 2018, 119, 3111–3117. [Google Scholar] [CrossRef]
- Luengo, E.; Buendia, I.; Fernandez-Mendivil, C.; Trigo-Alonso, P.; Negredo, P.; Michalska, P.; Hernandez-Garcia, B.; Sanchez-Ramos, C.; Bernal, J.A.; Ikezu, T.; et al. Pharmacological doses of melatonin impede cognitive decline in tau-related Alzheimer models, once tauopathy is initiated, by restoring the autophagic flux. J. Pineal Res. 2019, 67, e12578. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, O.V.; Radanovic, M.; Talib, L.L.; Gattaz, W.F. Clinical and biological effects of long-term lithium treatment in older adults with amnestic mild cognitive impairment: Randomised clinical trial. Br. J. Psych. 2019, 215, 668–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchsinger, J.A.; Perez, T.; Chang, H.; Mehta, P.; Steffener, J.; Pradabhan, G.; Ichise, M.; Manly, J.; Devanand, D.P.; Bagiella, E. Metformin in Amnestic Mild Cognitive Impairment: Results of a Pilot Randomized Placebo Controlled Clinical Trial. J. Alzheimers. Dis. 2016, 51, 501–514. [Google Scholar] [CrossRef] [Green Version]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib Effects in Parkinson’s disease and Dementia with Lewy bodies. J. Parkinsons. Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cen, X.; Chen, Y.; Xu, X.; Wu, R.; He, F.; Zhao, Q.; Sun, Q.; Yi, C.; Wu, J.; Najafov, A.; et al. Pharmacological targeting of MCL-1 promotes mitophagy and improves disease pathologies in an Alzheimer’s disease mouse model. Nat. Commun. 2020, 11, 5731. [Google Scholar] [CrossRef]
- Bauer, P.O.; Goswami, A.; Wong, H.K.; Okuno, M.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Matsumoto, G.; Kino, Y.; Nagai, Y.; et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 2010, 28, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, C.; Wang, Z.; Zhu, C.; Li, J.; Sha, T.; Ma, L.; Gao, C.; Yang, Y.; Sun, Y.; et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature 2019, 575, 203–209. [Google Scholar] [CrossRef]
- Dou, J.; Su, P.; Xu, C.; Wen, Z.; Mao, Z.; Li, W. Targeting Hsc70-based autophagy to eliminate amyloid β oligomers. Biochem Biophys Res Commun 2020, 524, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE 2011, 6, e25416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauckman, K.A.; Owusu-Boaitey, N.; Mysorekar, I.U. Selective autophagy: Xenophagy. Methods 2015, 75, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, V.; Verma, S.; Seranova, E.; Sarkar, S.; Kumar, D. Selective Autophagy and Xenophagy in Infection and Disease. Front. Cell Dev. Biol. 2018, 6, 147. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhou, P.; Cheng, S.; Lu, Q.; Nowak, K.; Hopp, A.K.; Li, L.; Shi, X.; Zhou, Z.; Gao, W.; et al. A Bacterial Effector Reveals the V-ATPase-ATG16L1 Axis that Initiates Xenophagy. Cell 2019, 178, 552–566. [Google Scholar] [CrossRef]
- Thurston, T.L.; Wandel, M.P.; von Muhlinen, N.; Foeglein, A.; Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012, 482, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.O.; Manzanillo, P.S.; Cox, J.S. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 2012, 150, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Chai, Q.; Wang, X.; Qiang, L.; Zhang, Y.; Ge, P.; Lu, Z.; Zhong, Y.; Li, B.; Wang, J.; Zhang, L.; et al. A Mycobacterium tuberculosis surface protein recruits ubiquitin to trigger host xenophagy. Nat. Commun. 2019, 10, 1973. [Google Scholar] [CrossRef]
- Nakagawa, I.; Amano, A.; Mizushima, N.; Yamamoto, A.; Yamaguchi, H.; Kamimoto, T.; Nara, A.; Funao, J.; Nakata, M.; Tsuda, K.; et al. Autophagy defends cells against invading group A Streptococcus. Science 2004, 306, 1037–1040. [Google Scholar] [CrossRef]
- Sakurai, A.; Maruyama, F.; Funao, J.; Nozawa, T.; Aikawa, C.; Okahashi, N.; Shintani, S.; Hamada, S.; Ooshima, T.; Nakagawa, I. Specific behavior of intracellular Streptococcus pyogenes that has undergone autophagic degradation is associated with bacterial streptolysin O and host small G proteins Rab5 and Rab7. J. Biol. Chem. 2010, 285, 22666–22675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozawa, T.; Aikawa, C.; Goda, A.; Maruyama, F.; Hamada, S.; Nakagawa, I. The small GTPases Rab9A and Rab23 function at distinct steps in autophagy during Group A Streptococcus infection. Cell Microbiol. 2012, 14, 1149–1165. [Google Scholar] [CrossRef]
- Losier, T.T.; Akuma, M.; McKee-Muir, O.C.; LeBlond, N.D.; Suk, Y.; Alsaadi, R.M.; Guo, Z.; Reshke, R.; Sad, S.; Campbell-Valois, F.X.; et al. AMPK Promotes Xenophagy through Priming of Autophagic Kinases upon Detection of Bacterial Outer Membrane Vesicles. Cell Rep. 2019, 26, 2150–2165. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, R.; Hos, N.J.; Gutierrez, S.; Fischer, J.; Stepek, J.M.; Daglidu, E.; Krönke, M.; Robinson, N. Salmonella Typhimurium disrupts Sirt1/AMPK checkpoint control of mTOR to impair autophagy. PLoS Pathog. 2017, 13, e1006227. [Google Scholar] [CrossRef] [Green Version]
- Choy, A.; Dancourt, J.; Mugo, B.; O’Connor, T.J.; Isberg, R.R.; Melia, T.J.; Roy, C.R. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 2012, 338, 1072–1076. [Google Scholar] [CrossRef] [Green Version]
- Dong, N.; Zhu, Y.; Lu, Q.; Hu, L.; Zheng, Y.; Shao, F. Structurally distinct bacterial TBC-like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell 2012, 150, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Ao, X.; Zou, L.; Wu, Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014, 21, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.Z.; Jiang, A.J.; Mao, A.W.; Feng, Y.; Wang, W.; Li, J.; Zhang, X.; Xing, K.; Peng, X. The. J. Biol. Chem. 2018, 293, 9662–9673. [Google Scholar] [CrossRef] [Green Version]
- Romagnoli, A.; Etna, M.P.; Giacomini, E.; Pardini, M.; Remoli, M.E.; Corazzari, M.; Falasca, L.; Goletti, D.; Gafa, V.; Simeone, R.; et al. ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy 2012, 8, 1357–1370. [Google Scholar] [CrossRef] [Green Version]
- Raju, D.; Hussey, S.; Ang, M.; Terebiznik, M.R.; Sibony, M.; Galindo-Mata, E.; Gupta, V.; Blanke, S.R.; Delgado, A.; Romero-Gallo, J.; et al. Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology 2012, 142, 1160–1171. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Brumell, J.H. Bacteria-autophagy interplay: a battle for survival. Nat. Rev. Microbiol. 2014, 12, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, L.; Mostowy, S.; Sancho-Shimizu, V. Autophagy-Virus Interplay: From Cell Biology to Human Disease. Front. Cell Dev. Biol. 2018, 6, 155. [Google Scholar] [CrossRef] [Green Version]
- Staring, J.; von Castelmur, E.; Blomen, V.A.; van den Hengel, L.G.; Brockmann, M.; Baggen, J.; Thibaut, H.J.; Nieuwenhuis, J.; Janssen, H.; van Kuppeveld, F.J.; et al. PLA2G16 represents a switch between entry and clearance of Picornaviridae. Nature 2017, 541, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kim, M.J.; Sung, P.S.; Bae, Y.C.; Shin, E.C.; Yoo, J.Y. Interferon-inducible protein SCOTIN interferes with HCV replication through the autolysosomal degradation of NS5A. Nat. Commun. 2016, 7, 10631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection - a double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Jackson, W.T.; Giddings, T.H.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [Green Version]
- Guévin, C.; Manna, D.; Bélanger, C.; Konan, K.V.; Mak, P.; Labonté, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Tian, Y.; Ou, J.H. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog. 2015, 11, e1004764. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Jiang, X.; Liu, D.; Fan, Z.; Hu, X.; Yan, J.; Wang, M.; Gao, G.F. Autophagy is involved in influenza A virus replication. Autophagy 2009, 5, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Lu, K.; Mao, B.; Liu, S.; Trilling, M.; Huang, A.; Lu, M.; Lin, Y. The interplay between emerging human coronavirus infections and autophagy. Emerg. Microbes. Infect. 2021, 10, 196–205. [Google Scholar] [CrossRef]
- Carmona-Gutierrez, D.; Bauer, M.A.; Zimmermann, A.; Kainz, K.; Hofer, S.J.; Kroemer, G.; Madeo, F. Digesting the crisis: Autophagy and coronaviruses. Microb. Cell 2020, 7, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, K.; Xing, Y.; Tu, J.; Yang, X.; Zhao, Q.; Li, K.; Chen, Z. Coronavirus membrane-associated papain-like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell 2014, 5, 912–927. [Google Scholar] [CrossRef] [Green Version]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mösbauer, K.; Zellner, A.; et al. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat. Commun. 2019, 10, 5770. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Axford, E.; Klionsky, D.J. Highlights in the fight against COVID-19: Does autophagy play a role in SARS-CoV-2 infection? Autophagy 2020, 16, 2123–2127. [Google Scholar] [CrossRef]
- Brest, P.; Benzaquen, J.; Klionsky, D.J.; Hofman, P.; Mograbi, B. Open questions for harnessing autophagy-modulating drugs in the SARS-CoV-2 war: Hope or hype? Autophagy 2020, 16, 2267–2270. [Google Scholar] [CrossRef]
- Singh, K.; Chen, Y.C.; Hassanzadeh, S.; Han, K.; Judy, J.T.; Seifuddin, F.; Tunc, I.; Sack, M.N.; Pirooznia, M. Network Analysis and Transcriptome Profiling Identify Autophagic and Mitochondrial Dysfunctions in SARS-CoV-2 Infection. Front Genet. 2021, 12, 599261. [Google Scholar] [CrossRef] [PubMed]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef]
- Hayn, M.; Hirschenberger, M.; Koepke, L.; Nchioua, R.; Straub, J.H.; Klute, S.; Hunszinger, V.; Zech, F.; Prelli Bozzo, C.; Aftab, W.; et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021, 35, 109126. [Google Scholar] [CrossRef]
- Miao, G.; Zhao, H.; Li, Y.; Ji, M.; Chen, Y.; Shi, Y.; Bi, Y.; Wang, P.; Zhang, H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 2021, 56, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, H.; Pei, R.; Mao, B.; Zhao, Z.; Li, H.; Lin, Y.; Lu, K. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. 2021, 7, 31. [Google Scholar] [CrossRef]
- Qu, Y.; Wang, X.; Zhu, Y.; Wang, W.; Wang, Y.; Hu, G.; Liu, C.; Li, J.; Ren, S.; Xiao, M.Z.X.; et al. ORF3a-Mediated Incomplete Autophagy Facilitates Severe Acute Respiratory Syndrome Coronavirus-2 Replication. Front Cell. Dev. Biol. 2021, 9, 716208. [Google Scholar] [CrossRef]
- Wang, R.; Simoneau, C.R.; Kulsuptrakul, J.; Bouhaddou, M.; Travisano, K.A.; Hayashi, J.M.; Carlson-Stevermer, J.; Zengel, J.R.; Richards, C.M.; Fozouni, P.; et al. Genetic Screens Identify Host Factors for SARS-CoV-2 and Common Cold Coronaviruses. Cell 2021, 184, 106–119. [Google Scholar] [CrossRef]
- Daniloski, Z.; Jordan, T.X.; Wessels, H.H.; Hoagland, D.A.; Kasela, S.; Legut, M.; Maniatis, S.; Mimitou, E.P.; Lu, L.; Geller, E.; et al. Identification of Required Host Factors for SARS-CoV-2 Infection in Human Cells. Cell 2021, 184, 92–105. [Google Scholar] [CrossRef]
- Schneider, W.M.; Luna, J.M.; Hoffmann, H.H.; Sánchez-Rivera, F.J.; Leal, A.A.; Ashbrook, A.W.; Le Pen, J.; Ricardo-Lax, I.; Michailidis, E.; Peace, A.; et al. Genome-Scale Identification of SARS-CoV-2 and Pan-coronavirus Host Factor Networks. Cell 2021, 184, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.L.; Diamond, M.S. A Crisp(r) New Perspective on SARS-CoV-2 Biology. Cell 2021, 184, 15–17. [Google Scholar] [CrossRef]
- Yuen, C.K.; Wong, W.M.; Mak, L.F.; Wang, X.; Chu, H.; Yuen, K.Y.; Kok, K.H. Suppression of SARS-CoV-2 infection in ex-vivo human lung tissues by targeting class III phosphoinositide 3-kinase. J. Med. Virol. 2021, 93, 2076–2083. [Google Scholar] [CrossRef]
- Trimarco, J.D.; Heaton, B.E.; Chaparian, R.R.; Burke, K.N.; Binder, R.A.; Gray, G.C.; Smith, C.M.; Menachery, V.D.; Heaton, N.S. TMEM41B is a host factor required for the replication of diverse coronaviruses including SARS-CoV-2. PLoS Pathog. 2021, 17, e1009599. [Google Scholar] [CrossRef]
- Morita, K.; Hama, Y.; Izume, T.; Tamura, N.; Ueno, T.; Yamashita, Y.; Sakamaki, Y.; Mimura, K.; Morishita, H.; Shihoya, W.; et al. Genome-wide CRISPR screen identifies. J. Cell Biol. 2018, 217, 3817–3828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, F.; Bergman, P.; Dodgson, S.; Marcellin, D.; Claerr, I.; Goodwin, J.M.; DeJesus, R.; Kang, Z.; Antczak, C.; Begue, D.; et al. TMEM41B is a novel regulator of autophagy and lipid mobilization. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Shoemaker, C.J.; Huang, T.Q.; Weir, N.R.; Polyakov, N.J.; Schultz, S.W.; Denic, V. CRISPR screening using an expanded toolkit of autophagy reporters identifies TMEM41B as a novel autophagy factor. PLoS Biol. 2019, 17, e2007044. [Google Scholar] [CrossRef] [Green Version]
- Snijder, E.J.; Limpens, R.W.A.L.; de Wilde, A.H.; de Jong, A.W.M.; Zevenhoven-Dobbe, J.C.; Maier, H.J.; Faas, F.F.G.A.; Koster, A.J.; Bárcena, M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020, 18, e3000715. [Google Scholar] [CrossRef]
- Wolff, G.; Limpens, R.W.A.L.; Zevenhoven-Dobbe, J.C.; Laugks, U.; Zheng, S.; de Jong, A.W.M.; Koning, R.I.; Agard, D.A.; Grünewald, K.; Koster, A.J.; et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science 2020, 369, 1395–1398. [Google Scholar] [CrossRef]
- Mészáros, B.; Sámano-Sánchez, H.; Alvarado-Valverde, J.; Čalyševa, J.; Martínez-Pérez, E.; Alves, R.; Shields, D.C.; Kumar, M.; Rippmann, F.; Chemes, L.B.; et al. Short linear motif candidates in the cell entry system used by SARS-CoV-2 and their potential therapeutic implications. Sci. Signal 2021, 14, abd0334. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Hui, X.; Zhang, L.; Cao, L.; Huang, K.; Zhao, Y.; Zhang, Y.; Chen, X.; Lin, X.; Chen, M.; Jin, M. SARS-CoV-2 promote autophagy to suppress type I interferon response. Signal Transduct. Target Ther. 2021, 6, 180. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Li, Y.; Huang, F.; Luo, B.; Yuan, Y.; Xia, B.; Ma, X.; Yang, T.; Yu, F.; et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-I. Proc. Natl. Acad. Sci. USA 2021, 118, e2024202118. [Google Scholar] [CrossRef]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. β-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535. [Google Scholar] [CrossRef]
- Gassen, N.C.; Papies, J.; Bajaj, T.; Emanuel, J.; Dethloff, F.; Chua, R.L.; Trimpert, J.; Heinemann, N.; Niemeyer, C.; Weege, F.; et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat. Commun. 2021, 12, 3818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Vicario, C.; Alcaraz-Quiles, J.; García-Alonso, V.; Rius, B.; Hwang, S.H.; Titos, E.; Lopategi, A.; Hammock, B.D.; Arroyo, V.; Clària, J. Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: Role for omega-3 epoxides. Proc. Natl. Acad. Sci. USA 2015, 112, 536–541. [Google Scholar] [CrossRef] [Green Version]
- Kovsan, J.; Blüher, M.; Tarnovscki, T.; Klöting, N.; Kirshtein, B.; Madar, L.; Shai, I.; Golan, R.; Harman-Boehm, I.; Schön, M.R.; et al. Altered autophagy in human adipose tissues in obesity. J. Clin. Endocrinol. Metab. 2011, 96, E268–E277. [Google Scholar] [CrossRef] [PubMed]
- Kosacka, J.; Kern, M.; Klöting, N.; Paeschke, S.; Rudich, A.; Haim, Y.; Gericke, M.; Serke, H.; Stumvoll, M.; Bechmann, I.; et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol. Cell Endocrinol. 2015, 409, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Aijälä, M.; Malo, E.; Ukkola, O.; Bloigu, R.; Lehenkari, P.; Autio-Harmainen, H.; Santaniemi, M.; Kesäniemi, Y.A. Long-term fructose feeding changes the expression of leptin receptors and autophagy genes in the adipose tissue and liver of male rats: A possible link to elevated triglycerides. Genes Nutr. 2013, 8, 623–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.; Nguyen, H.Q.; Hwang, J.S.; Zada, S.; Lai, T.H.; Kang, S.S.; Kim, D.R. Systematic characterization of autophagy-related genes during the adipocyte differentiation using public-access data. Oncotarget 2018, 9, 15526–15541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baerga, R.; Zhang, Y.; Chen, P.H.; Goldman, S.; Jin, S. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy 2009, 5, 1118–1130. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; He, Y.; Okutsu, M.; Ong, L.C.; Jin, Y.; Zheng, L.; Chow, P.; Yu, S.; Zhang, M.; Yan, Z. Autophagy is involved in adipogenic differentiation by repressesing proteasome-dependent PPARγ2 degradation. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E530–E539. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Huang, J.X.; Liu, Y.; Li, X.; Zhou, S.R.; Qian, S.W.; Zhu, H.; Huang, H.Y.; Dang, Y.J.; Tang, Q.Q. Transactivation of Atg4b by C/EBPβ promotes autophagy to facilitate adipogenesis. Mol. Cell Biol. 2013, 33, 3180–3190. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Wang, X.; Jin, X.; Xue, Z.; Zhu, J.; Wang, C.; Jin, Y.; Fu, Z. β -Cypermethrin promotes the adipogenesis of 3T3-L1 cells via inducing autophagy and shaping an adipogenesis-friendly microenvironment. Acta Biochim. Biophys. Sin. 2020, 52, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Skop, V.; Cahova, M.; Dankova, H.; Papackova, Z.; Palenickova, E.; Svoboda, P.; Zidkova, J.; Kazdova, L. Autophagy inhibition in early but not in later stages prevents 3T3-L1 differentiation: Effect on mitochondrial remodeling. Differentiation 2014, 87, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Yamamuro, T.; Kawabata, T.; Fukuhara, A.; Saita, S.; Nakamura, S.; Takeshita, H.; Fujiwara, M.; Enokidani, Y.; Yoshida, G.; Tabata, K.; et al. Age-dependent loss of adipose Rubicon promotes metabolic disorders via excess autophagy. Nat. Commun. 2020, 11, 4150. [Google Scholar] [CrossRef]
- Zoico, E.; Rubele, S.; De Caro, A.; Nori, N.; Mazzali, G.; Fantin, F.; Rossi, A.; Zamboni, M. Brown and Beige Adipose Tissue and Aging. Front. Endocrinol. (Lausanne) 2019, 10, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altshuler-Keylin, S.; Shinoda, K.; Hasegawa, Y.; Ikeda, K.; Hong, H.; Kang, Q.; Yang, Y.; Perera, R.M.; Debnath, J.; Kajimura, S. Beige Adipocyte Maintenance Is Regulated by Autophagy-Induced Mitochondrial Clearance. Cell Metab. 2016, 24, 402–419. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Guo, Y.; Yuan, F.; Chen, S.; Yin, H.; Jiang, X.; Jiao, F.; Wang, F.; Ji, H.; Hu, G.; et al. Autophagy inhibition prevents glucocorticoid-increased adiposity via suppressing BAT whitening. Autophagy 2020, 16, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Altshuler-Keylin, S.; Wang, Q.; Chen, Y.; Henrique Sponton, C.; Ikeda, K.; Maretich, P.; Yoneshiro, T.; Kajimura, S. Mitophagy controls beige adipocyte maintenance through a Parkin-dependent and UCP1-independent mechanism. Sci. Signal 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.; Gottlieb, R.A. Parkin-mediated mitophagy is downregulated in browning of white adipose tissue. Obesity 2017, 25, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Cairó, M.; Villarroya, J.; Cereijo, R.; Campderrós, L.; Giralt, M.; Villarroya, F. Thermogenic activation represses autophagy in brown adipose tissue. Int. J. Obes. 2016, 40, 1591–1599. [Google Scholar] [CrossRef]
- Ro, S.H.; Jang, Y.; Bae, J.; Kim, I.M.; Schaecher, C.; Shomo, Z.D. Autophagy in Adipocyte Browning: Emerging Drug Target for Intervention in Obesity. Front Physiol. 2019, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.M.; Lim, H.; Hur, K.Y.; Quan, W.; Lee, H.Y.; Cheon, H.; Ryu, D.; Koo, S.H.; Kim, H.L.; Kim, J.; et al. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat. Commun. 2014, 5, 4934. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Takahashi, Y.; Desai, N.; Zhang, J.; Serfass, J.M.; Shi, Y.G.; Lynch, C.J.; Wang, H.G. Bif-1 deficiency impairs lipid homeostasis and causes obesity accompanied by insulin resistance. Sci. Rep. 2016, 6, 20453. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Wei, Y.; Sun, K.; Li, B.; Dong, X.; Zou, Z.; Liu, Y.; Kinch, L.N.; Khan, S.; Sinha, S.; et al. Beclin 2 functions in autophagy, degradation of G protein-coupled receptors, and metabolism. Cell 2013, 154, 1085–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, Á.; Bárcena, C.; Martínez-García, G.G.; Tamargo-Gómez, I.; Suárez, M.F.; Pietrocola, F.; Castoldi, F.; Esteban, L.; Sierra-Filardi, E.; Boya, P.; et al. Autophagy couteracts weight gain, lipotoxicity and pancreatic β-cell death upon hypercaloric pro-diabetic regimens. Cell. Death. Dis. 2017, 8, e2970. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S.; Kuramoto, K.; Wang, N.; Situ, X.; Priyadarshini, M.; Zhang, W.; Cordoba-Chacon, J.; Layden, B.T.; He, C. Autophagy Differentially Regulates Insulin Production and Insulin Sensitivity. Cell. Rep. 2018, 23, 3286–3299. [Google Scholar] [CrossRef]
- Zhang, Y.; Goldman, S.; Baerga, R.; Zhao, Y.; Komatsu, M.; Jin, S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 19860–19865. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J.Clin. Invest. 2009, 119, 3329–3339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wu, R.; Liu, Y.; Zhao, Y.; Bi, Z.; Yao, Y.; Liu, Q.; Shi, H.; Wang, F.; Wang, Y. m6A mRNA methylation controls autophagy and adipogenesis by targeting Atg5 and Atg7. Autophagy 2020, 16, 1221–1235. [Google Scholar] [CrossRef]
- Cai, J.; Pires, K.M.; Ferhat, M.; Chaurasia, B.; Buffolo, M.A.; Smalling, R.; Sargsyan, A.; Atkinson, D.L.; Summers, S.A.; Graham, T.E.; et al. Autophagy Ablation in Adipocytes Induces Insulin Resistance and Reveals Roles for Lipid Peroxide and Nrf2 Signaling in Adipose-Liver Crosstalk. Cell. Rep. 2018, 25, 1708–1717. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Ji, Y.; Song, Y.; Choe, S.S.; Jeon, Y.G.; Na, H.; Nam, T.W.; Kim, H.J.; Nahmgoong, H.; Kim, S.M.; et al. Depletion of Adipocyte. Diabetes 2021, 70, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Jaber, N.; Dou, Z.; Chen, J.S.; Catanzaro, J.; Jiang, Y.P.; Ballou, L.M.; Selinger, E.; Ouyang, X.; Lin, R.Z.; Zhang, J.; et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc. Natl. Acad. Sci. USA 2012, 109, 2003–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyagi, K.; Ohara-Imaizumi, M.; Itakura, M.; Torii, S.; Akimoto, Y.; Nishiwaki, C.; Nakamichi, Y.; Kishimoto, T.; Kawakami, H.; Harada, A.; et al. VAMP7 Regulates Autophagy to Maintain Mitochondrial Homeostasis and to Control Insulin Secretion in Pancreatic β-Cells. Diabetes 2016, 65, 1648–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, W.; Kim, H.K.; Moon, E.Y.; Kim, S.S.; Choi, C.S.; Komatsu, M.; Jeong, Y.T.; Lee, M.K.; Kim, K.W.; Kim, M.S.; et al. Role of hypothalamic proopiomelanocortin neuron autophagy in the control of appetite and leptin response. Endocrinology 2012, 153, 1817–1826. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Kim, J.; Quan, W.; Lee, J.C.; Kim, M.S.; Kim, S.H.; Bae, J.W.; Hur, K.Y.; Lee, M.S. Autophagy deficiency in myeloid cells increases susceptibility to obesity-induced diabetes and experimental colitis. Autophagy 2016, 12, 1390–1403. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers. 2015, 1, 15019. [Google Scholar] [CrossRef]
- Yao, D.; GangYi, Y.; QiNan, W. Autophagic dysfunction of β cell dysfunction in type 2 diabetes, a double-edged sword. Genes Dis. 2021, 8, 438–447. [Google Scholar] [CrossRef]
- Shigihara, N.; Fukunaka, A.; Hara, A.; Komiya, K.; Honda, A.; Uchida, T.; Abe, H.; Toyofuku, Y.; Tamaki, M.; Ogihara, T.; et al. Human IAPP-induced pancreatic β cell toxicity and its regulation by autophagy. J. Clin. Invest. 2014, 124, 3634–3644. [Google Scholar] [CrossRef]
- Rivera, J.F.; Costes, S.; Gurlo, T.; Glabe, C.G.; Butler, P.C. Autophagy defends pancreatic β cells from human islet amyloid polypeptide-induced toxicity. J. Clin. Investig. 2014, 124, 3489–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Park, K.; Kim, M.J.; Lim, H.; Kim, K.H.; Kim, S.W.; Lee, E.S.; Kim, H.H.; Kim, S.J.; Hur, K.Y.; et al. An autophagy enhancer ameliorates diabetes of human IAPP-transgenic mice through clearance of amyloidogenic oligomer. Nat. Commun. 2021, 12, 183. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.J.; Wu, J.H.; Sun, S.Y.; Zhou, J.Q. The endoplasmic reticulum stress/autophagy pathway is involved in cholesterol-induced pancreatic β-cell injury. Sci. Rep. 2017, 7, 44746. [Google Scholar] [CrossRef] [Green Version]
- Bugliani, M.; Mossuto, S.; Grano, F.; Suleiman, M.; Marselli, L.; Boggi, U.; De Simone, P.; Eizirik, D.L.; Cnop, M.; Marchetti, P.; et al. Modulation of Autophagy Influences the Function and Survival of Human Pancreatic Beta Cells Under Endoplasmic Reticulum Stress Conditions and in Type 2 Diabetes. Front Endocrinol. 2019, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Quan, W.; Hur, K.Y.; Lim, Y.; Oh, S.H.; Lee, J.C.; Kim, K.H.; Kim, G.H.; Kim, S.W.; Kim, H.L.; Lee, M.K.; et al. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia 2012, 55, 392–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leloup, C.; Tourrel-Cuzin, C.; Magnan, C.; Karaca, M.; Castel, J.; Carneiro, L.; Colombani, A.L.; Ktorza, A.; Casteilla, L.; Pénicaud, L. Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes 2009, 58, 673–681. [Google Scholar] [CrossRef] [Green Version]
- Sidarala, V.; Pearson, G.L.; Parekh, V.S.; Thompson, B.; Christen, L.; Gingerich, M.A.; Zhu, J.; Stromer, T.; Ren, J.; Reck, E.C.; et al. Mitophagy protects β cells from inflammatory damage in diabetes. JCI Insight 2020, 5, e141138. [Google Scholar] [CrossRef]
- Bhansali, S.; Bhansali, A.; Walia, R.; Saikia, U.N.; Dhawan, V. Alterations in Mitochondrial Oxidative Stress and Mitophagy in Subjects with Prediabetes and Type 2 Diabetes Mellitus. Front Endocrinol. 2017, 8, 347. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121. [Google Scholar] [CrossRef] [Green Version]
- Hernández, M.G.; Aguilar, A.G.; Burillo, J.; Oca, R.G.; Manca, M.A.; Novials, A.; Alcarraz-Vizan, G.; Guillén, C.; Benito, M. Pancreatic β cells overexpressing hIAPP impaired mitophagy and unbalanced mitochondrial dynamics. Cell Death Dis. 2018, 9, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soleimanpour, S.A.; Ferrari, A.M.; Raum, J.C.; Groff, D.N.; Yang, J.; Kaufman, B.A.; Stoffers, D.A. Diabetes Susceptibility Genes Pdx1 and Clec16a Function in a Pathway Regulating Mitophagy in β-Cells. Diabetes 2015, 64, 3475–3484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, K.; Polonsky, K.S. Pdx1 and other factors that regulate pancreatic beta-cell survival. Diabetes Obes. Metab. 2009, 11 (Suppl. 4), 30–37. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Liu, T.; Sun, Y.; Liu, X.; Xiong, R.; Li, H.; Li, Z.; Zhang, Z.; Tian, Z.; Tian, Y. Sonodynamic therapy inhibits palmitate-induced beta cell dysfunction via PINK1/Parkin-dependent mitophagy. Cell Death Dis. 2019, 10, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muralidharan, C.; Conteh, A.M.; Marasco, M.R.; Crowder, J.J.; Kuipers, J.; de Boer, P.; Linnemann, A.K. Pancreatic beta cell autophagy is impaired in type 1 diabetes. Diabetologia 2021, 64, 865–877. [Google Scholar] [CrossRef]
- Liu, Y.; Palanivel, R.; Rai, E.; Park, M.; Gabor, T.V.; Scheid, M.P.; Xu, A.; Sweeney, G. Adiponectin stimulates autophagy and reduces oxidative stress to enhance insulin sensitivity during high-fat diet feeding in mice. Diabetes 2015, 64, 36–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zhu, Y.Y.; Wang, L.; Teng, T.; Zhou, M.; Wang, S.G.; Tian, Y.Z.; Du, L.; Yin, X.X.; Sun, Y. Mangiferin ameliorates fatty liver via modulation of autophagy and inflammation in high-fat-diet induced mice. Biomed. Pharmacother. 2017, 96, 328–335. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Sha, S.; Sun, L.; Zhang, J.; Dong, M. GLP-1 analogue improves hepatic lipid accumulation by inducing autophagy via AMPK/mTOR pathway. Biochem. Biophys. Res. Commun. 2016, 476, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Lim, Y.M.; Kim, K.H.; Jeon, Y.E.; Park, K.; Kim, J.; Hwang, H.Y.; Lee, D.J.; Pagire, H.; Kwon, H.J.; et al. A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes. Nat. Commun. 2018, 9, 1438. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Zhou, Z.C.; Niu, R.; Su, Y.T.; Sun, Y.; Liu, H.L.; Teng, J.L.; Ye, J.N.; Shi, H.; Yang, C.D.; et al. Hydroxychloroquine Improves Obesity-Associated Insulin Resistance and Hepatic Steatosis by Regulating Lipid Metabolism. Front. Pharmacol. 2019, 10, 855. [Google Scholar] [CrossRef] [Green Version]
- Wasko, M.C.; McClure, C.K.; Kelsey, S.F.; Huber, K.; Orchard, T.; Toledo, F.G. Antidiabetogenic effects of hydroxychloroquine on insulin sensitivity and beta cell function: A randomised trial. Diabetologia 2015, 58, 2336–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kounakis, K.; Chaniotakis, M.; Markaki, M.; Tavernarakis, N. Emerging Roles of Lipophagy in Health and Disease. Front. Cell Dev. Biol. 2019, 7, 185. [Google Scholar] [CrossRef]
- Fu, Y.; Chen, N.; Wang, Z.; Luo, S.; Ding, Y.; Lu, B. Degradation of lipid droplets by chimeric autophagy-tethering compounds. Cell Res. 2021, 31, 965–979. [Google Scholar] [CrossRef]
- Baumeier, C.; Kaiser, D.; Heeren, J.; Scheja, L.; John, C.; Weise, C.; Eravci, M.; Lagerpusch, M.; Schulze, G.; Joost, H.G.; et al. Caloric restriction and intermittent fasting alter hepatic lipid droplet proteome and diacylglycerol species and prevent diabetes in NZO mice. Biochim. Biophys. Acta 2015, 1851, 566–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anson, R.M.; Guo, Z.; de Cabo, R.; Iyun, T.; Rios, M.; Hagepanos, A.; Ingram, D.K.; Lane, M.A.; Mattson, M.P. Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake. Proc. Natl. Acad. Sci. USA 2003, 100, 6216–6220. [Google Scholar] [CrossRef] [Green Version]
- Sutton, E.F.; Beyl, R.; Early, K.S.; Cefalu, W.T.; Ravussin, E.; Peterson, C.M. Early Time-Restricted Feeding Improves Insulin Sensitivity, Blood Pressure, and Oxidative Stress Even without Weight Loss in Men with Prediabetes. Cell Metab. 2018, 27, 1212–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, S.; Seok, S.; Kim, Y.C.; Zhang, Y.; Yau, P.; Iwamori, N.; Xu, H.E.; Ma, J.; Kemper, B.; Kemper, J.K. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat. Commun. 2020, 11, 807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Javaheri, A.; Godar, R.J.; Murphy, J.; Ma, X.; Rohatgi, N.; Mahadevan, J.; Hyrc, K.; Saftig, P.; Marshall, C.; et al. Intermittent fasting preserves beta-cell mass in obesity-induced diabetes via the autophagy-lysosome pathway. Autophagy 2017, 13, 1952–1968. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Tarabra, E.; Toledo, M.; Garcia-Macia, M.; Sahu, S.; Coletto, L.; Batista-Gonzalez, A.; Barzilai, N.; Pessin, J.E.; Schwartz, G.J.; et al. System-wide Benefits of Intermeal Fasting by Autophagy. Cell Metab. 2017, 26, 856–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
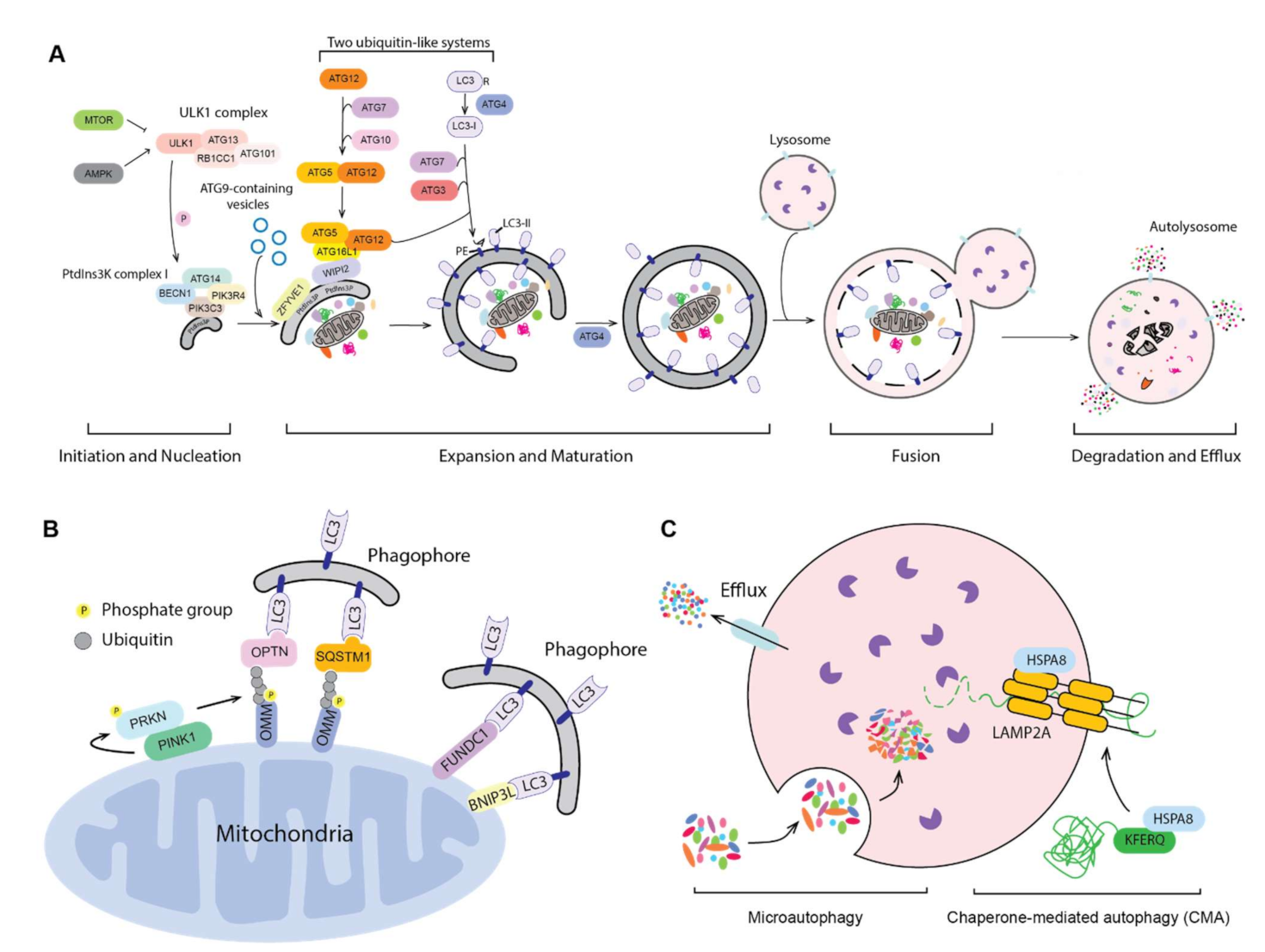
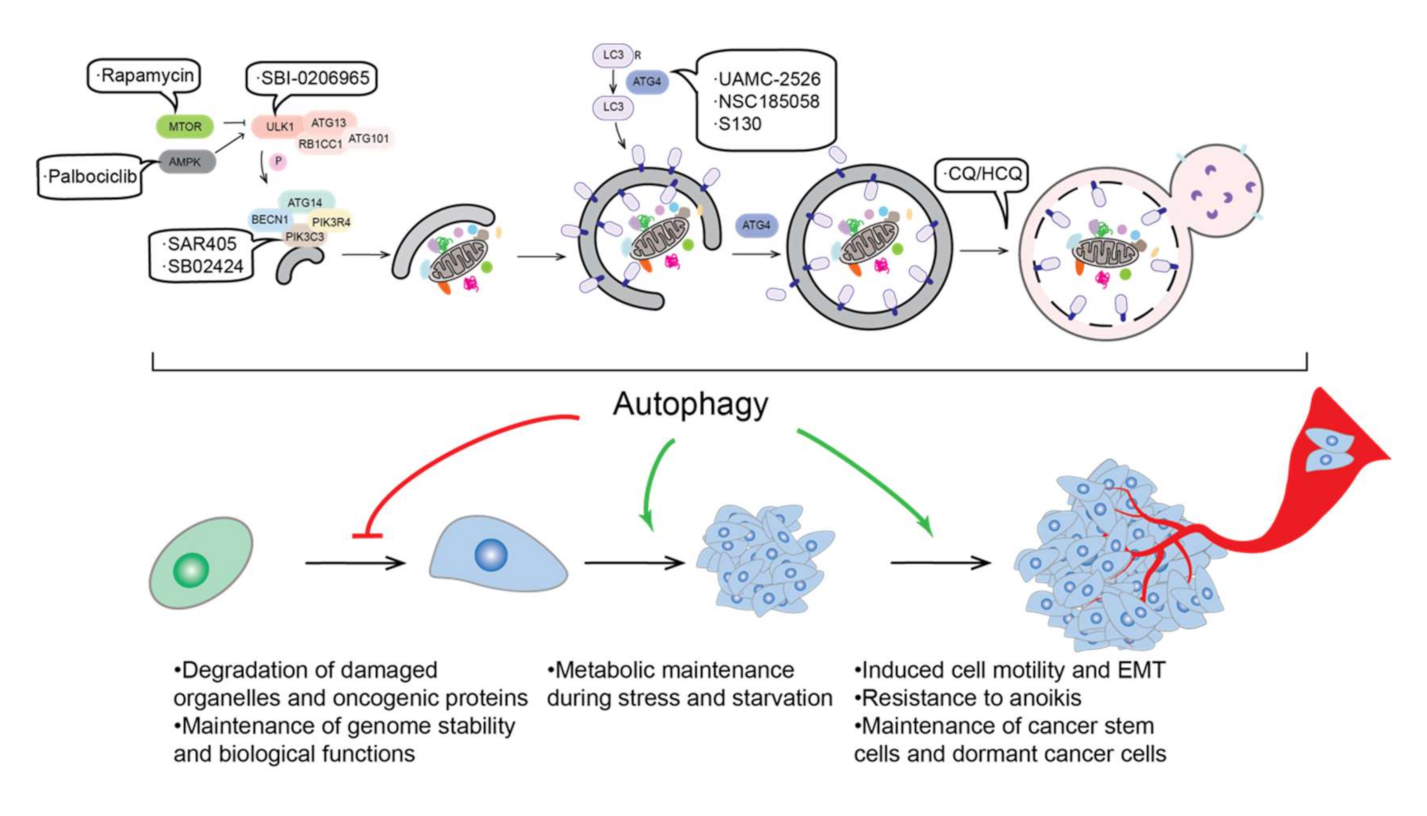
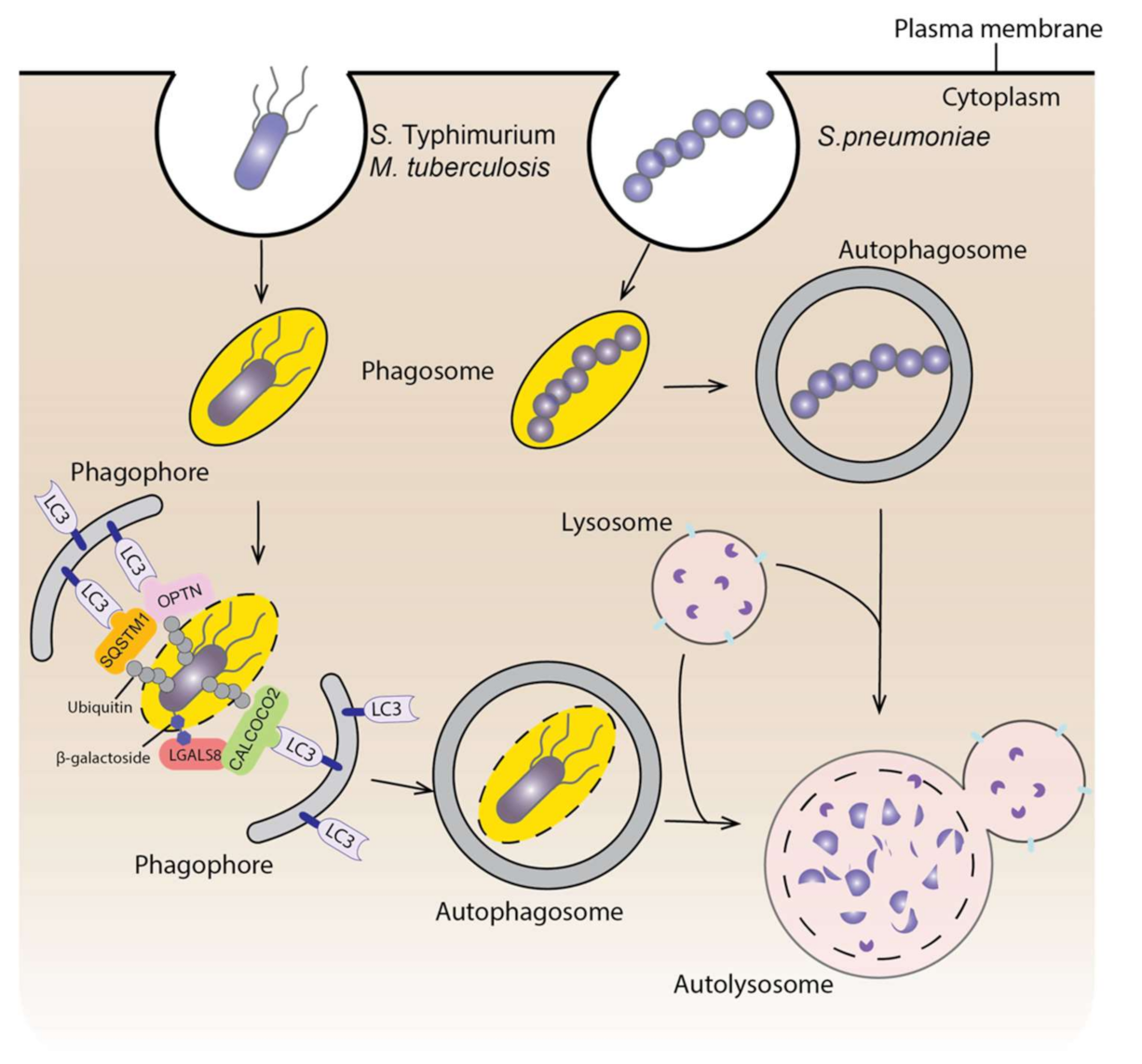
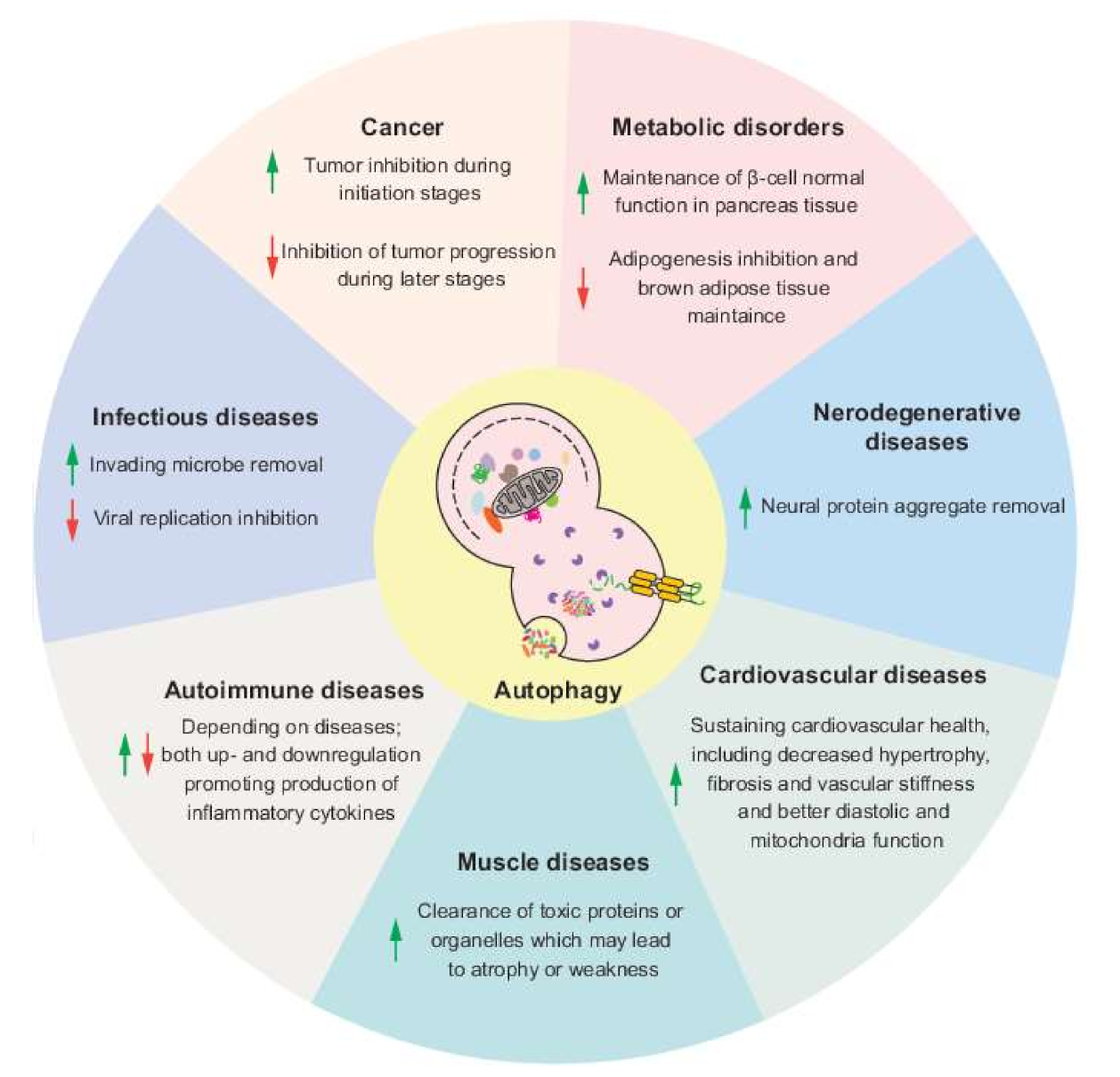

{kind=link}
{kind=link}
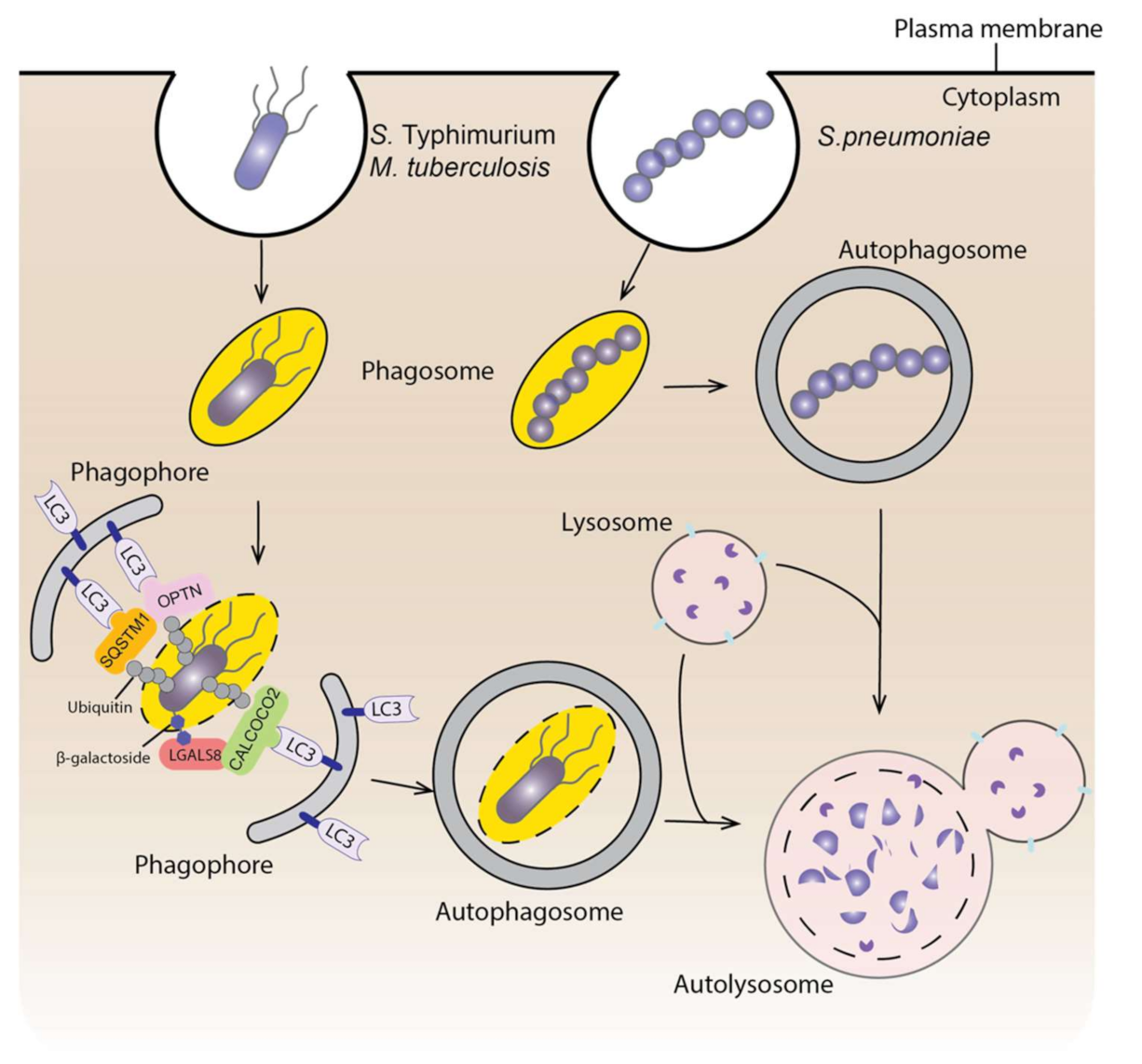{kind=link}
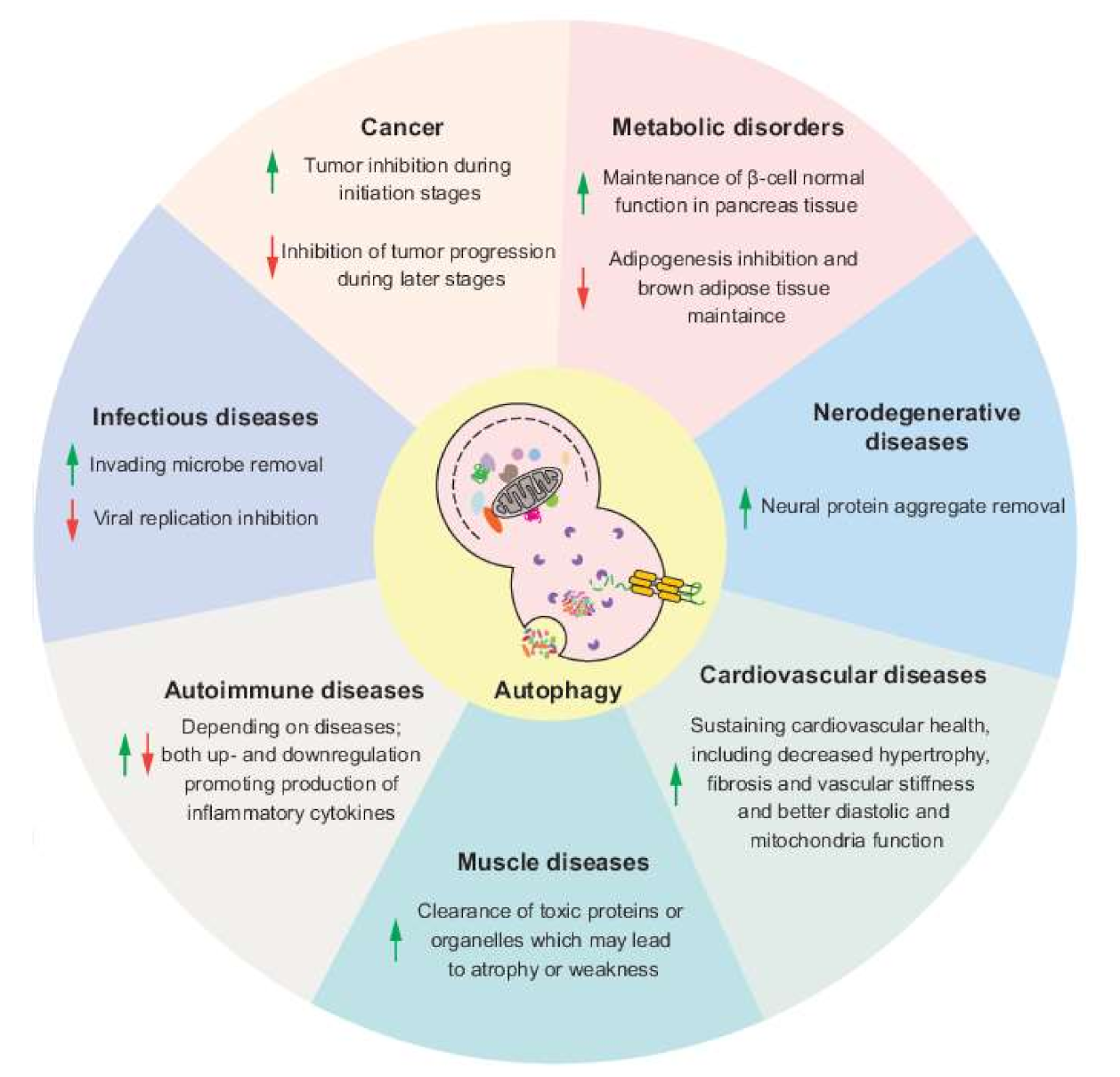{kind=link}
| Gene Name | Description | Reference |
|---|---|---|
| GBA | Loss of GBA function impairs autophagy via PPP2/PP2A inactivation. PD-associated mutation L444P heterozygote impairs autophagy, mitochondria priming and autophagy-lysosome degradation. | [214,215] |
| VPS13C | Deletion of VPS13C is correlated with impaired mitochondrial morphology and upregulate PINK1-PRKN-dependent mitophagy, but the study does not show the connection between PD-associated mutations with mitophagy. | [216] |
| VPS35 | VPS35D620N causes autosomal-dominant Parkinson disease. VPS35D620N has a reduced affinity for WASH and impairs ATG9A trafficking and localization, thus compromising autophagosome formation. | [217] |
| VPS35D620N impairs endosome-to-Golgi retrieval of LAMP2A and accelerates LAMP2A degradation, thus inhibiting SNCA degradation through CMA. | [218] | |
| VPS35D620N hampers PINK1 and PRKN recruitment to mitochondria thus impairing mitophagy. | [219] | |
| PARK7 | PARK7 knockdown impairs autophagy and the SNCA uptake and degradation in microglia. | [220] |
| PARK7 deficiency downregulates HSPA8 expression level and accelerates the degradation of LAMP2A, inhibiting SNCA degradation through CMA. | [221] | |
| Park7 may function in mitophagy because it is important for proper mitochondria function and Park7 upregulation can rescue the phenotype in pink1 mutant Drosophila. | [222] | |
| SREBF1 | SREBF1 knockdown inhibits PRKN translocation to mitochondria, thus inhibiting mitophagy. | [223] |
| FBXO7 | FBXO7T22M inhibits its interaction with PRKN and impairs PRKN translocation to mitochondria. | [224] |
| FBXO7R378G mutation impairs ubiquitination of MFN1. | ||
| FBXO7R498X truncation inhibits PRKN recruitment to mitochondria. | ||
| T22M, R378G and R498X mutations aggravate aggregation of FBXO7 in mitochondria, which may inhibit mitophagy. | [225] | |
| TMEM175 | TMEM175 deficiency leads to the impaired autophagosome degradation in the lysosome. | [226] |
| TMEM175M393T shows similar autophagosome clearance phenotype as a knockout. | [227] |
| Genotype | Target Organ | Modulation on Autophagy | Model | Phenotype | Reference |
|---|---|---|---|---|---|
| Atg7+/− | Whole body | Suppression | Cross with ob/ob mice | Resistance to insulin. Increased intracellular lipid content. | [371] |
| sh3glb1−/− | Whole body | Suppression | HFD or NCD | Higher body weight under both diets. Insulin resistance under HFD. | [372] |
| Becn2+/− | Whole body | Suppression | HFD or NCD | Obesity. Insulin resistance. | [373] |
| atg4b−/− | Whole body | Suppression | Fasting, HFD or sucrose fed | Reduced starvation-induced weight loss. Larger adipocytes in visceral fat tissue and increased hepatic steatosis. Reduced glucose tolerance and attenuated insulin sensitivity. | [374] |
| Atg5 overexpression | Whole body | Enhancement | NCD and aging | Lower body weight. Enhanced insulin sensitivity. Resistance to age-associated obesity. | [375] |
| Becn1F121Aknockin | Whole body | Enhancement | HFD | Impaired glucose tolerance but improved insulin sensitivity. | [376] |
| atg7−/− | Adipose tissue | Suppression | HFD or NCD | Lower body weight. Lower white adipose tissue mass. Resistance to HFD-induced obesity. Enhanced insulin sensitivity. | [377,378] |
| fto−/− | Adipose tissue | Suppression | HFD or NCD | Lower white fat mass. Lower body weight. | [379] |
| atg3−/− | Adipose tissue | Suppression | HFD or NCD | Induced insulin resistance. | [380] |
| atg16l1−/− | Adipose tissue | Suppression | NCD | Induced insulin resistance. | [380] |
| becn1−/− | Adipose tissue | Suppression | HFD or NCD | Adipose tissue inflammation. Lower white adipose tissue mass. Lower body weight when fed with HFD. | [381] |
| rubcn−/− | Adipose tissue | Enhancement | NCD | Lower body weight. Lower adipose tissue mass. Glucose intolerance. | [363] |
| Atg7 knockdown | Liver | Suppression | NCD | Increased insulin resistance. | [352] |
| tfeb−/− | Liver | Suppression | HFD | Large and pale liver filled with lipid vacuoles. Impaired lipid degradation. | [382] |
| pik3c3−/− | Liver | Suppression | NCD | Lack of glycogen deposits in liver. Accumulation of lipid in liver. | [383] |
| atg7−/− | Pancreas | Suppression | HFD or NCD | Impaired glucose tolerance. Decreased serum insulin level. | [384] |
| vma7−/− | Pancreas | Suppression | HFD or NCD | Impaired insulin secretion. Glucose intolerance on HFD. | [385] |
| atg7−/− | Skeletal muscle | Suppression | HFD or NCD | Lower body weight on NCD. Enhanced lipid catabolism and browning of white adipose tissue when fed with HFD. Protected from HFD-induced obesity and insulin resistance. | [386] |
| atg7−/− | Hypothalamic POMC (proopiomelanocortin) neurons | Suppression | HFD or NCD | Higher body weight on NCD. Higher blood glucose level on HFD. | [387] |
| atg7−/− | Myeloid cells | Suppression | Cross with ob/ob mice | Insulin resistance. Enhanced adipose tissue inflammation. | [388] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, Y.; Klionsky, D.J. The Emerging Roles of Autophagy in Human Diseases. Biomedicines 2021, 9, 1651. https://doi.org/10.3390/biomedicines9111651
Lei Y, Klionsky DJ. The Emerging Roles of Autophagy in Human Diseases. Biomedicines. 2021; 9(11):1651. https://doi.org/10.3390/biomedicines9111651
Chicago/Turabian StyleLei, Yuchen, and Daniel J. Klionsky. 2021. "The Emerging Roles of Autophagy in Human Diseases" Biomedicines 9, no. 11: 1651. https://doi.org/10.3390/biomedicines9111651
APA StyleLei, Y., & Klionsky, D. J. (2021). The Emerging Roles of Autophagy in Human Diseases. Biomedicines, 9(11), 1651. https://doi.org/10.3390/biomedicines9111651