
A UHPLC–MS/MS Method for Therapeutic Drug Monitoring of Aciclovir and Ganciclovir in Plasma and Dried Plasma Spots

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Calibration Curve, Quality Control, and Stock Solution Preparation
2.3. Human Samples
2.4. Sample Preparation
2.4.1. Extraction from Plasma
2.4.2. Extraction from Dried Plasma Spots (DPS)
2.5. Chromatographic Conditions
2.6. MS/MS Conditions
2.7. Method Validation
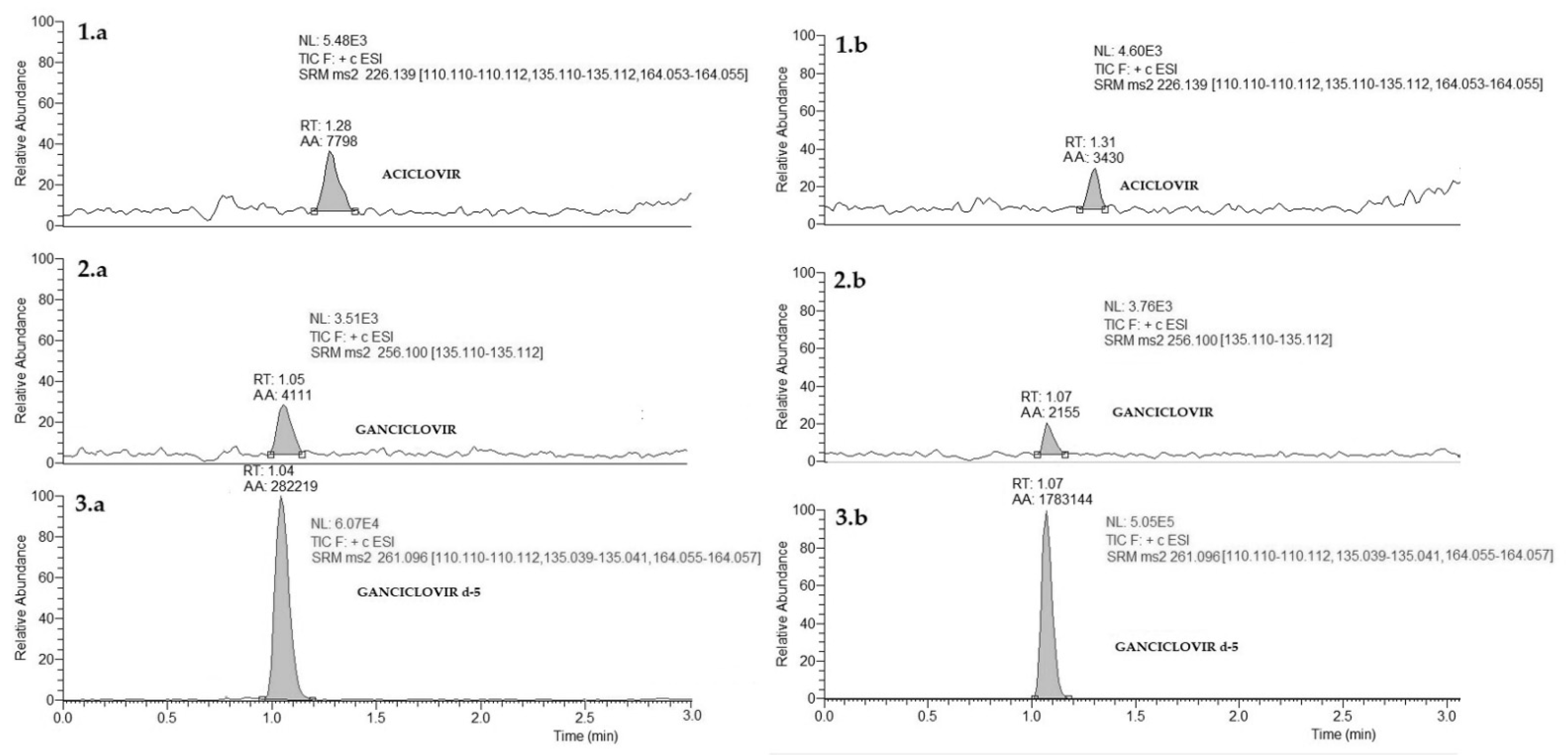
2.7.1. Selectivity
2.7.2. Carry-Over
2.7.3. Matrix Effects and Extraction Recoveries
2.7.4. Linearity
2.7.5. Precision, Accuracy, and LLOQ
2.7.6. Stability
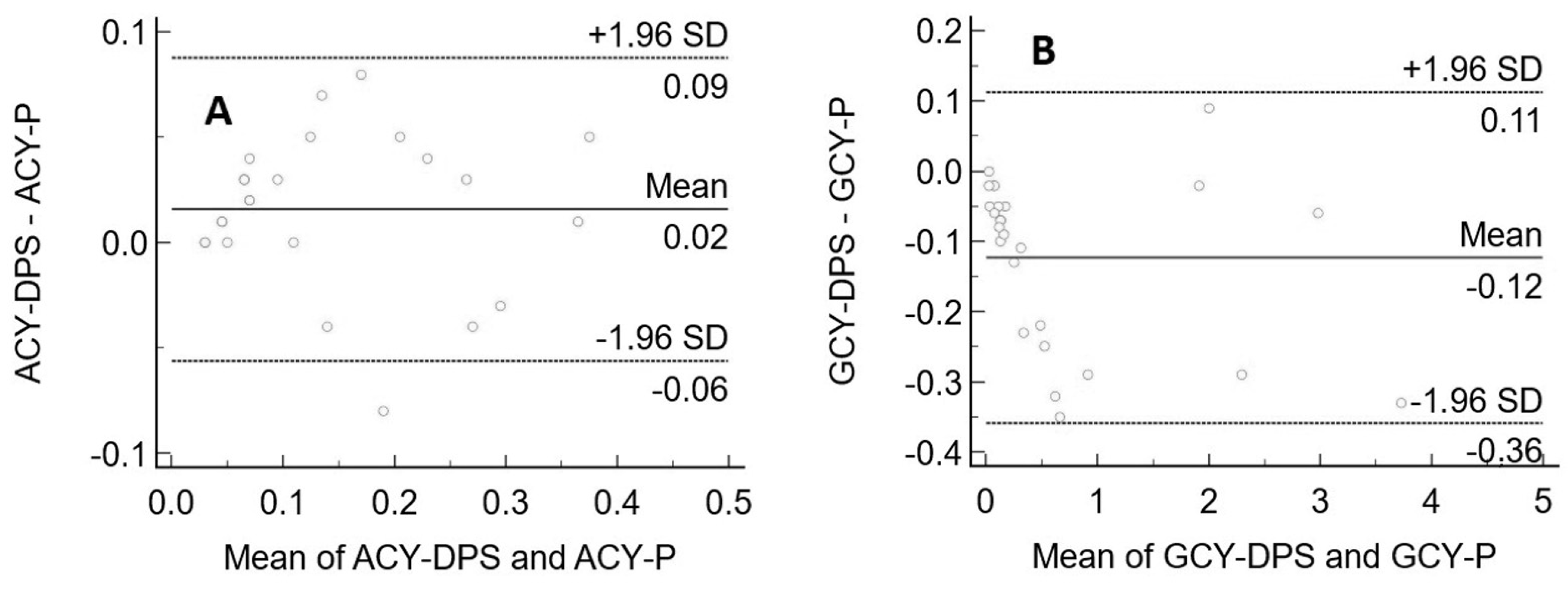
2.8. Statistical Analyses
3. Results
3.1. Methods Development
3.2. Method Validation
3.3. Clinical Application and Method Comparison
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abdul-Aziz, M.H.; Alffenaar, J.W.C.; Bassetti, M.; Bracht, H.; Dimopoulos, G.; Marriott, D.; Neely, M.N.; Paiva, J.A.; Pea, F.; Sjovall, F.; et al. Antimicrobial therapeutic drug monitoring in critically ill adult patients: A Position Paper#. Intensive Care Med. 2020, 46, 1127–1153. [Google Scholar] [CrossRef]
- Roberts, J.A.; Abdul-Aziz, M.H.; Lipman, J.; Mouton, J.W.; Vinks, A.A.; Felton, T.W.; Hope, W.W.; Farkas, A.; Neely, M.N.; Schentag, J.J.; et al. Individualised antibiotic dosing for patients who are critically ill: Challenges and potential solutions. Lancet Infect. Dis. 2014, 14, 498–509. [Google Scholar] [CrossRef] [Green Version]
- Tabah, A.; de Waele, J.; Lipman, J.; Zahar, J.R.; Cotta, M.O.; Barton, G.; Timsit, J.F.; Roberts, J.A. The ADMIN-ICU survey: A survey on antimicrobial dosing and monitoring in ICUs. J. Antimicrob. Chemother. 2015, 70, 2671–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, G.; Brinkman, A.; Benefield, R.J.; Carlier, M.; De Waele, J.J.; El Helali, N.; Frey, O.; Harbarth, S.; Huttner, A.; McWhinney, B.; et al. An international, multicentre survey of β-lactam antibiotic therapeutic drug monitoring practice in intensive care units. J. Antimicrob. Chemother. 2014, 69, 1416–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustinetti, G.; Cangemi, G.; Bandettini, R.; Castagnola, E. Pharmacokinetic/pharmacodynamic parameters for treatment optimization of infection due to antibiotic resistant bacteria: A summary for practical purposes in children and adults. J. Chemother. 2018, 30, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Reitano, M.; Tyring, S.; Lang, W.; Thoming, C.; Worm, A.M.; Borelli, S.; Chambers, L.O.; Robinson, J.M.; Corey, L. Valaciclovir for the suppression of recurrent genital herpes simplex virus infection: A large-scale dose range-finding study. J. Infect. Dis. 1998, 178, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Corr Guideline on Bioanalytical Method Validation. 1922, 44, pp. 1–23. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 24 August 2021).
- Gu, H.; Liu, G.; Wang, J.; Aubry, A.F.; Arnold, M.E. Selecting the correct weighting factors for linear and quadratic calibration curves with least-squares regression algorithm in bioanalytical LC-MS/MS assays and impacts of using incorrect weighting factors on curve stability, data quality, and assay perfo. Anal. Chem. 2014, 86, 8959–8966. [Google Scholar] [CrossRef]
- Passing, H.; Bablok, W. Comparison of Several Regression Procedures for Method Comparison Studies and Determination of Sample Sizes Application of linear regression procedures for method comparison studies in Clinical Chemistry, Part II. Clin. Chem. Lab. Med. 2009, 22, 431–445. [Google Scholar] [CrossRef] [Green Version]
- Martin Bland, J.; Altman, D.G. Statistical Methods for Assessing Agreement Between Two Methods of Clinical Measurement. Lancet 1986, 327, 307–310. [Google Scholar] [CrossRef]
- Rigo-Bonnin, R.; Padullés, A.; Corral-Comesaña, S.; Cerezo, G.; Grinyó, J.M.; Colom, H.; Alía-Ramos, P.; Lloberas, N. Measurement of ganciclovir concentration in human plasma by ultra-performance liquid chromatography-tandem mass spectrometry. Clin. Chim. Acta 2014, 427, 58–64. [Google Scholar] [CrossRef]
- Spruance, S.L.; Tyring, S.K.; DeGregorio, B.; Miller, C.; Beutner, K. A large-scale, placebo-controlled, dose-ranging trial of peroral valaciclovir for episodic treatment of recurrent herpes genitalis. Arch. Intern. Med. 1996, 156, 1729–1735. [Google Scholar] [CrossRef]
- Weller, S.; Blum, M.R.; Doucette, M.; Burnette, T.; Cederberg, D.M.; De Miranda, P.; Smiley, M.L. Pharmacolunetics of the acvclovir pro- drug valaciclovir after iscalati and multiple-dose adrninis tration normal volunteers. Clin. Pharmacol. Ther. 2000, 595–605. [Google Scholar]
- Lisboa, L.F.; Tong, Y.; Kumar, D.; Pang, X.L.; Åsberg, A.; Hartmann, A.; Rollag, H.; Jardine, A.G.; Pescovitz, M.D.; Humar, A. Analysis and clinical correlation of genetic variation in cytomegalovirus. Transpl. Infect. Dis. 2012, 14, 132–140. [Google Scholar] [CrossRef]
- Lombardi, G.; Garofoli, F.; Villani, P.; Tizzoni, M.; Angelini, M.; Cusato, M.; Bollani, L.; De Silvestri, A.; Regazzi, M.; Stronati, M. Oral valganciclovir treatment in newborns with symptomatic congenital cytomegalovirus infection. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 1465–1470. [Google Scholar] [CrossRef]
- Luck, S.; Lovering, A.; Griffiths, P.; Sharland, M. Ganciclovir treatment in children: Evidence of subtherapeutic levels. Int. J. Antimicrob. Agents 2011, 37, 445–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinig, K.; Wirz, T.; Gajate-Perez, A.; Belli, S. Determination of Ganciclovir and its prodrug Valganciclovir by hydrophilic interaction liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Kanneti, R.; Rajesh, R.; Aravinda Raj, J.R.; Bhatt, P.A. An LC-MS-MS method for the simultaneous quantitation of acyclovir and valacyclovir in human plasma. Chromatographia 2009, 70, 407–414. [Google Scholar] [CrossRef]
- Yadav, M.; Upadhyay, V.; Singhal, P.; Goswami, S.; Shrivastav, P.S. Stability evaluation and sensitive determination of antiviral drug, valacyclovir and its metabolite acyclovir in human plasma by a rapid liquid chromatography-tandem mass spectrometry method. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 680–688. [Google Scholar] [CrossRef]
- Weller, D.R.; Balfour, H.H.; Vezina, H.E. Simultaneous determination of acyclovir, ganciclovir, and (R)-9-[4-hydroxy-2-(hydroxymethyl)butyl]guanine in human plasma using high-performance liquid chromatography. Biomed. Chromatogr. 2009, 23, 822–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Lanuti, M.; Lambright, E.S.; Force, S.D.; Albelda, S.M.; Blair, I.A. A rapid and sensitive method for the quantification of ganciclovir in plasma using liquid chromatography/selected reaction monitoring/mass spectrometry. Biomed. Chromatogr. 2000, 14, 93–98. [Google Scholar] [CrossRef]
- Xu, H.R.; Li, X.N.; Chen, W.L.; Liu, G.Y.; Chu, N.N.; Yu, C. A sensitive assay for simultaneous determination of plasma concentrations of valganciclovir and its active metabolite ganciclovir by LC/MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 848, 329–334. [Google Scholar] [CrossRef]
- Rower, J.E.; Nielson, C.; Shi, K.; Park, A.H. Validation of an assay for quantifying ganciclovir in dried blood spots. J. Pharm. Biomed. Anal. 2020, 184, 113181. [Google Scholar] [CrossRef]
- D’Avolio, A.; Simiele, M.; Siccardi, M.; Baietto, L.; Sciandra, M.; Bonora, S.; Di Perri, G. HPLC-MS method for the quantification of nine anti-HIV drugs from dry plasma spot on glass filter and their long term stability in different conditions. J. Pharm. Biomed. Anal. 2010, 52, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Baietto, L.; D’avolio, A.; Marra, C.; Simiele, M.; Cusato, J.; Pace, S.; Ariaudo, A.; De rosa, F.G.; Di perri, G. Development and validation of a new method to simultaneously quantify triazoles in plasma spotted on dry sample spot devices and analysed by HPLC-MS. J. Antimicrob. Chemother. 2012, 67, 2645–2649. [Google Scholar] [CrossRef] [PubMed]
- Cangemi, G.; Barco, S.; Castagnola, E.; Tripodi, G.; Favata, F.; D’Avolio, A. Development and validation of UHPLC–MS/MS methods for the quantification of colistin in plasma and dried plasma spots. J. Pharm. Biomed. Anal. 2016, 129, 551–557. [Google Scholar] [CrossRef] [PubMed]
- D’urso, A.; Cangemi, G.; Barco, S.; Striano, P.; D’avolio, A.; De Grazia, U. LC-MS/MS-Based Quantification of 9 Antiepileptic Drugs From a Dried Sample Spot Device. Ther. Drug Monit. 2019, 41, 331–339. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasma | ||||||
| INTER-DAY | ||||||
| Ganciclovir | Aciclovir | |||||
| SD (σ) | CV% | Accuracy% | SD (σ) | CV% | Accuracy% | |
| LLOQ | 0.02 | 14% | 9% | 0.02 | 13% | 13% |
| QC low | 0.00 | 13% | 8% | 0.00 | 13% | −14% |
| QC medium | 0.03 | 14% | −6% | 0.03 | 8% | −12% |
| QC high | 0.42 | 9% | −6% | 0.42 | 6% | −11% |
| INTRA-DAY | ||||||
| Ganciclovir | Aciclovir | |||||
| SD (σ) | CV% | Accuracy% | SD (σ) | CV% | Accuracy% | |
| LLOQ | 0.01 | 5% | 10% | 0.01 | 9% | 13% |
| QC low | 0.00 | 6% | 15% | 0.00 | 8% | −14% |
| QC medium | 0.02 | 7% | 2% | 0.02 | 5% | −11% |
| QC high | 0.19 | 4% | −3% | 0.34 | 4% | −12% |
| DPS | ||||||
| INTER-DAY | ||||||
| Ganciclovir | Aciclovir | |||||
| SD (σ) | CV% | Accuracy% | SD (σ) | CV% | Accuracy% | |
| LLOQ | 0.08 | 14% | 12% | 0.02 | 13% | 15% |
| QC low | 0.09 | 13% | 14% | 001 | 15% | 13% |
| QC medium | 0.01 | 2% | 1% | 006 | 1% | −15% |
| QC high | 0.01 | 4% | −9% | 001 | 4% | −12% |
| INTRA-DAY | ||||||
| Ganciclovir | Aciclovir | |||||
| SD (σ) | CV% | Accuracy% | SD (σ) | CV% | Accuracy% | |
| LLOQ | 0.05 | 14% | 13% | 0.04 | 10% | 12% |
| QC low | 0.07 | 13% | 13% | 0.08 | 9% | 15% |
| QC medium | 0.05 | 6% | 2% | 0.07 | 6% | −15% |
| QC high | 0.01 | 2% | −6% | 0.01 | 3% | −15% |
| Ganciclovir | Aciclovir | ||
|---|---|---|---|
| T+25 °C | |||
| 15 days | 15 days | ||
| QC low | 10% (7%) | QC low | 11% (2%) |
| QC medium | 0.5% (2%) | QC medium | 4% (2%) |
| QC high | 2% (2%) | QC high | 5% (4%) |
| 30 days | 30 days | ||
| QC low | 10% (9%) | QC low | 11% (10%) |
| QC medium | 6% (5%) | QC medium | 4% (6%) |
| QC high | 5% (4%) | QC high | 9% (5%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pigliasco, F.; Cafaro, A.; Simeoli, R.; Barco, S.; Magnasco, A.; Faraci, M.; Tripodi, G.; Goffredo, B.M.; Cangemi, G. A UHPLC–MS/MS Method for Therapeutic Drug Monitoring of Aciclovir and Ganciclovir in Plasma and Dried Plasma Spots. Biomedicines 2021, 9, 1379. https://doi.org/10.3390/biomedicines9101379
Pigliasco F, Cafaro A, Simeoli R, Barco S, Magnasco A, Faraci M, Tripodi G, Goffredo BM, Cangemi G. A UHPLC–MS/MS Method for Therapeutic Drug Monitoring of Aciclovir and Ganciclovir in Plasma and Dried Plasma Spots. Biomedicines. 2021; 9(10):1379. https://doi.org/10.3390/biomedicines9101379
Chicago/Turabian StylePigliasco, Federica, Alessia Cafaro, Raffaele Simeoli, Sebastiano Barco, Alberto Magnasco, Maura Faraci, Gino Tripodi, Bianca Maria Goffredo, and Giuliana Cangemi. 2021. "A UHPLC–MS/MS Method for Therapeutic Drug Monitoring of Aciclovir and Ganciclovir in Plasma and Dried Plasma Spots" Biomedicines 9, no. 10: 1379. https://doi.org/10.3390/biomedicines9101379
APA StylePigliasco, F., Cafaro, A., Simeoli, R., Barco, S., Magnasco, A., Faraci, M., Tripodi, G., Goffredo, B. M., & Cangemi, G. (2021). A UHPLC–MS/MS Method for Therapeutic Drug Monitoring of Aciclovir and Ganciclovir in Plasma and Dried Plasma Spots. Biomedicines, 9(10), 1379. https://doi.org/10.3390/biomedicines9101379