Highlighting Human Enzymes Active in Different Metabolic Pathways and Diseases: The Case Study of EC 1.2.3.1 and EC 2.3.1.9


Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Computational Methods
3. Results
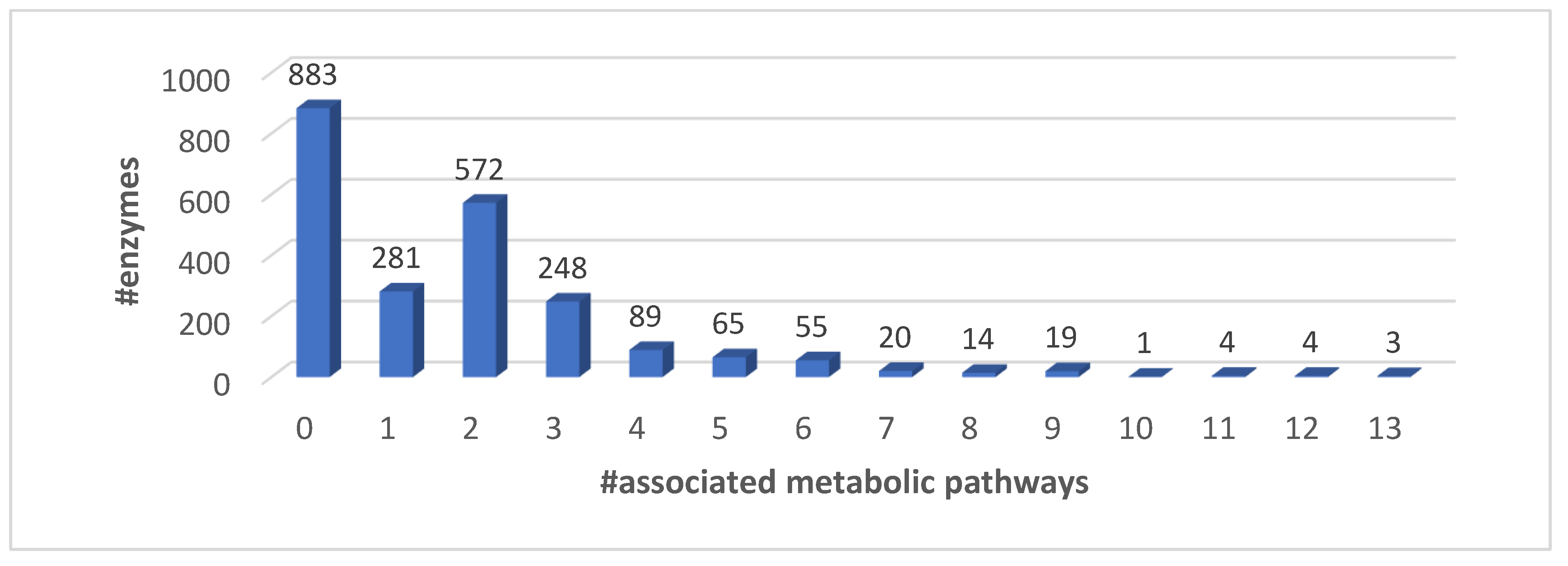
3.1. EC Proteins and KEGG Metabolic Pathways
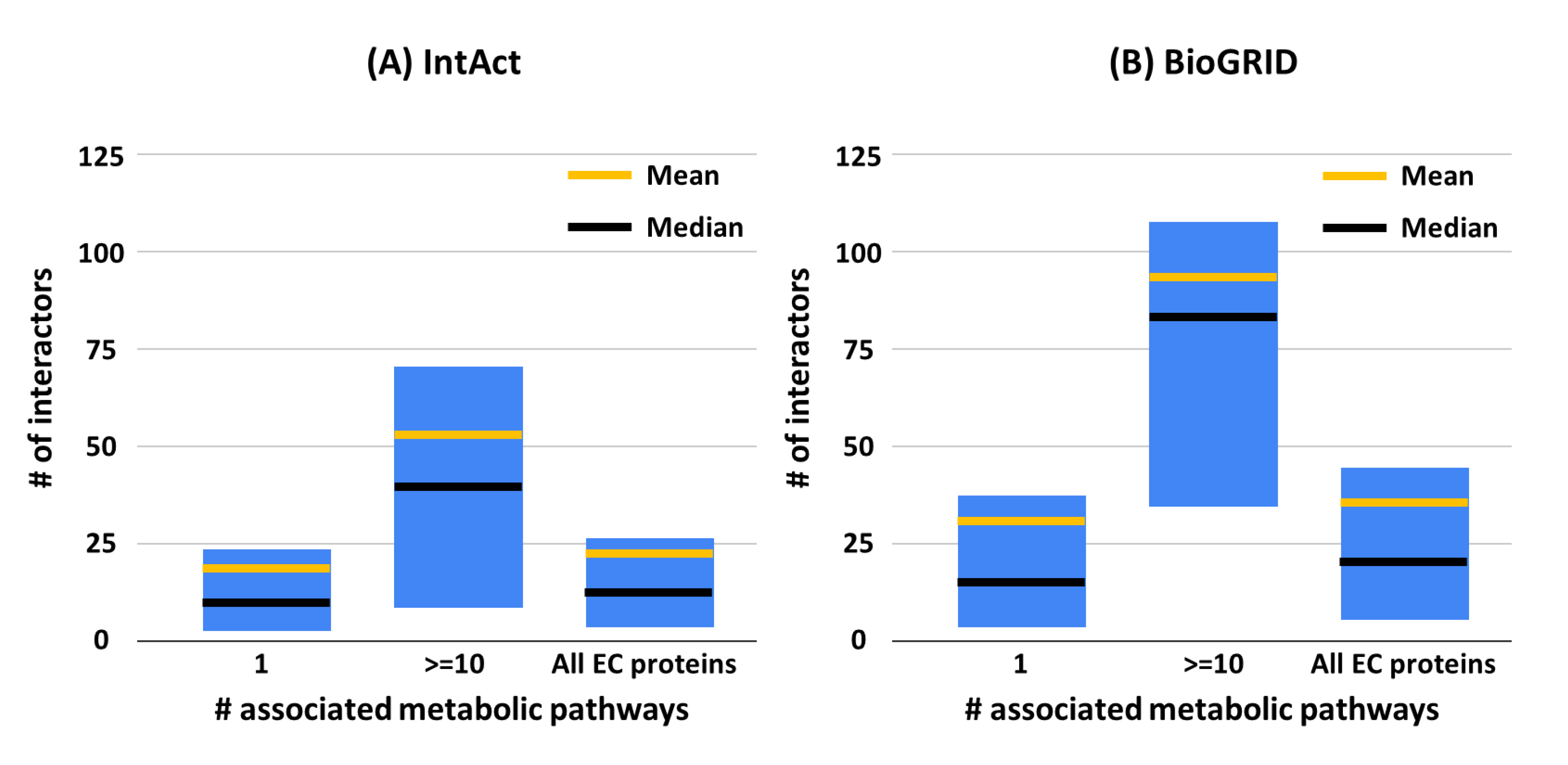
3.2. EC Proteins and Their Interactions
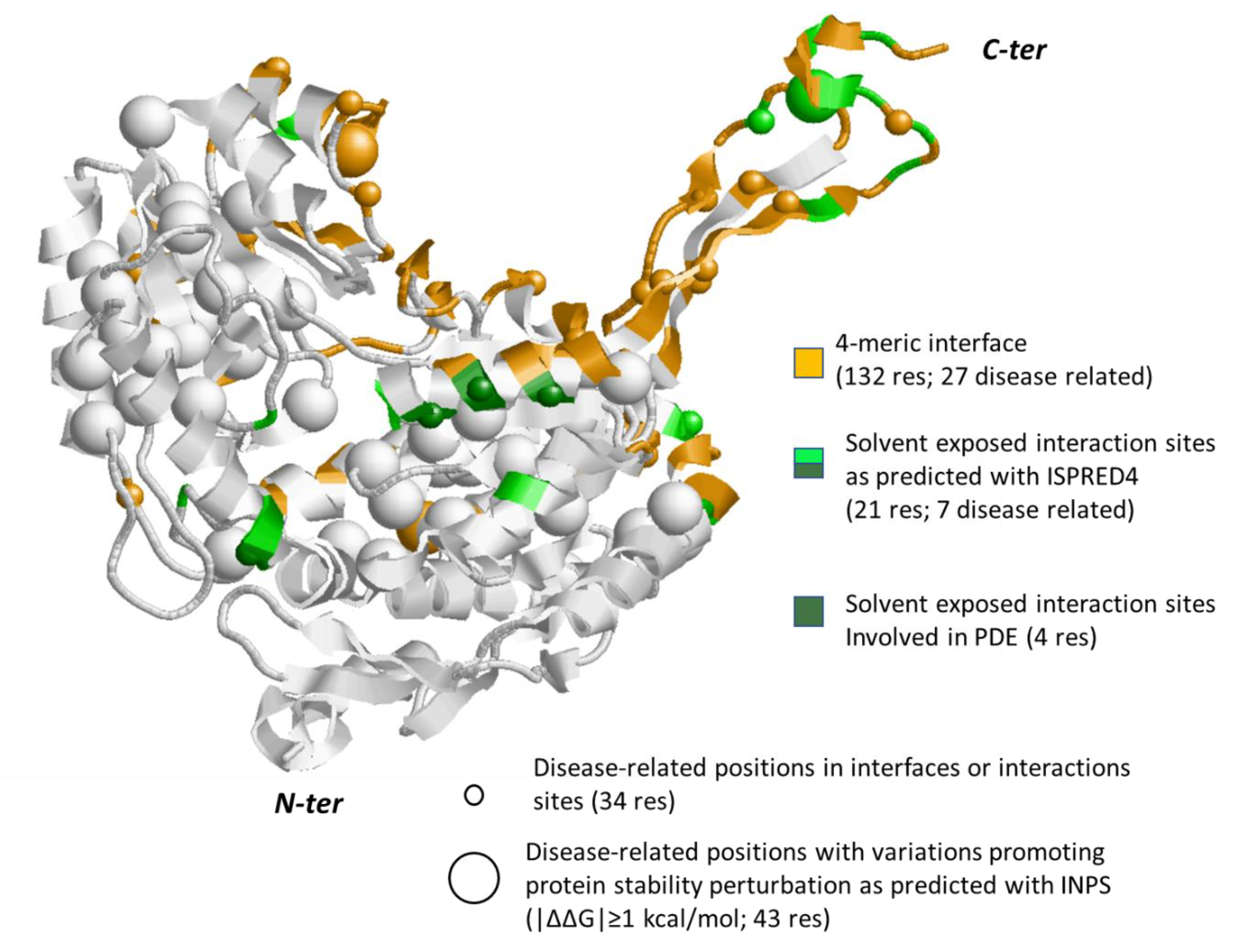
3.3. The Case Study of Alpha-Aminoadipic Semialdehyde Dehydrogenase
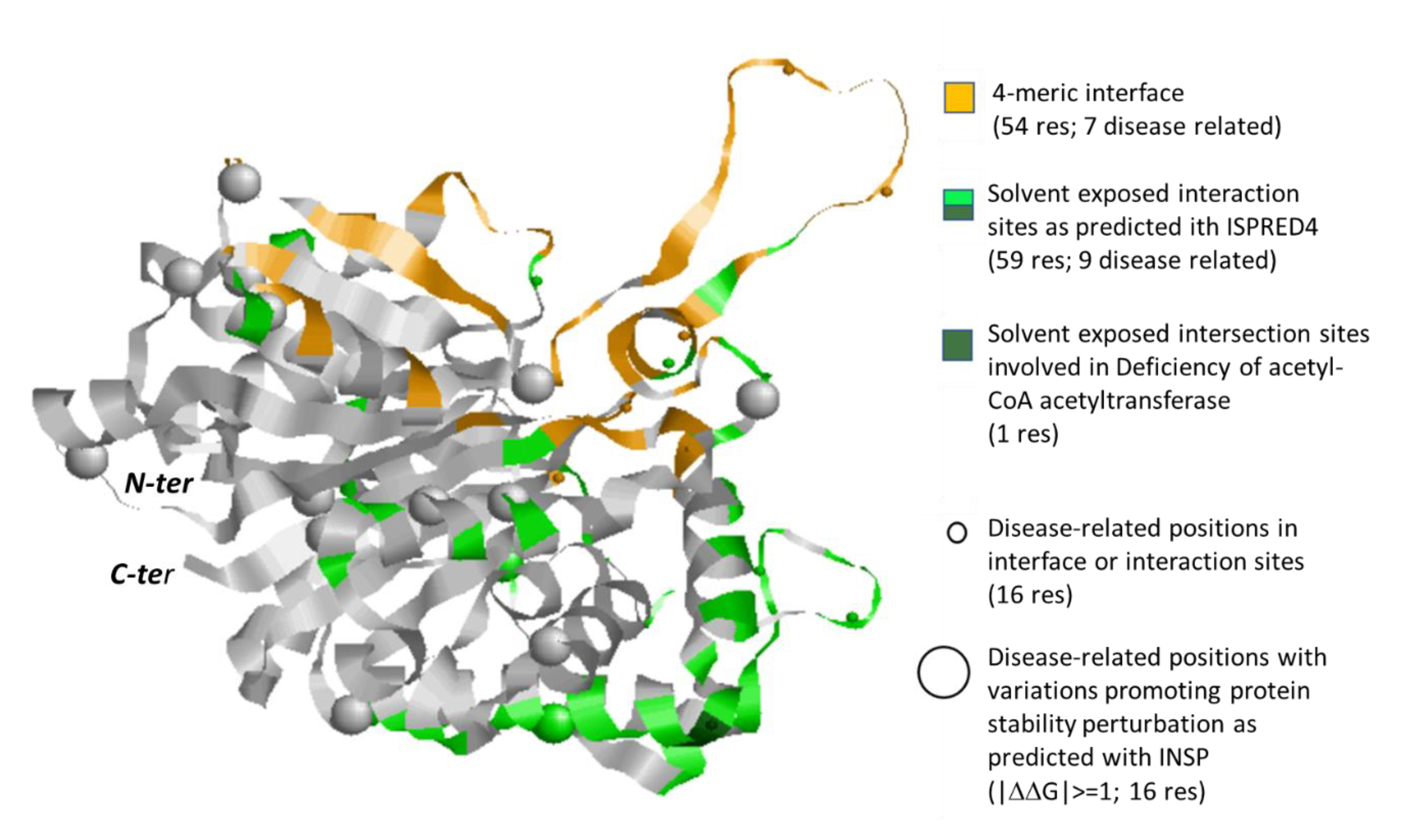
3.4. The Case Study of Acetyl-CoA C-Acetyltransferase
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bugg, T.D.H. Introduction to Enzyme and Coenzyme Chemistry, 3rd ed.; Wiley: New York, NY, USA, 2012. [Google Scholar]
- Savojardo, C.; Martelli, P.L.; Casadio, R. Protein–Protein Interaction Methods and Protein Phase Separation. Ann. Rev. Biom. Data Sci. 2020, 89–112. [Google Scholar] [CrossRef]
- Wolfinbarger, L., Jr. Enzyme Regulation in Metabolic Pathways; Wiley: New York, NY, USA, 2017. [Google Scholar]
- Kegg. Available online: https://www.genome.jp/kegg (accessed on 1 June 2020).
- Reactome. Available online: https://reactome.org (accessed on 1 June 2020).
- Enzyme Nomenclature. Available online: https://www.qmul.ac.uk/sbcs/iubmb/enzyme (accessed on 1 June 2020).
- Protein Data Bank. Available online: https://www.rcsb.org (accessed on 1 June 2020).
- UniProt. Available online: https://www.uniprot.org (accessed on 1 June 2020).
- OMIM. Available online: https://omim.org (accessed on 1 June 2020).
- BioMuta. Available online: https://hive.biochemistry.gwu.edu/biomuta (accessed on 1 June 2020).
- DisGeNet. Available online: https://www.disgenet.org (accessed on 1 June 2020).
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar (accessed on 1 June 2020).
- MalaCards. Available online: https://www.malacards.org (accessed on 1 June 2020).
- IntAct. Available online: https://www.ebi.ac.uk/intact (accessed on 1 June 2020).
- BioGrid. Available online: https://thebiogrid.org (accessed on 1 June 2020).
- Brenda. Available online: https://www.brenda-enzymes.org (accessed on 1 June 2020).
- Enzyme Portal. Available online: https://www.ebi.ac.uk/enzymeportal (accessed on 1 June 2020).
- Mechanism and Catalytic Site Atlas. Available online: https://www.ebi.ac.uk/thornton-srv/m-csa (accessed on 1 June 2020).
- GeneCards. Available online: https://www.genecards.org (accessed on 1 June 2020).
- Mughal, F.; Caetano-Anollés, G. MANET 3.0: Hierarchy and modularity in evolving metabolic networks. PLoS ONE 2019, 14, e0224201. [Google Scholar] [CrossRef] [PubMed]
- Manet. Available online: http://manet.illinois.edu (accessed on 1 June 2020).
- Babbi, G.; Martelli, P.L.; Profiti, G.; Bovo, S.; Savojardo, C.; Casadio, R. eDGAR: a database of Disease-Gene Associations with annotated Relationships among genes. BMC Genom. 2017, 18, 554. [Google Scholar] [CrossRef] [PubMed]
- eDGAR. Available online: http://edgar.biocomp.unibo.it/gene_disease_db (accessed on 1 June 2020).
- ISPRED4. Available online: https://ispred4.biocomp.unibo.it/welcome/default/index (accessed on 1 June 2020).
- Savojardo, C.; Fariselli, P.; Martelli, P.L.; Casadio, R. ISPRED4: interaction sites PREDiction in protein structures with a refining grammar model. Bioinformatics 2017, 33, 1656. [Google Scholar] [CrossRef] [PubMed]
- INPS-Md. Available online: https://inpsmd.biocomp.unibo.it/inpsSuite (accessed on 1 June 2020).
- Savojardo, C.; Fariselli, P.; Martelli, P.L.; Casadio, R. INPS-MD: A web server to predict stability of protein variants from sequence and structure. Bioinformatics 2016, 32, 2542. [Google Scholar] [CrossRef] [PubMed]
- Rhea-DB. Available online: https://www.rhea-db.org (accessed on 1 June 2020).
- Human Phenotype Ontology. Available online: https://hpo.jax.org/app (accessed on 1 June 2020).
- Gene Organizer. Available online: http://geneorganizer.huji.ac.il/browse/?GENE_IDs=ALDH7A1&FullSite=T#btn_organs-browse (accessed on 1 June 2020).
- Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000164904-ALDH7A1/tissue (accessed on 1 June 2020).
- Luo, M.; Tanner, J.J. Structural Basis of Substrate Recognition by Aldehyde Dehydrogenase 7A1. Biochemistry 2015, 54, 5513. [Google Scholar] [CrossRef] [PubMed]
- Laciak, A.R.; Korasick, D.A.; Wyatt, J.W.; Gates, K.S.; Tanner, J.J. Structural and biochemical consequences of pyridoxine-dependent epilepsy mutations that target the aldehyde binding site of aldehyde dehydrogenase ALDH7A1. FEBS J. 2020, 287, 173. [Google Scholar] [CrossRef] [PubMed]
- MobiDB. Available online: https://mobidb.bio.unipd.it/P49419/db (accessed on 1 June 2020).
- Haapalainen, A.M.; Meriläinen, G.; Pirilä, P.L.; Kondo, N.; Fukao, T.; Wierenga, R.K. Crystallographic and Kinetic Studies of Human Mitochondrial Acetoacetyl-CoA Thiolase: The Importance of Potassium and Chloride Ions for Its Structure and Function. Biochemistry 2007, 46, 4305–4321. [Google Scholar] [CrossRef] [PubMed]
- Kursula, P.; Sikkilä, H.; Fukao, T.; Kondo, N.; Wierenga, R.K. High Resolution Crystal Structures of Human Cytosolic Thiolase (CT): A Comparison of the Active Sites of Human CT, Bacterial Thiolase, and Bacterial KAS I. J. Mol. Biol. 2005, 347, 189–201. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Set | # Human Proteins |
|---|---|
| In SwissProt/UniProt | 20365 |
| Proteins with four-digit EC (EC proteins) | 3428 (1411 EC) * |
| Proteins associated with genetic diseases | 7316 (5788 diseases) |
| EC proteins with genetic disease associations | 1669 (955 EC and 1900 diseases) |
| Set | Human Proteins with KEGG Pathways | Human Proteins with KEGG Metabolic Pathways | ||
|---|---|---|---|---|
| #Proteins | #Pathways | #Proteins | #Pathways | |
| In SwissProt | 6904 | 320 | 1642 | 90 |
| EC proteins | 2258 | 317 | 1375 | 90 |
| Proteins associated to genetic diseases | 3391 | 320 | 895 | 90 |
| EC proteins associated to genetic diseases | 1255 | 314 | 770 | 90 |
| EC Number 1 | KEGG 2 | UniProt 3 | PDB 4 | IntAct 5 | BioGRID 6 | Int. Sites 7 | Other EC 8 |
|---|---|---|---|---|---|---|---|
| 1.2.1.3 | 13: hsa00010 hsa00053 hsa00071 hsa00260 hsa00280 hsa00310 hsa00330 hsa00340 hsa00380 hsa00410 hsa00561 hsa00620 hsa01100 | P49419 (13) | 4ZUL (homo 4-mer) | 23 | 62 | 78/235; 34/83 (21/132; 7/34) | 1.2.1.8 (1) 1.2.1.31 (1) |
| P49189 (12) -hsa00260 | 6QAP (homo 4-mer) | 10 | 38 | 48/223; 0/2 (10/139; none) | 1.2.1.19 (0) 1.2.1.47 (1) | ||
| P05091 (12) -hsa00260 | 1O02 (homo 4-mer) | 45 | 75 | 88/223; 16/41 (19/133; 4/25) | − | ||
| P51648 (12) -hsa00260 | 4QGK (homo 2-mer) | 91 | 107 | 82/238; 19/54 (20/139; 4/32) | 1.2.1.94 (0) | ||
| P30837 (12)-hsa00260 | - | 41 | 93 | 111/517; 1/1 | − | ||
| 2.3.1.9 | 13: hsa00071 hsa00072 hsa00280 hsa00310 hsa00380 hsa00620 hsa00630 hsa00640 hsa00650 hsa00900 hsa01100 hsa01200 hsa01212 | Q9BWD1 (13) | 1WL4 (homo 4-mer) | 20 | 46 | 94/175; 20/37 (66/113; 9/16) | − |
| P24752 (13) | 2IBY (homo 4-mer) | 32 | 108 | 117/185; 35/59 (65/121;17/37) | − | ||
| 2.6.1.1 | 11: hsa00220 hsa00250 hsa00270 hsa00330 hsa00350 hsa00360 hsa00400 hsa01100 hsa01200 hsa01210 hsa01230 | P00505 (11) | 5AX8 (homo 2-mer) | 37 | 42 | 38/192; 5/32 (4/126; 0/21) | 2.6.1.7 (0) |
| P17174 (11) | 6DND (1-mer) | 12 | 73 | 46/200; 10/42 | 2.6.1.3 (0) | ||
| 4.2.1.17 | 11: hsa00062 hsa00071 hsa00280 hsa00310 hsa00380 hsa00410 hsa00640 hsa00650 hsa01100 hsa01200 hsa01212 | P40939 (11) | 6DV2 (hetero 4-mer) | 116 | 254 | 24/375; 6/65 (23/337; 5/57) | 1.1.1.211 (2) |
| P30084 (11) | 2HW5 (homo 6-mer) | 65 | 112 | 45/163; 9/37 (2/68; 2/16) | − | ||
| Q08426 (10) - hsa00062 | − | 123 | 109 | 234/723; 53/150 | 5.3.3.8 (1) 1.1.1.35 (8) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babbi, G.; Baldazzi, D.; Savojardo, C.; Martelli, P.L.; Casadio, R. Highlighting Human Enzymes Active in Different Metabolic Pathways and Diseases: The Case Study of EC 1.2.3.1 and EC 2.3.1.9. Biomedicines 2020, 8, 250. https://doi.org/10.3390/biomedicines8080250
Babbi G, Baldazzi D, Savojardo C, Martelli PL, Casadio R. Highlighting Human Enzymes Active in Different Metabolic Pathways and Diseases: The Case Study of EC 1.2.3.1 and EC 2.3.1.9. Biomedicines. 2020; 8(8):250. https://doi.org/10.3390/biomedicines8080250
Chicago/Turabian StyleBabbi, Giulia, Davide Baldazzi, Castrense Savojardo, Pier Luigi Martelli, and Rita Casadio. 2020. "Highlighting Human Enzymes Active in Different Metabolic Pathways and Diseases: The Case Study of EC 1.2.3.1 and EC 2.3.1.9" Biomedicines 8, no. 8: 250. https://doi.org/10.3390/biomedicines8080250
APA StyleBabbi, G., Baldazzi, D., Savojardo, C., Martelli, P. L., & Casadio, R. (2020). Highlighting Human Enzymes Active in Different Metabolic Pathways and Diseases: The Case Study of EC 1.2.3.1 and EC 2.3.1.9. Biomedicines, 8(8), 250. https://doi.org/10.3390/biomedicines8080250