Targeting NF-κB Signaling by Calebin A, a Compound of Turmeric, in Multicellular Tumor Microenvironment: Potential Role of Apoptosis Induction in CRC Cells


 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies and Chemicals
2.2. Cells and Cell Culture Conditions
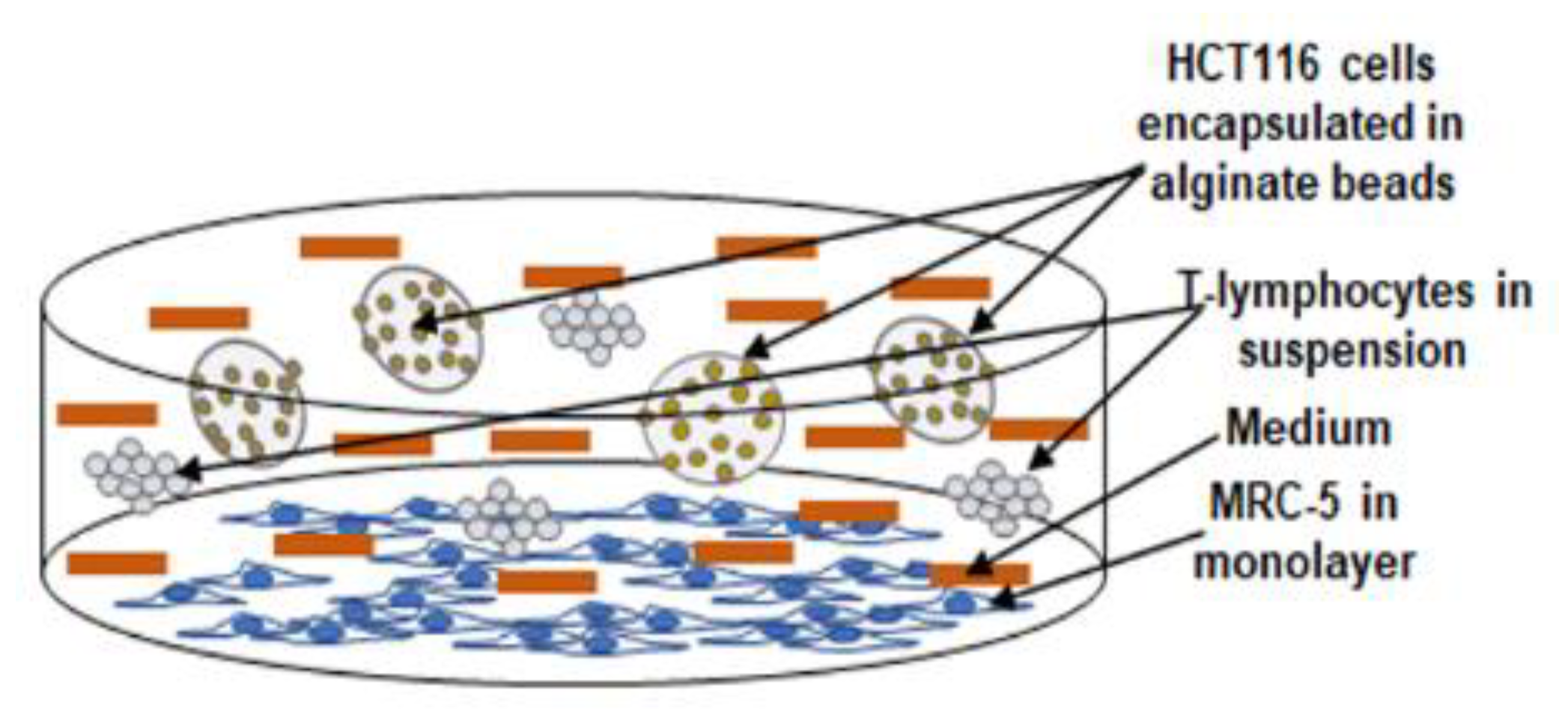
2.3. Experimental Study Design
2.4. Alginate Culture
2.5. Vitality and Proliferation
2.6. Colonosphere Formation and Invasion
2.7. Immunofluorescence
2.8. Western Blotting Investigations
2.9. DNA-Binding Assay
2.10. Statistical Evaluation
3. Results
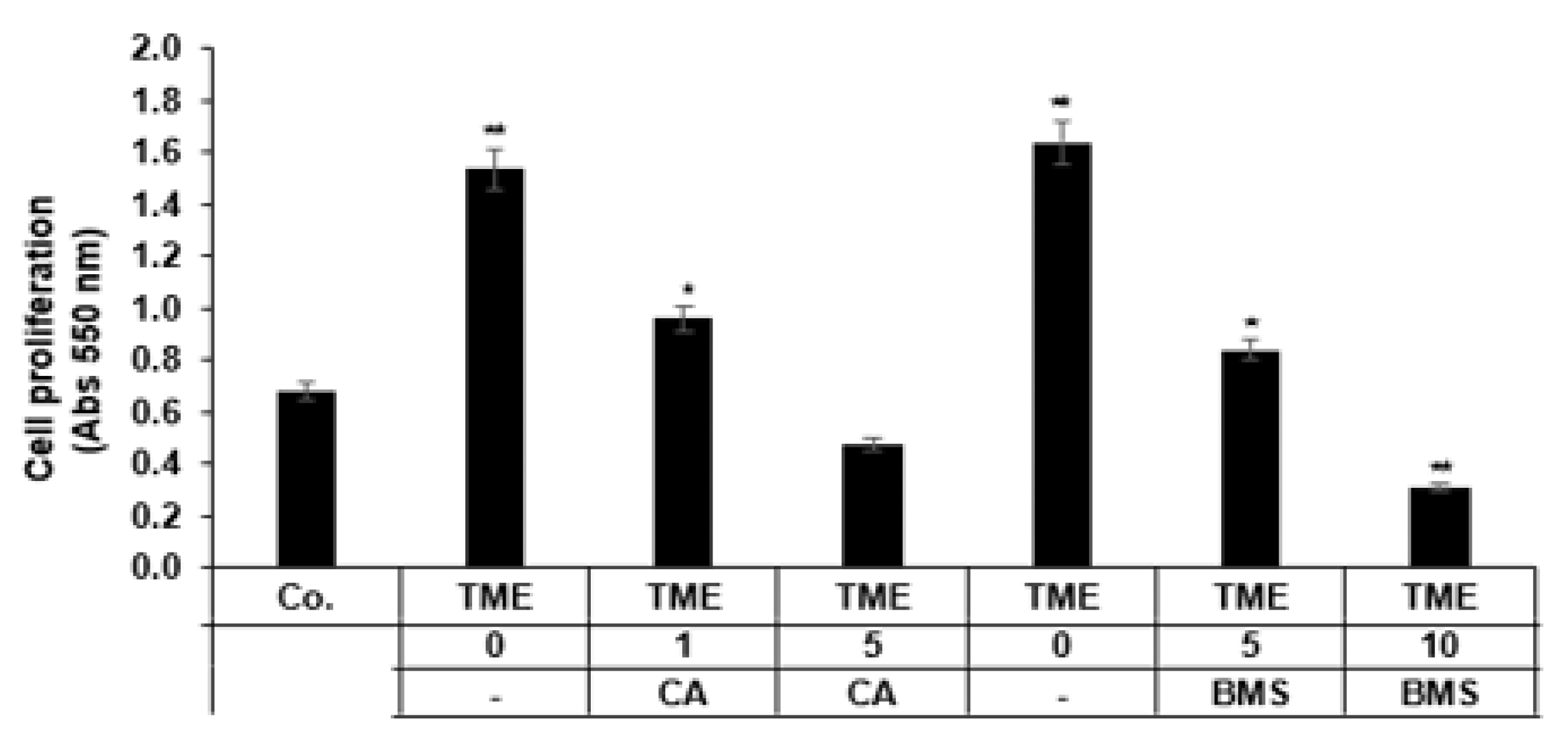
3.1. Calebin A Suppresses Proliferation Promoted by TME Cultures in 3D-Alginate CRC Cells
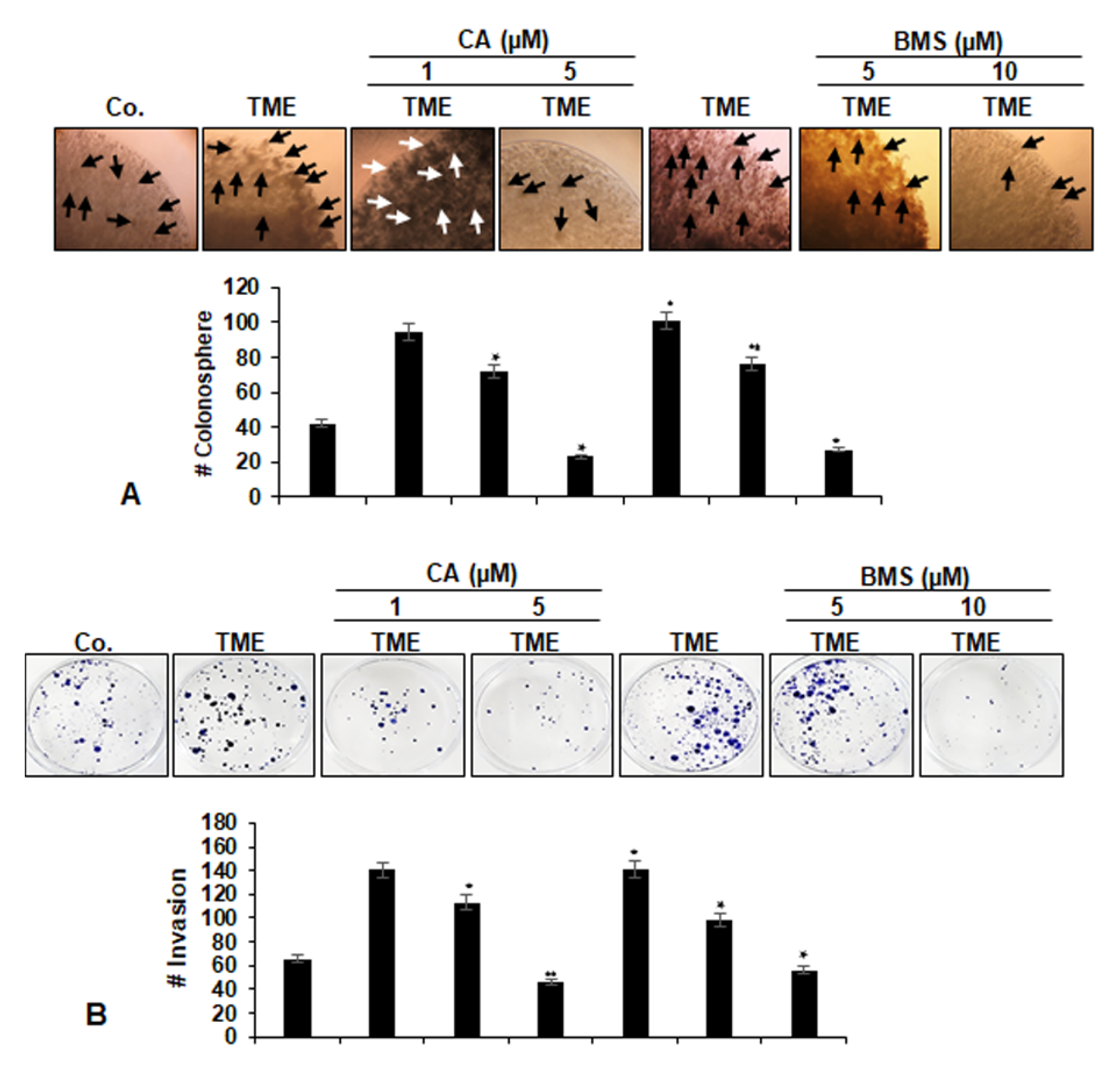
3.2. Calebin A Suppresses Invasion and Colony Formation Ability, Promoted by TME in 3D-Alginate CRC Cells
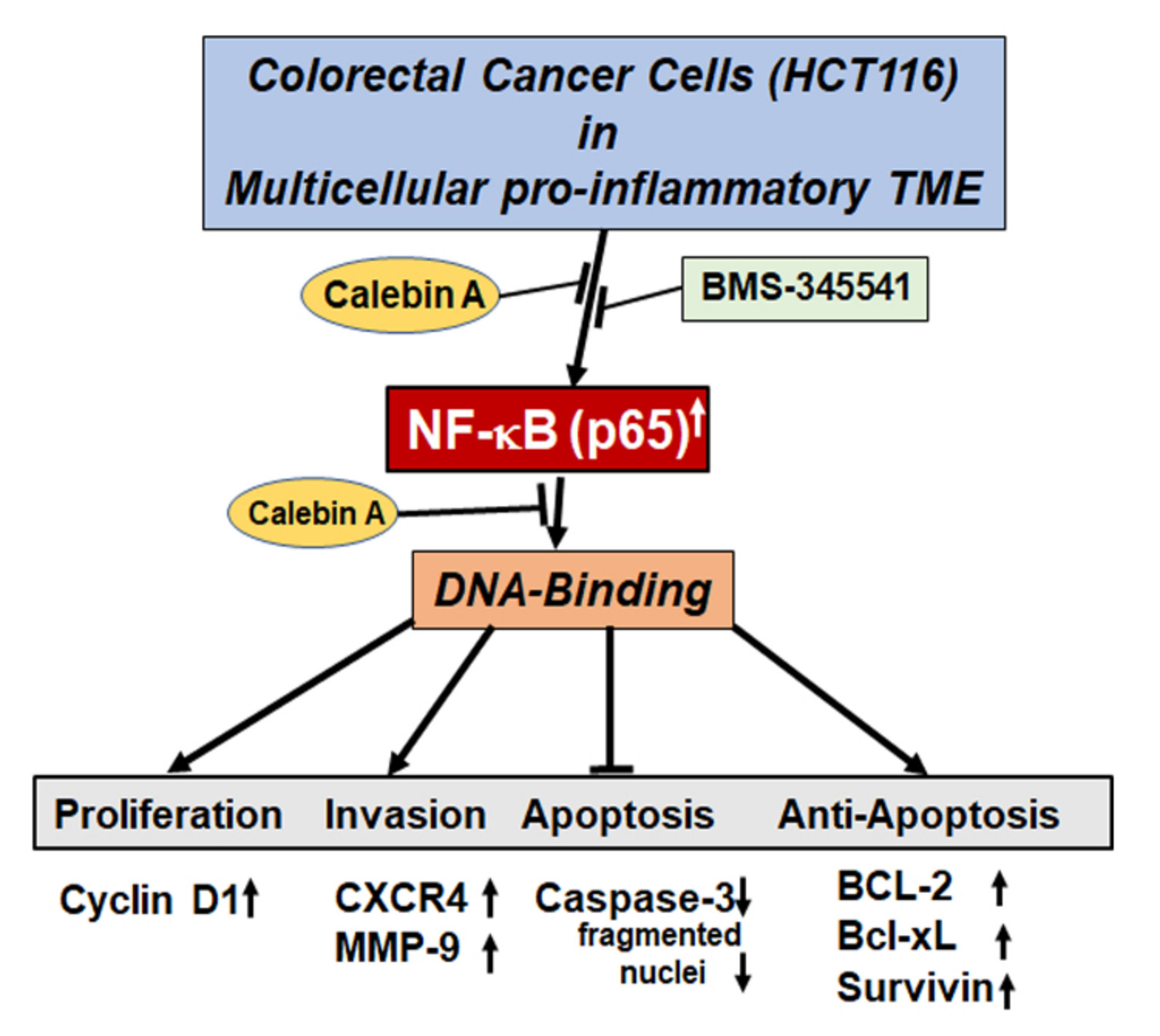
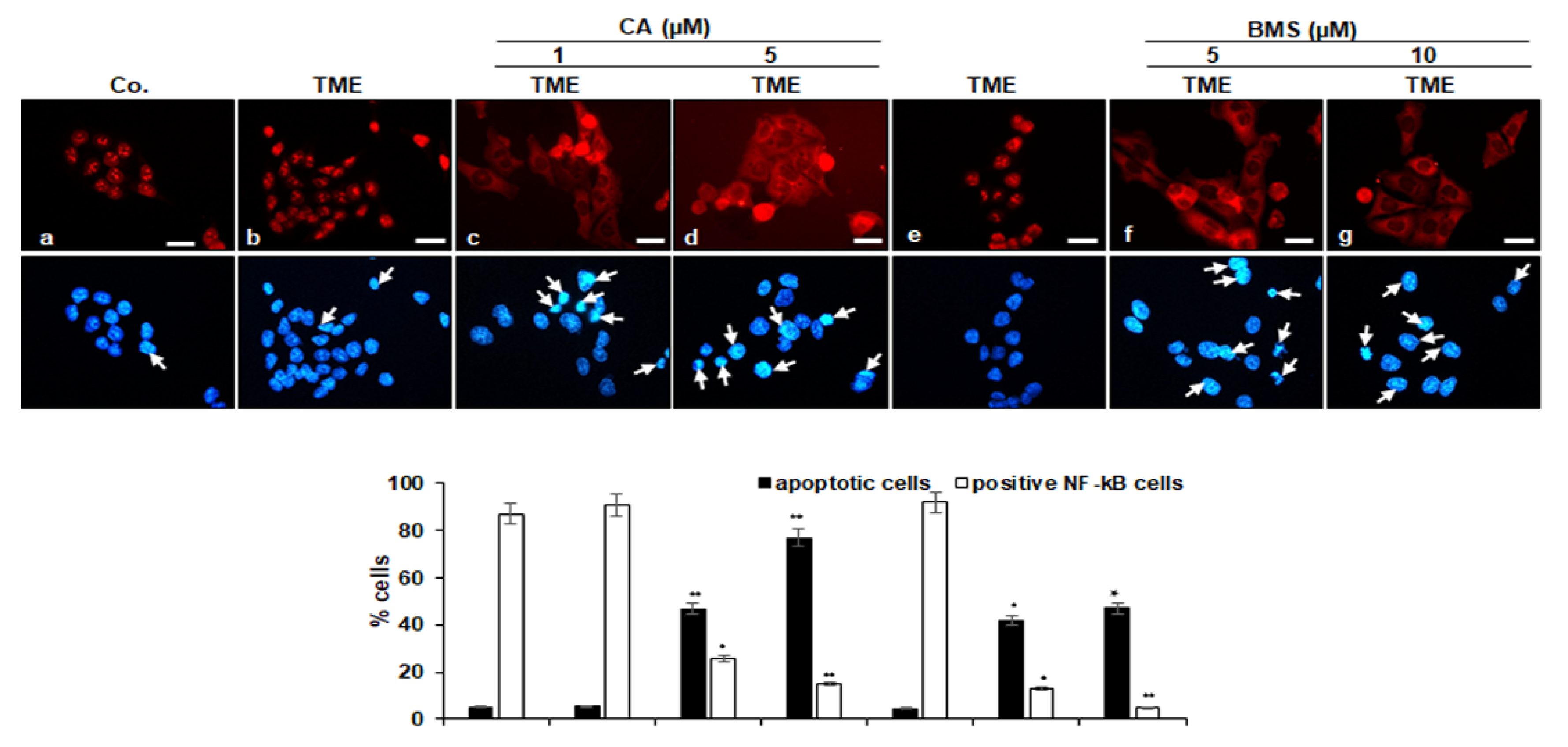
3.3. Calebin A Decreases TME-Induced Activation and Nuclear Translocation of p65-NF-κB in CRC Cells
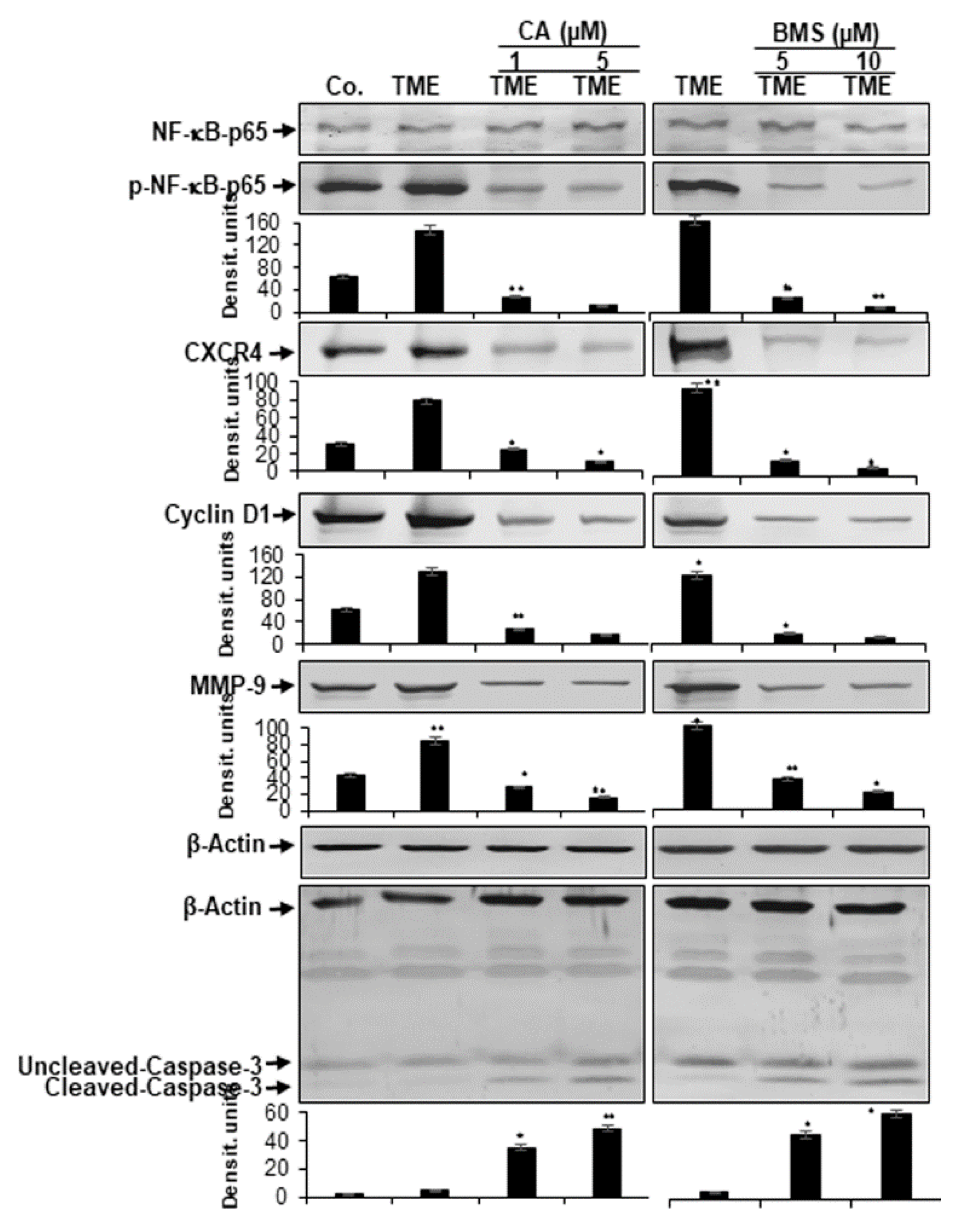
3.4. Calebin A Represses TME-Induced NF-κB Activation and NF-κB-Dependent Gene End-Products Involved in Cell Proliferation, Invasion, and Apoptosis in CRC Cells
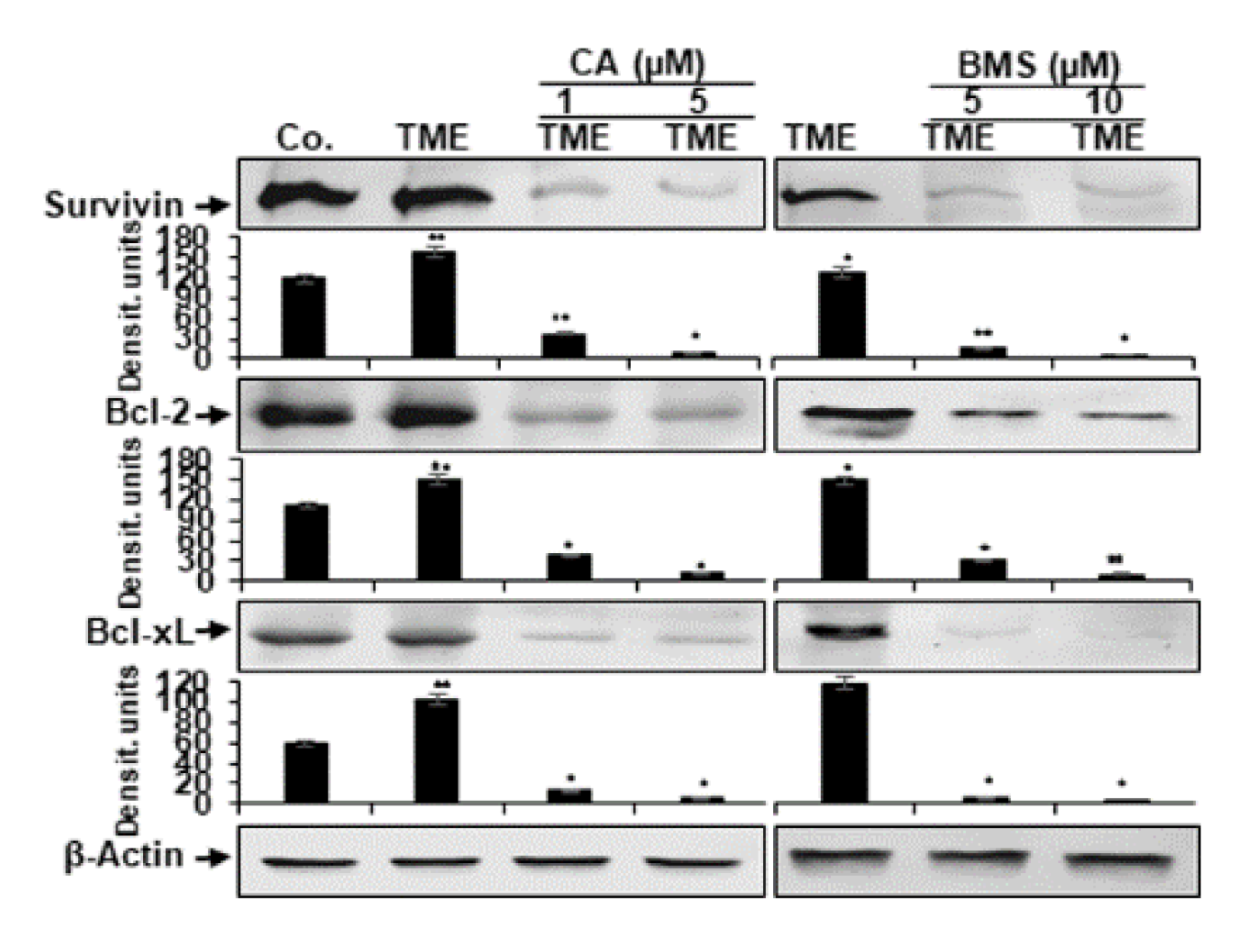
3.5. Calebin A Reduces TME-Induced NF-κB-Dependent Anti-Apoptotic Gene Biomarkers in CRC Cells
3.6. Calebin A Disturbs the Direct Interaction of NF-κB with DNA in CRC Cells
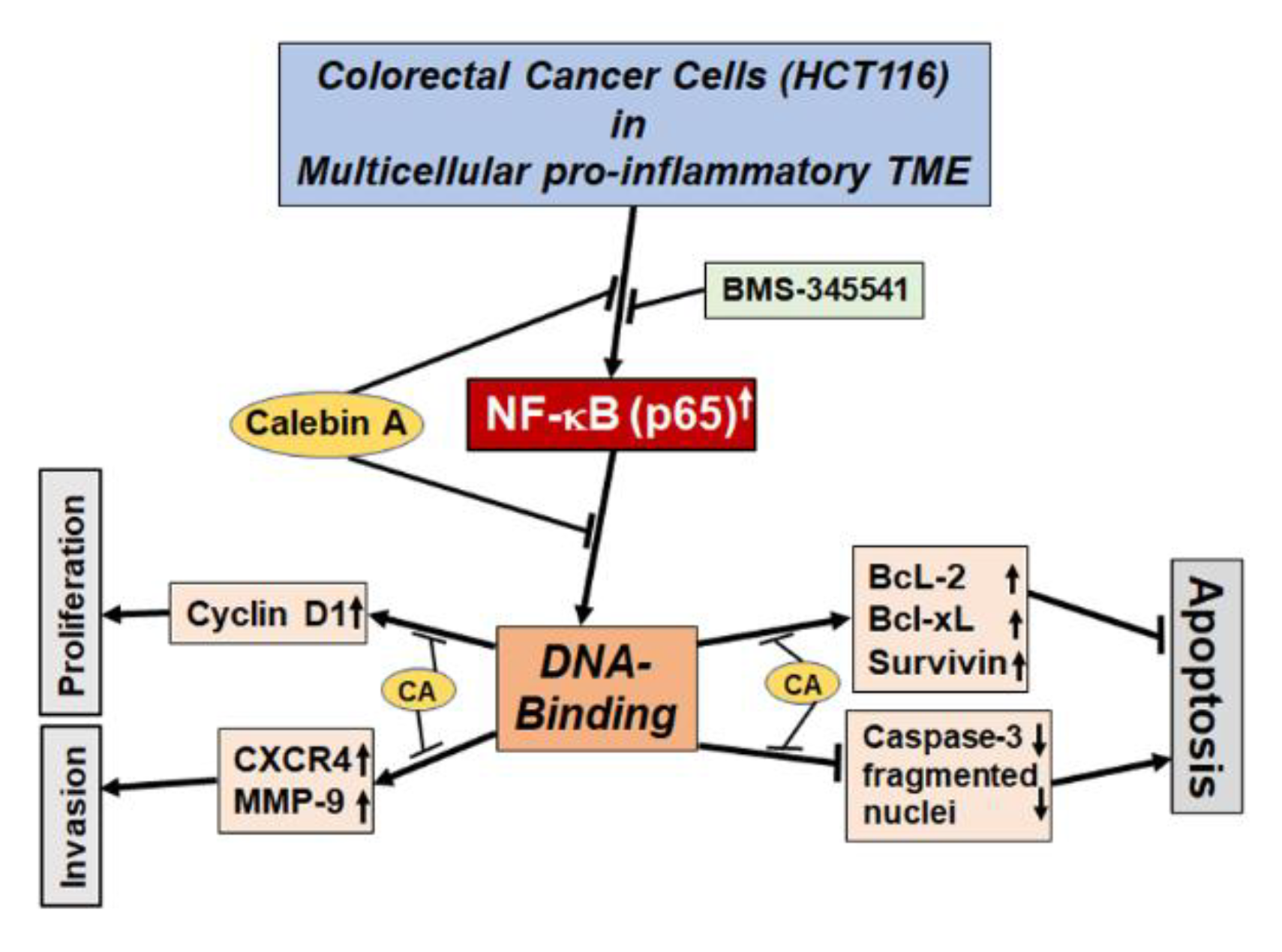
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers. 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Emon, B.; Bauer, J.; Jain, Y.; Jung, B.; Saif, T. Biophysics of Tumor Microenvironment and Cancer Metastasis: A Mini Review. Comput. Struct. Biotechnol. J. 2018, 16, 279–287. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [Green Version]
- Storz, P.; Crawford, H.C. Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterol. 2020, 158, 2072–2081. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Cortese, N.; Donadon, M.; Rigamonti, A.; Marchesi, F. Macrophages at the crossroads of anticancer strategies. Front. Biosci. (Landmark Ed.) 2019, 24, 1271–1283. [Google Scholar]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, F.; Zambrano, S.; Agresti, A. NF-kappaB, the Importance of Being Dynamic: Role and Insights in Cancer. Biomedicines 2018, 6, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFkappaB-signaling pathway in cancer. Oncotargets Ther. 2018, 11, 2063–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.H.; Hong, J.T. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef]
- Aggarwal, B.B. Nuclear factor-kappaB: The enemy within. Cancer Cell. 2004, 6, 203–208. [Google Scholar] [CrossRef] [Green Version]
- Kearns, J.D.; Basak, S.; Werner, S.L.; Huang, C.S.; Hoffmann, A. IkappaBepsilon provides negative feedback to control NF-kappaB oscillations, signaling dynamics, and inflammatory gene expression. J. Cell Biol. 2006, 173, 659–664. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Inoue, J.; Gohda, J.; Akiyama, T.; Semba, K. NF-kappaB activation in development and progression of cancer. Cancer Sci. 2007, 98, 268–274. [Google Scholar] [CrossRef]
- Schottelius, A.J.; Dinter, H. Cytokines, NF-kappaB, microenvironment, intestinal inflammation and cancer. Cancer Treat. Res. 2006, 130, 67–87. [Google Scholar] [CrossRef]
- Bharti, A.C.; Aggarwal, B.B. Nuclear factor-kappa B and cancer: Its role in prevention and therapy. Biochem. Pharmacol. 2002, 64, 883–888. [Google Scholar] [CrossRef]
- Yu, L.L.; Yu, H.G.; Yu, J.P.; Luo, H.S.; Xu, X.M.; Li, J.H. Nuclear factor-kappaB p65 (RelA) transcription factor is constitutively activated in human colorectal carcinoma tissue. World J. Gastroenterol. 2004, 10, 3255–3260. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Lin, A. NF-kappaB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Jani, T.S.; DeVecchio, J.; Mazumdar, T.; Agyeman, A.; Houghton, J.A. Inhibition of NF-kappaB signaling by quinacrine is cytotoxic to human colon carcinoma cell lines and is synergistic in combination with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) or oxaliplatin. J. Biol. Chem. 2010, 285, 19162–19172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, B.B.; Gehlot, P. Inflammation and cancer: How friendly is the relationship for cancer patients? Curr. Opin. Pharmacol. 2009, 9, 351–369. [Google Scholar] [CrossRef] [Green Version]
- Buhrmann, C.; Popper, B.; Kunnumakkara, A.B.; Aggarwal, B.B.; Shakibaei, M. Evidence That Calebin A, a Component of Curcuma Longa Suppresses NF-B Mediated Proliferation, Invasion and Metastasis of Human Colorectal Cancer Induced by TNF-beta (Lymphotoxin). Nutrients 2019, 11, 2904. [Google Scholar] [CrossRef] [Green Version]
- Buhrmann, C.; Yazdi, M.; Popper, B.; Kunnumakkara, A.B.; Aggarwal, B.B.; Shakibaei, M. Induction of the Epithelial-to-Mesenchymal Transition of Human Colorectal Cancer by Human TNF-beta (Lymphotoxin) and its Reversal by Resveratrol. Nutrients 2019, 11, 704. [Google Scholar] [CrossRef] [Green Version]
- Buhrmann, C.; Yazdi, M.; Popper, B.; Shayan, P.; Goel, A.; Aggarwal, B.B.; Shakibaei, M. Evidence that TNF-beta induces proliferation in colorectal cancer cells and resveratrol can down-modulate it. Exp. Biol. Med. 2019, 244, 1–12. [Google Scholar] [CrossRef]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Yang, J.G.; Wang, W.M.; Xia, H.F.; Yu, Z.L.; Li, H.M.; Ren, J.G.; Chen, G.; Wang, B.K.; Jia, J.; Zhang, W.; et al. Lymphotoxin-alpha promotes tumor angiogenesis in HNSCC by modulating glycolysis in a PFKFB3-dependent manner. Int. J. Cancer 2019, 145, 1358–1370. [Google Scholar] [CrossRef]
- Soleimani, A.; Rahmani, F.; Ferns, G.A.; Ryzhikov, M.; Avan, A.; Hassanian, S.M. Role of the NF-κB signaling pathway in the pathogenesis of colorectal cancer. Gene 2020, 726, 144132. [Google Scholar] [CrossRef]
- Huang, X.M.; Yang, Z.J.; Xie, Q.; Zhang, Z.K.; Zhang, H.; Ma, J.Y. Natural products for treating colorectal cancer: A mechanistic review. Biomed. Pharm. 2019, 117, 109142. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.T.; Yang, C.C.; Shyur, L.F. Phytomedicine-Modulating oxidative stress and the tumor microenvironment for cancer therapy. Pharm. Res. 2016, 114, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Amedei, A.; Aquilano, K.; Azmi, A.S.; Benencia, F.; Bhakta, D.; Bilsland, A.E.; Boosani, C.S.; Chen, S.; Ciriolo, M.R.; et al. Cancer prevention and therapy through the modulation of the tumor microenvironment. Semin. Cancer Biol. 2015, 35, s199–s223. [Google Scholar] [CrossRef]
- Nair, A.; Amalraj, A.; Jacob, J.; Kunnumakkara, A.B.; Gopi, S. Non-Curcuminoids from Turmeric and Their Potential in Cancer Therapy and Anticancer Drug Delivery Formulations. Biomolecules. 2019, 9, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, B.B.; Yuan, W.; Li, S.; Gupta, S.C. Curcumin-free turmeric exhibits anti-inflammatory and anticancer activities: Identification of novel components of turmeric. Mol. Nutr. Food Res. 2013, 57, 1529–1542. [Google Scholar] [CrossRef]
- Gupta, S.C.; Sung, B.; Kim, J.H.; Prasad, S.; Li, S.; Aggarwal, B.B. Multitargeting by turmeric, the golden spice: From kitchen to clinic. Mol. Nutr. Food Res. 2013, 57, 1510–1528. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.K.; Prasad, S.; Majeed, M.; Aggarwal, B.B. Calebin A, a novel component of turmeric, suppresses NF-kappaB regulated cell survival and inflammatory gene products leading to inhibition of cell growth and chemosensitization. Phytomedicine: Int. J. Phytother. Phytopharm. 2017, 34, 171–181. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Han, Y.; Liu, J.; Zhang, J.; Li, F.; Wang, Y.; Liu, X.; Yao, L. Calebin-A induces apoptosis and modulates MAPK family activity in drug resistant human gastric cancer cells. Eur. Pharmacol. 2008, 591, 252–258. [Google Scholar] [CrossRef]
- Lee, M.J.; Tsai, Y.J.; Lin, M.Y.; You, H.L.; Kalyanam, N.; Ho, C.T.; Pan, M.H. Calebin-A induced death of malignant peripheral nerve sheath tumor cells by activation of histone acetyltransferase. Phytomedicine: Int. J. Phytother. Phytopharm. 2019, 57, 377–384. [Google Scholar] [CrossRef]
- Buhrmann, C.; Kunnumakkara, A.B.; Popper, B.; Majeed, M.; Aggarwal, B.B.; Shakibaei, M. Calebin A Potentiates the Effect of 5-FU and TNF-beta (Lymphotoxin alpha) against Human Colorectal Cancer Cells: Potential Role of NF-kappaB. Int. J. Mol. Sci. 2020, 21, 2393. [Google Scholar] [CrossRef] [Green Version]
- Shakibaei, M.; Kraehe, P.; Popper, B.; Shayan, P.; Goel, A.; Buhrmann, C. Curcumin potentiates antitumor activity of 5-fluorouracil in a 3D alginate tumor microenvironment of colorectal cancer. BMC. Cancer 2015, 15, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakibaei, M.; Sung, B.; Sethi, G.; Aggarwal, B.B. TNF-alpha-induced mitochondrial alterations in human T cells requires FADD and caspase-8 activation but not RIP and caspase-3 activation. Antioxid. Redox Signal. 2010, 13, 821–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakibaei, M.; De Souza, P. Differentiation of mesenchymal limb bud cells to chondrocytes in alginate beads. Cell Biol. Int. 1997, 21, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Baichwal, V.R.; Baeuerle, P.A. Activate NF-kappa B or die? Curr. Biol. 1997, 7, R94–R96. [Google Scholar] [CrossRef] [Green Version]
- Shishodia, S.; Aggarwal, B.B. Nuclear factor-kappaB activation: A question of life or death. J. Biochem. Mol. Biol. 2002, 35, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Pattoli, M.A.; Gregor, K.R.; Brassil, P.J.; MacMaster, J.F.; McIntyre, K.W.; Yang, X.; Iotzova, V.S.; Clarke, W.; Strnad, J.; et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J. Biol. Chem. 2003, 278, 1450–1456. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef]
- Duyao, M.P.; Kessler, D.J.; Spicer, D.B.; Bartholomew, C.; Cleveland, J.L.; Siekevitz, M.; Sonenshein, G.E. Transactivation of the c-myc promoter by human T cell leukemia virus type 1 tax is mediated by NF kappa B. J. Biol. Chem. 1992, 267, 16288–16291. [Google Scholar]
- Estève, P.O.; Chicoine, E.; Robledo, O.; Aoudjit, F.; Descoteaux, A.; Potworowski, E.F.; St-Pierre, Y. Protein kinase C-zeta regulates transcription of the matrix metalloproteinase-9 gene induced by IL-1 and TNF-alpha in glioma cells via NF-kappa B. J. Biol. Chem. 2002, 277, 35150–35155. [Google Scholar] [CrossRef] [Green Version]
- Guttridge, D.C.; Albanese, C.; Reuther, J.Y.; Pestell, R.G.; Baldwin, A.S., Jr. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol. 1999, 19, 5785–5799. [Google Scholar] [CrossRef] [Green Version]
- Van de Stolpe, A.; Caldenhoven, E.; Stade, B.G.; Koenderman, L.; Raaijmakers, J.A.; Johnson, J.P.; van der Saag, P.T. 12-O-tetradecanoylphorbol-13-acetate- and tumor necrosis factor alpha-mediated induction of intercellular adhesion molecule-1 is inhibited by dexamethasone. Functional analysis of the human intercellular adhesion molecular-1 promoter. J. Biol. Chem. 1994, 269, 6185–6192. [Google Scholar] [PubMed]
- Yamamoto, K.; Arakawa, T.; Ueda, N.; Yamamoto, S. Transcriptional roles of nuclear factor kappa B and nuclear factor-interleukin-6 in the tumor necrosis factor alpha-dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J. Biol. Chem. 1995, 270, 31315–31320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Kuo, M.T. NF-kappaB-mediated induction of mdr1b expression by insulin in rat hepatoma cells. J. Biol. Chem. 1997, 272, 15174–15183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Fukuda, S.; Cordis, G.; Das, D.K.; Maulik, N. Anti-apoptotic protein survivin plays a significant role in tubular morphogenesis of human coronary arteriolar endothelial cells by hypoxic preconditioning. FEBS. Lett. 2001, 508, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Schwenzer, R.; Siemienski, K.; Liptay, S.; Schubert, G.; Peters, N.; Scheurich, P.; Schmid, R.M.; Wajant, H. The human tumor necrosis factor (TNF) receptor-associated factor 1 gene (TRAF1) is up-regulated by cytokines of the TNF ligand family and modulates TNF-induced activation of NF-kappaB and c-Jun N-terminal kinase. J. Biol. Chem. 1999, 274, 19368–19374. [Google Scholar] [CrossRef] [Green Version]
- Chu, Z.L.; McKinsey, T.A.; Liu, L.; Gentry, J.J.; Malim, M.H.; Ballard, D.W. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc. Natl. Acad. Sci. USA 1997, 94, 10057–10062. [Google Scholar] [CrossRef] [Green Version]
- You, M.; Ku, P.T.; Hrdlicková, R.; Bose, H.R., Jr. ch-IAP1, a member of the inhibitor-of-apoptosis protein family, is a mediator of the antiapoptotic activity of the v-Rel oncoprotein. Mol. Cell. Biol. 1997, 17, 7328–7341. [Google Scholar] [CrossRef] [Green Version]
- Stehlik, C.; de Martin, R.; Kumabashiri, I.; Schmid, J.A.; Binder, B.R.; Lipp, J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J. Exp. Med. 1998, 188, 211–216. [Google Scholar] [CrossRef]
- Grumont, R.J.; Rourke, I.J.; Gerondakis, S. Rel-dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation-induced apoptosis. Genes Dev. 1999, 13, 400–411. [Google Scholar] [CrossRef]
- Kreuz, S.; Siegmund, D.; Scheurich, P.; Wajant, H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol. Cell. Biol. 2001, 21, 3964–3973. [Google Scholar] [CrossRef] [Green Version]
- Tamatani, M.; Che, Y.H.; Matsuzaki, H.; Ogawa, S.; Okado, H.; Miyake, S.; Mizuno, T.; Tohyama, M. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J. Biol. Chem. 1999, 274, 8531–8538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahon, T.M.; O’Neill, L.A. Studies into the effect of the tyrosine kinase inhibitor herbimycin A on NF-kappa B activation in T lymphocytes. Evidence for covalent modification of the p50 subunit. J. Biol. Chem. 1995, 270, 28557–28564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, K.; Singh, S.; Burke, T.R., Jr.; Grunberger, D.; Aggarwal, B.B. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc. Natl. Acad. Sci. USA 1996, 93, 9090–9095. [Google Scholar] [CrossRef] [Green Version]
- Sandur, S.K.; Ichikawa, H.; Sethi, G.; Ahn, K.S.; Aggarwal, B.B. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) suppresses NF-kappaB activation and NF-kappaB-regulated gene products through modulation of p65 and IkappaBalpha kinase activation, leading to potentiation of apoptosis induced by cytokine and chemotherapeutic agents. J. Biol. Chem. 2006, 281, 17023–17033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef]
- Ding, G.J.; Fischer, P.A.; Boltz, R.C.; Schmidt, J.A.; Colaianne, J.J.; Gough, A.; Rubin, R.A.; Miller, D.K. Characterization and quantitation of NF-kappaB nuclear translocation induced by interleukin-1 and tumor necrosis factor-alpha. Development and use of a high capacity fluorescence cytometric system. J. Biol. Chem. 1998, 273, 28897–28905. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Pagès, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [CrossRef]
- Balkwill, F.; Coussens, L.M. Cancer: An inflammatory link. Nature 2004, 431, 405–406. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Vilimas, T.; Mascarenhas, J.; Palomero, T.; Mandal, M.; Buonamici, S.; Meng, F.; Thompson, B.; Spaulding, C.; Macaroun, S.; Alegre, M.L.; et al. Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat. Med. 2007, 13, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Gupta, S.C.; Park, B.; Yadav, V.R.; Aggarwal, B.B. Turmeric (Curcuma longa) inhibits inflammatory nuclear factor (NF)-κB and NF-κB-regulated gene products and induces death receptors leading to suppressed proliferation, induced chemosensitization, and suppressed osteoclastogenesis. Mol. Nutr. Food Res. 2012, 56, 454–465. [Google Scholar] [CrossRef] [Green Version]
- Buhrmann, C.; Popper, B.; Aggarwal, B.B.; Shakibaei, M. Resveratrol downregulates inflammatory pathway activated by lymphotoxin alpha (TNF-beta) in articular chondrocytes: Comparison with TNF-alpha. PLoS ONE 2017, 12, e0186993. [Google Scholar] [CrossRef]
- Buhrmann, C.; Yazdi, M.; Popper, B.; Shayan, P.; Goel, A.; Aggarwal, B.B.; Shakibaei, M. Resveratrol Chemosensitizes TNF-beta-Induced Survival of 5-FU-Treated Colorectal Cancer Cells. Nutrients 2018, 10, 888. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Takada, Y.; Boriek, A.M.; Aggarwal, B.B. Nuclear factor-kappaB: Its role in health and disease. J. Mol. Med. 2004, 82, 434–448. [Google Scholar] [CrossRef]
- García-Piñeres, A.J.; Castro, V.; Mora, G.; Schmidt, T.J.; Strunck, E.; Pahl, H.L.; Merfort, I. Cysteine 38 in p65/NF-kappaB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J. Biol. Chem. 2001, 276, 39713–39720. [Google Scholar] [CrossRef] [Green Version]
- Sethi, G.; Ahn, K.S.; Aggarwal, B.B. Targeting nuclear factor-kappa B activation pathway by thymoquinone: Role in suppression of antiapoptotic gene products and enhancement of apoptosis. Mol. Cancer Res. 2008, 6, 1059–1070. [Google Scholar] [CrossRef] [Green Version]
- Gali-Muhtasib, H.; Diab-Assaf, M.; Boltze, C.; Al-Hmaira, J.; Hartig, R.; Roessner, A.; Schneider-Stock, R. Thymoquinone extracted from black seed triggers apoptotic cell death in human colorectal cancer cells via a p53-dependent mechanism. Int. J. Oncol. 2004, 25, 857–866. [Google Scholar] [PubMed]
- Sethi, G.; Sung, B.; Aggarwal, B.B. TNF: A master switch for inflammation to cancer. Front. Biosci. 2008, 13, 5094–5107. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buhrmann, C.; Shayan, P.; Banik, K.; Kunnumakkara, A.B.; Kubatka, P.; Koklesova, L.; Shakibaei, M. Targeting NF-κB Signaling by Calebin A, a Compound of Turmeric, in Multicellular Tumor Microenvironment: Potential Role of Apoptosis Induction in CRC Cells. Biomedicines 2020, 8, 236. https://doi.org/10.3390/biomedicines8080236
Buhrmann C, Shayan P, Banik K, Kunnumakkara AB, Kubatka P, Koklesova L, Shakibaei M. Targeting NF-κB Signaling by Calebin A, a Compound of Turmeric, in Multicellular Tumor Microenvironment: Potential Role of Apoptosis Induction in CRC Cells. Biomedicines. 2020; 8(8):236. https://doi.org/10.3390/biomedicines8080236
Chicago/Turabian StyleBuhrmann, Constanze, Parviz Shayan, Kishore Banik, Ajaikumar B. Kunnumakkara, Peter Kubatka, Lenka Koklesova, and Mehdi Shakibaei. 2020. "Targeting NF-κB Signaling by Calebin A, a Compound of Turmeric, in Multicellular Tumor Microenvironment: Potential Role of Apoptosis Induction in CRC Cells" Biomedicines 8, no. 8: 236. https://doi.org/10.3390/biomedicines8080236
APA StyleBuhrmann, C., Shayan, P., Banik, K., Kunnumakkara, A. B., Kubatka, P., Koklesova, L., & Shakibaei, M. (2020). Targeting NF-κB Signaling by Calebin A, a Compound of Turmeric, in Multicellular Tumor Microenvironment: Potential Role of Apoptosis Induction in CRC Cells. Biomedicines, 8(8), 236. https://doi.org/10.3390/biomedicines8080236