Novel Blood-Derived Extracellular Vesicle-Based Biomarkers in Alzheimer’s Disease Identified by Proximity Extension Assay

 , , ,
, , , 
Abstract
1. Introduction
2. Experimental Section
2.1. Study Participants
2.2. Sample Collection
2.3. Biochemical Analysis
2.4. Extracellular Vesicle Enrichment and Preparation
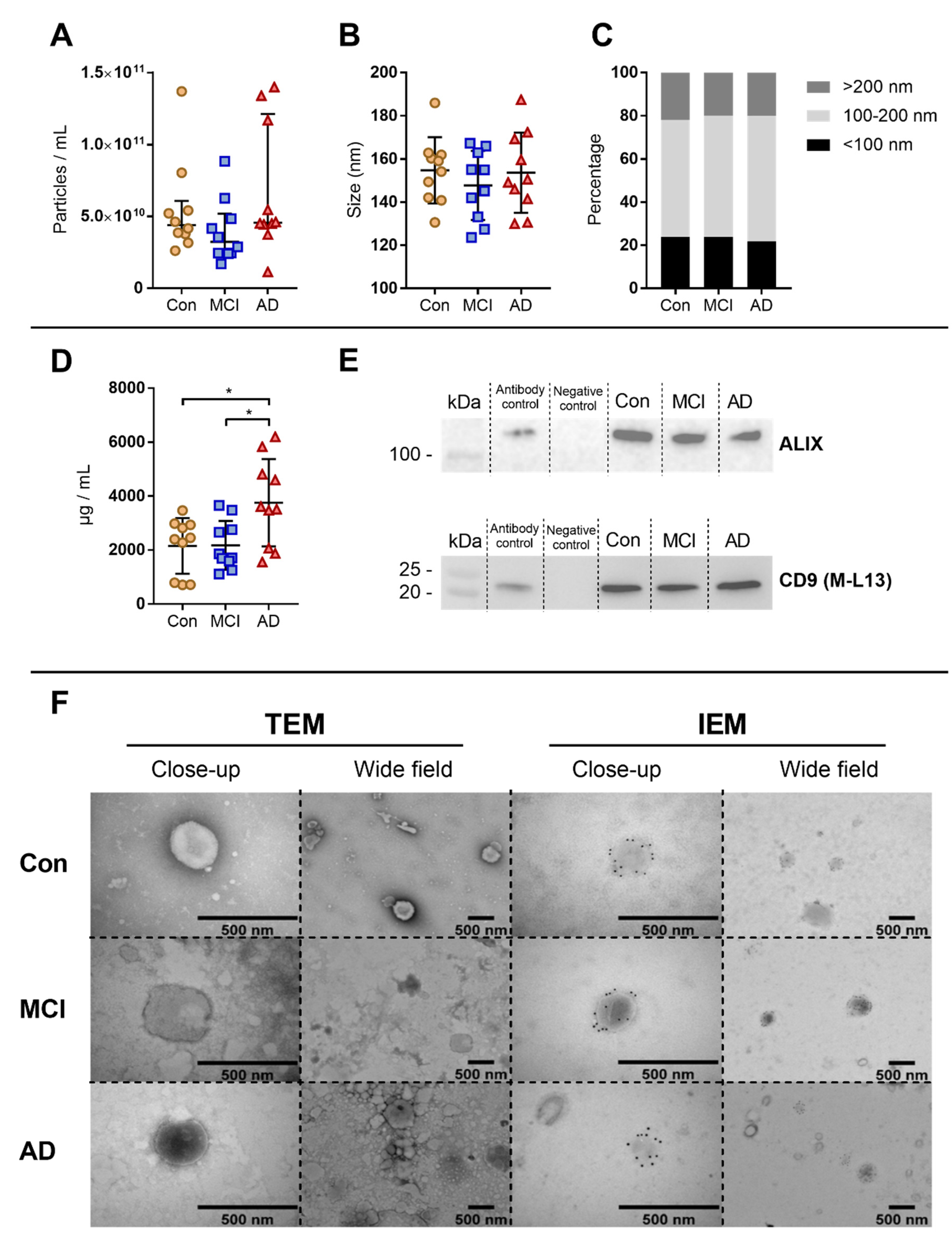
2.5. Characterization of Extracellular Vesicles
2.5.1. Nanoparticle Tracking Analysis
2.5.2. Western Blotting
2.5.3. Transmission Electron Microscopy with Immunogold Labelling
2.6. Proximity Extension Assay
2.7. Statistical Analysis
3. Results
3.1. Characteristics of Study Participants
3.2. Characterisation of Extracellular Vesicles Enrichment
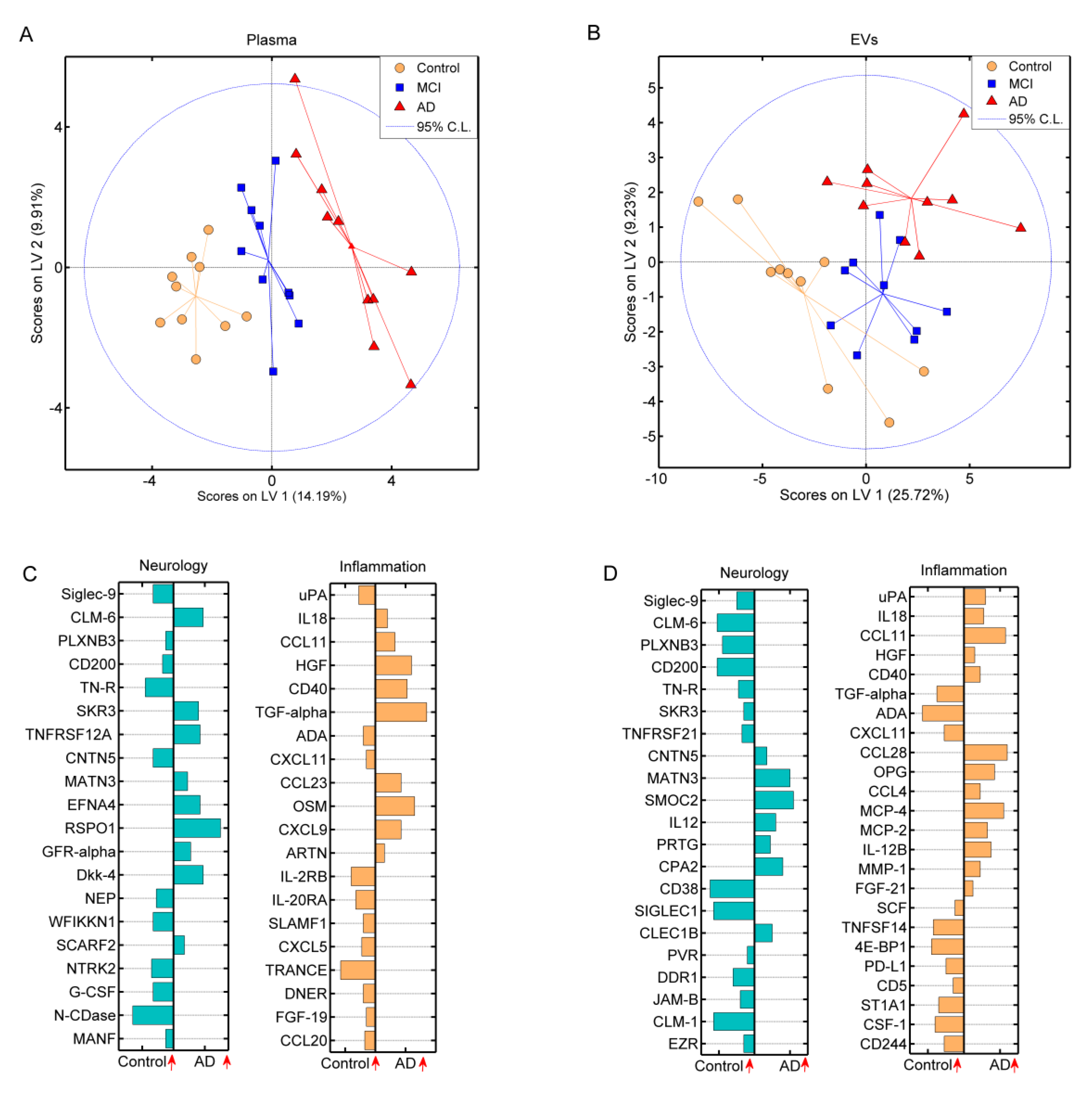
3.3. Multiplex Detection of Plasma and Extracellular Vesicle Protein Profiles by the Proximity Extension Assay
3.4. Differentially Expressed Proteins Related to Alzheimer’s Dementia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Association, A. 2020 Alzheimer’s disease facts and figures. J. Alzheimer’s Assoc. 2020, 16, 391–460. [Google Scholar]
- Alzheimer’s Disease International: World Alzheimer Report 2019 Attitudes to dementia. Available online: https://www.alz.co.uk/research/WorldAlzheimerReport2019.pdf (accessed on 6 July 2020).
- Judge, D.; Roberts, J.; Khandker, R.K.; Ambegaonkar, B.; Black, C.M. Physician Practice Patterns Associated with Diagnostic Evaluation of Patients with Suspected Mild Cognitive Impairment and Alzheimer’s Disease. Int. J. Alzheimers. Dis. 2019, 2019, 4942562. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 1–13. [Google Scholar] [CrossRef]
- Riphagen, J.M.; Ramakers, I.H.G.M.; Freeze, W.M.; Pagen, L.H.G.; Hanseeuw, B.J.; Verbeek, M.M.; Verhey, F.R.J.; Jacobs, H.I.L. Linking APOE-ε4, blood-brain barrier dysfunction, and inflammation to Alzheimer’s pathology. Neurobiol. Aging 2020, 85, 96–103. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Orazio, L.M.D.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Piazza, F.; Galimberti, G.; Conti, E.; Isella, V.; Perlangeli, M.V.; Speranza, T.; Borroni, B.; Pogliani, E.M.; Padovani, A.; Ferrarese, C. Increased tissue factor pathway inhibitor and homocysteine in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L. The Human Plasma Proteome: History, Character, and Diagnostic Prospects. Mol. Cell. Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Gézsi, A.; Kovács, Á.; Visnovitz, T.; Buzás, E.I. Systems biology approaches to investigating the roles of extracellular vesicles in human diseases. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef]
- Garcia-Romero, N.; Esteban-Rubio, S.; Rackov, G.; Carrión-Navarro, J.; Belda-Iniesta, C.; Ayuso-Sacido, A. Extracellular vesicles compartment in liquid biopsies: Clinical application. Mol. Asp. Med. 2018, 60, 27–37. [Google Scholar] [CrossRef]
- Court, F.A.; Midha, R.; Cisterna, B.A.; Grochmal, J.; Shakhbazau, A.; Hendriks, W.T.; Van Minnen, J. Morphological evidence for a transport of ribosomes from Schwann cells to regenerating axons. Glia 2011, 59, 1529–1539. [Google Scholar] [CrossRef]
- Lachenal, G.; Pernet-Gallay, K.; Chivet, M.; Hemming, F.J.; Belly, A.; Bodon, G.; Blot, B.; Haase, G.; Goldberg, Y.; Sadoul, R. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell. Neurosci. 2011, 46, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.G.; Gray, E.; Heman-Ackah, S.M.; Mäger, I.; Talbot, K.; El Andaloussi, S.; Wood, M.J.; Turner, M.R. Extracellular vesicles in neurodegenerative disease-pathogenesis to biomarkers. Nat. Rev. Neurol. 2016, 12, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D.; Schwartz, J.B. Declining levels of functionally specialized synaptic proteins in plasma neuronal exosomes with progression of Alzheimer’s disease. Faseb J. 2018, 32, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Nogueras-Ortiz, C.; Mustapic, M.; Mullins, R.J.; Abner, E.L.; Schwartz, J.B.; Kapogiannis, D. Deficient neurotrophic factors of CSPG4-type neural cell exosomes in Alzheimer disease. Faseb J. 2019, 33, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D. High complement levels inastrocyte-derived exosomes of Alzheimer’s disease. Ann. Neurol. 2018, 83, 544–552. [Google Scholar] [CrossRef]
- Hosseinzadeh, S.; Noroozian, M.; Mortaz, E.; Mousavizadeh, K. Plasma microparticles in alzheimer’s disease: The role of vascular dysfunction. Metab. Brain Dis. 2018, 33, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Cai, X.; Li, W.; Zhang, Z.; Dong, W.; Hui, G. Elevated plasma endothelial microparticles in alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2012, 34, 174–180. [Google Scholar] [CrossRef]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Plaque Load in the 5XFAD Mouse Model of Alzheimer ’ s Disease. Neurobiol. Aging 2014, 35, 1792–1800. [Google Scholar] [CrossRef]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Götz, J. Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Mitsutake, S.; Igarashi, Y. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-B by microglia. J. Biol. Chem. 2012, 287, 10977–10989. [Google Scholar] [CrossRef] [PubMed]
- Simón, D.; García-García, E.; Royo, F.; Falcón-Pérez, J.M.; Avila, J. Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. Febs Lett. 2012, 586, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xia, W. Proteomic Profiling of Plasma and Brain Tissue from Alzheimer’s Disease Patients Reveals Candidate Network of Plasma Biomarkers. J. Alzheimer’s Dis. 2020, 76, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Westwood, S.; Baird, A.L.; Winchester, L.; Dobricic, V.; Kilpert, F.; Hong, S.; Franke, A.; Hye, A.; Ashton, N.J.; et al. Discovery and validation of plasma proteomic biomarkers relating to brain amyloid burden by SOMAscan assay. Alzheimer’s Dement. 2019, 15, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Perrotte, M.; Haddad, M.; Le, A.; Frost, E.H.; Fulöp, T.; Ramassamy, C. Profile of pathogenic proteins in total circulating extracellular vesicles in mild cognitive impairment and during the progression of Alzheimer’s disease. Neurobiol. Aging 2020, 86, 102–111. [Google Scholar] [CrossRef]
- Hurwitz, S.N.; Sun, L.; Cole, K.Y.; Ford, C.R.; Olcese, J.M.; Meckes, D.G. An optimized method for enrichment of whole brain-derived extracellular vesicles reveals insight into neurodegenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. Methods 2018, 307, 210–220. [Google Scholar] [CrossRef]
- Kreimer, S.; Belov, A.M.; Ghiran, I.; Murthy, S.K.; Frank, D.A.; Ivanov, A.R. Mass-spectrometry-based molecular characterization of extracellular vesicles: Lipidomics and proteomics. J. Proteome Res. 2015, 14, 2367–2384. [Google Scholar] [CrossRef]
- Assarsson, E.; Lundberg, M.; Holmquist, G.; Björkesten, J.; Thorsen, S.B.; Ekman, D.; Eriksson, A.; Dickens, E.R.; Ohlsson, S.; Edfeldt, G.; et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 2014, 9, e95192. [Google Scholar] [CrossRef]
- Lundberg, M.; Eriksson, A.; Tran, B.; Assarsson, E.; Fredriksson, S. Homogeneous antibody-based proximity extension assays provide sensitive and specific detection of low-abundant proteins in human blood. Nucleic Acids Res. 2011, 39, e102. [Google Scholar] [CrossRef] [PubMed]
- WHO International Classification of Diseases (ICD). Available online: http://www.who.int/classifications/icd/en/ (accessed on 22 June 2020).
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939. [Google Scholar] [CrossRef]
- Petersen, R.C. Mild cognitive impairment as a diagnostic entity. J. Intern. Med. 2004, 256, 183–194. [Google Scholar] [CrossRef]
- Lacroix, R.; Judicone, C.; Poncelet, P.; Robert, S.; Arnaud, L.; Sampol, J.; Dignat-George, F. Impact of pre-analytical parameters on the measurement of circulating microparticles: Towards standardization of protocol. J. Thromb. Haemost. 2011, 10, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Thermo Scientific, M-PER® Mammalian Protein Extraction Reagent. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0011378_MPER_Mammal_Protein_Extract_Reag_UG.pdf (accessed on 6 July 2020).
- Nielsen, T.; Kristensen, S.R.; Gregersen, H.; Teodorescu, E.M.; Christiansen, G.; Pedersen, S. Extracellular vesicle-associated procoagulant phospholipid and tissue factor activity in multiple myeloma. PLoS ONE 2019, 14, e0210835. [Google Scholar] [CrossRef] [PubMed]
- Thermo Scientific, PierceTM BCA Protein Assay Kit. Available online: https://tools.thermofisher.com/content/sfs/manuals/MAN0011430_Pierce_BCA_Protein_Asy_UG.pdf (accessed on 20 February 2020).
- Olink Proteomics. Strategies for Design of Protein Biomarker Studies; Olink Proteomics: Uppsala, Sweden, 2018. [Google Scholar]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Youden, W.J. Index for rating diagnostic tests. Cancer 1950, 3, 32–35. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014, 21, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Agosta, F.; Libera, D.D.; Spinelli, E.G.; Finardi, A.; Canu, E.; Bergami, A.; Chiavetto, L.B.; Baronio, M.; Comi, G.; Martino, G.; et al. Myeloid microvesicles in cerebrospinal fluid are associated with myelin damage and neuronal loss in mild cognitive impairment and alzheimer disease. Ann. Neurol. 2014, 76, 813–825. [Google Scholar] [CrossRef]
- Gidlöf, O.; Evander, M.; Rezeli, M.; Marko-Varga, G.; Laurell, T.; Erlinge, D. Proteomic profiling of extracellular vesicles reveals additional diagnostic biomarkers for myocardial infarction compared to plasma alone. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Blacher, E.; Dadali, T.; Bespalko, A.; Haupenthal, V.J.; Grimm, M.O.W.; Hartmann, T.; Lund, F.E.; Stein, R.; Levy, A. Alzheimer’s disease pathology is attenuated in a CD38-deficient mouse model. Ann. Neurol. 2015, 78, 88–103. [Google Scholar] [CrossRef]
- Ejarque-Ortiz, A.; Solà, C.; Martínez-Barriocanal, Á.; Schwartz, S.; Martín, M.; Peluffo, H.; Sayós, J. The receptor CMRF35-like molecule-1 (CLM-1) enhances the production of LPS-induced pro-inflammatory mediators during microglial activation. PLoS ONE 2015, 10, 1–17. [Google Scholar] [CrossRef][Green Version]
- Xi, H.; Katschke, K.J.; Helmy, K.Y.; Wark, P.A.; Kljavin, N.; Clark, H.; Eastham-Anderson, J.; Shek, T.; Roose-Girma, M.; Ghilardi, N.; et al. Negative regulation of autoimmune demyelination by the inhibitory receptor CLM-1. J. Exp. Med. 2010, 207, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Simhadri, V.R.; Mariano, J.L.; Gil-Krzewska, A.; Zhou, Q.; Borrego, F. CD300c is an activating receptor expressed on human monocytes. J. Innate Immun. 2013, 5, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Su, M.; Lin, Y.; Lai, L. A CD300c-Fc fusion protein inhibits t cell immunity. Front. Immunol. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.S.; Matar, R.; Merheb, M.; Hodeify, R.; Vazhappilly, C.G.; Marton, J.; Shamsuddin, S.A.; Al Zouabi, H. Siglecs in Brain Function and Neurological Disorders. Cells 2019, 8, 1125. [Google Scholar] [CrossRef]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene expression profiles and transcriptional regulatory pathways underlying mouse tissue macrophage identity and diversity. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef]
- Claude, J.; Linnartz-Gerlach, B.; Kudin, A.P.; Kunz, W.S.; Neumann, H. Microglial CD33-related siglec-E Inhibits neurotoxicity by preventing the phagocytosis-associated oxidative burst. J. Neurosci. 2013, 33, 18270–18276. [Google Scholar] [CrossRef]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; De Lima, K.A.; Gutiérrez-Vázquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef]
- Teixeira, A.L.; Gama, C.S.; Rocha, N.P.; Teixeira, M.M. Revisiting the role of eotaxin-1/CCL11 in psychiatric disorders. Front. Psychiatry 2018, 9, 1–6. [Google Scholar] [CrossRef]
- Zhu, C.; Xu, B.; Sun, X.; Zhu, Q.; Sui, Y. Targeting CCR3 to Reduce Amyloid-β Production, Tau Hyperphosphorylation, and Synaptic Loss in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 7964–7978. [Google Scholar] [CrossRef]
- Chasaide, C.N.; Lynch, M.A. The role of the immune system in driving neuroinflammation. Brain Neurosci. Adv. 2020, 4, 1–8. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213. [Google Scholar] [CrossRef]
- Yu, H.; Gonzalez-gil, A.; Wei, Y.; Fernandes, S.M.; Porell, R.N.; Vajn, K.; Paulson, J.C.; Nycholat, C.M.; Schnaar, R.L. Siglec-8 and Siglec-9 binding speci fi cities and endogenous airway ligand distributions and properties. Glycobiology 2017, 27, 657–668. [Google Scholar] [CrossRef]
- Shik, D.; Moshkovits, I.; Reichman, H.; Munitz, A. Interleukin-33 requires CMRF35-like molecule-1 expression for induction of myeloid cell activation. Allergy 2014, 69, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Whelan, C.D.; Mattsson, N.; Nagle, M.W.; Vijayaraghavan, S.; Hyde, C.; Janelidze, S.; Stomrud, E.; Lee, J.; Fitz, L.; Samad, T.A.; et al. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 7, 1–14. [Google Scholar] [CrossRef]
- Tietz, S.; Périnat, T.; Greene, G.; Enzmann, G.; Deutsch, U.; Adams, R.; Imhof, B.; Aurrand-Lions, M.; Engelhardt, B. Lack of junctional adhesion molecule (JAM)-B ameliorates experimental autoimmune encephalomyelitis. Brain. Behav. Immun. 2018, 73, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Baird, A.L.; Westwood, S.; Lovestone, S. Blood-based proteomic biomarkers of Alzheimer’s disease pathology. Front. Neurol. 2015, 6, 236. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Léger, C.A.; Aurrand-Lions, M.; Beltraminelli, N.; Fasel, N.; Imhof, B.A. Junctional adhesion molecule-2 (JAM-2) promotes lymphocyte transendothelial migration. Blood 2002, 100, 2479–2486. [Google Scholar] [CrossRef]
- Weber, C.; Fraemohs, L.; Dejana, E. The role of junctional adhesion molecules in vascular inflammation. Nat. Rev. Immunol. 2007, 7, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Merino, P.; Diaz, A.; Jeanneret, V.; Wu, F.; Torre, E.; Cheng, L.; Yepes, M. Urokinase-type plasminogen activator (uPA) binding to the uPA receptor (uPAR) promotes axonal regeneration in the central nervous system. J. Biol. Chem. 2017, 292, 2741–2753. [Google Scholar] [CrossRef]
- Merino, P.; Diaz, A.; Manrique, L.G.; Cheng, L.; Yepes, M. Urokinase-type plasminogen activator (uPA) promotes ezrin-mediated reorganization of the synaptic cytoskeleton in the ischemic brain. J. Biol. Chem. 2018, 293, 9234–9247. [Google Scholar] [CrossRef] [PubMed]
- Tucker, H.M.; Kihiko-Ehmann, M.; Estus, S. Urokinase-type plasminogen activator inhibits amyloid-β neurotoxicity and fibrillogenesis via plasminogen. J. Neurosci. Res. 2002, 70, 249–255. [Google Scholar] [CrossRef]
- Cunningham, O.; Campion, S.; Perry, V.H.; Murray, C.; Sidenius, N.; Docagne, F.; Cunningham, C. Microglia and the Urokinase Plasminogen Activator Receptor/uPA System in Innate Brain Inflammation. Glia 2009, 1814, 1802–1814. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Wagner, M.R.; Zhang, W.; Xu, F.; Nostrand, W.E. Van Amyloid B-Protein Stimulates the Expression of Urokinase-type Plasminogen Activator (uPA) and Its Receptor (uPAR) in Human Cerebrovascular Smooth Muscle Cells. J. Biol. Chem. 2003, 278, 19054–19061. [Google Scholar] [CrossRef]
- Varnum, M.M.; Kiyota, T.; Ingraham, K.L.; Ikezu, S.; Ikezu, T. The Anti-inflammatory glycoprotein, CD200, Restores Neurogenesis and Enhances Amyloid Phagocytosis in a Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2015, 36, 2995–3007. [Google Scholar] [CrossRef] [PubMed]
- Pulliam, L.; Sun, B.; Mustapic, M.; Chawla, S.; Kapogiannis, D. Plasma neuronal exosomes serve as biomarkers of cognitive impairment in HIV infection and Alzheimer’s disease. J. Neurovirol. 2019, 25, 702–709. [Google Scholar] [CrossRef]
- Tsuboi, Y.; Kakimoto, K.; Nakajima, M.; Akatsu, H.; Yamamoto, T.; Ogawa, K.; Ohnishi, T.; Daikuhara, Y.; Yamada, T. Increased hepatocyte growth factor level in cerebrospinal fluid in Alzheimer’s disease. Acta Neurol. Scand. 2003, 107, 81–86. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject Group | ||||
|---|---|---|---|---|
| Con (n = 10) | MCI (n = 10) | AD (n = 10) | Reference Interval * | |
| Demographics | ||||
| Age (years) b | 65 (65–66) | 72 (69–76) | 72 (68–74) | - |
| Male/female (n) | 4/6 | 2/8 | 4/6 | - |
| Biochemical parameters | ||||
| Alanine transaminase (U/I) b | 19.1 (18.0–34.7) | 20.1 (15.4–45.4) | 20.0 (18.0–29.5) | 10.0–50.0 |
| Albumin (g/L) a | 39.9 ± 2.3 | 39.4 ± 2.2 | 40.6 ± 2.5 | 34.0–45.0 |
| Carbamide (mmol/L) b | 4.6 (4.3–6.1) | 5.6 (5.2–7.1) | 5.2 (4.8–6.4) | 3.1–8.1 |
| Cholesterol (mmol/L) a | 5.3 ± 0.7 | 5.8 ± 1.5 | 4.5 ± 0.9 | 4.1–8.5 |
| Creatinine (µmol/L) b | 72.0 (60.8–86.1) | 65.7 (60.5–80.1) | 72.4 (66.9–88.9) | 45.0–105.0 |
| C-reactive protein (mg/L) b | 1.3 (0.5–3.2) | 1.4 (0.6–3.7) | 0.7 (0.3–2.6) | ≤ 8.0 |
| Glucose (mmol/L) b | 5.4 (4.8–5.6) | 5.0 (4.5–5.6) | 5.1 (4.9–5.4) | 4.2–7.8 |
| Haemoglobin (mmol/L) a | 9.0 ± 0.9 | 8.4 ± 0.8 (n = 8) | 9.0 ± 0.5 (n = 9) | 7.3–10.5 |
| High density lipoprotein cholesterol (mmol/L) a | 1.5 ± 0.4 | 1.8 ± 0.5 | 1.7 ± 0.4 | 0.8–2.0 |
| Lactate dehydrogenase (U/I) a | 164.2 ± 22.0 | 179.6 ± 32.6 | 181.4 ± 9.9 | 105.0–255.0 |
| Low density lipoprotein cholesterol (mmol/L) a | 3.0 ± 0.7 | 3.3 ± 1.1 | 2.2 ± 0.6 | 1.8–4.5 |
| Triglyceride (mmol/L) b | 1.5 (1.0–2.4) | 1.3 (0.9–2.0) | 1.1 (1.0–1.6) | ≤ 2.0 |
| Clinical parameters | ||||
| Mini Mental State Examination b | - | 27.5 (26.0–30.0) | 25.5 (21.3–27.3) | - |
| Functional Activities Questionnaire a | - | 4.0 ± 2.0 (n = 3) | 10.4 ± 4.6 (n = 5) | - |
| Addenbrooke’s Cognitive Examinationa | - | 85.0 ± 5.6 (n = 6) | 58.7 ± 16.5 (n = 3) | - |
| CSF Aβ (ng/L) a | - | 998.5 ± 482.6 (n = 4) | 626.3 ± 260.9 (n = 6) | > 500 |
| CSF phospho-tau (ng/L) a | - | 98.0 ± 61.3 (n = 4) | 80.5 ± 29.5 (n = 6) | < 61 |
| CSF tau (ng/L) a | - | 563.0 ± 363.9 (n = 4) | 628.2 ± 288.9 (n = 6) | < 450 (51–70 years) < 500 (71–90 years) |
| Neurology Panel—Plasma | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Protein | NPX | Fold Change | AD│Con | Con │MCI│AD | ||||||
| Con | MCI | AD | MCI│Con | AD│Con | AD│MCI | p-Value | FDR | p-Value | FDR | |
| N-CDase | 4.42 ± 0.58 | 4.49 ± 0.62 | 3.73 ± 0.39 | 1.07 | 0.60 | 0.56 | 0.006 | 0.212 | 0.006 | 0.436 |
| RSPO1 | 3.83 ± 0.23 | 4.08 ± 0.24 | 4.20 ± 0.30 | 1.19 | 1.30 | 1.10 | 0.006 | 0.424 | 0.010 | 0.336 |
| TNFRSF12A | 6.00 ± 0.24 | 6.27 ± 0.28 | 6.27 ± 0.28 | 1.21 | 1.22 | 1.01 | 0.029 | 0.596 | 0.043 | 0.877 |
| TN-R | 4.24 ± 0.47 | 4.29 ± 0.47 | 3.81 ± 0.30 | 1.03 | 0.72 | 0.70 | 0.026 | 0.728 | 0.031 | 0.829 |
| NTRK2 | 5.85 ± 0.24 | 5.96 ± 0.14 | 5.74 ± 0.17 | 1.07 | 0.92 | 0.86 | 0.247 | 1.000 | <0.050 | 0.817 |
| Neurology Panel—EVs | ||||||||||
| CD38 | 0.94 ± 0.73 | 0.27 ± 0.20 | 0.24 ± 0.38 | 0.56 | 0.56 | 1.00 | 0.015 | 0.260 | 0.005 | 0.022 |
| CLM-1 | 2.64 ± 0.72 | 1.70 ± 0.43 | 1.65 ± 0.64 | 0.49 | 0.50 | 1.00 | 0.004 | 0.136 | 0.001 | 0.024 |
| CLM-6 | 2.33 ± 0.59 | 1.70 ± 0.23 | 1.75 ± 0.39 | 0.61 | 0.64 | 1.06 | 0.018 | 0.207 | 0.004 | 0.027 |
| JAM-B | 5.08 (4.52–5.42) | 4.13 (3.87–4.64) | 4.33 (3.99–4.87) | 0.51 | 0.60 | 1.16 | 0.029 a | 0.194 | 0.029 b | 0.101 |
| Siglec-9 | 3.76 ± 0.69 | 2.85 ± 0.31 | 3.06 ± 0.50 | 0.49 | 0.59 | 1.20 | 0.019 | 0.132 | 0.002 | 0.016 |
| SIGLEC1 | 3.10 ± 1.29 | 2.29 ± 0.48 | 1.89 ± 0.70 | 0.41 | 0.33 | 0.81 | 0.019 | 0.164 | 0.017 | 0.089 |
| SKR3 | 5.73 ± 1.05 | 4.78 ± 0.46 | 4.83 ± 0.75 | 0.43 | 0.47 | 1.09 | 0.041 | 0.218 | 0.020 | 0.067 |
| CD200 | 1.12 ± 0.83 | 0.36 ± 0.38 | 0.65 ± 0.51 | 0.54 | 0.67 | 1.25 | 0.148 | 0.245 | 0.030 | 0.080 |
| CLEC1B | 8.52 ± 1.16 | 7.78 ± 0.62 | 7.50 ± 0.97 | 0.51 | 0.48 | 0.95 | 0.048 | 0.225 | 0.062 | 0.143 |
| EZR | 4.30 (3.24–4.63) | 3.20 (2.30–3.70) | 3.66 (3.37–4.10) | 0.48 | 0.64 | 1.34 | 0.350 | 0.435 | 0.021b | 0.075 |
| gal-8 | 5.06 ± 0.87 | 4.17 ± 0.43 | 4.49 ± 0.73 | 0.48 | 0.65 | 1.37 | 0.131 | 0.229 | 0.020 | 0.067 |
| PLXNB3 | 1.42 ± 0.52 | 0.91 ± 0.30 | 1.02 ± 0.33 | 0.68 | 0.73 | 1.08 | 0.052 | 0.212 | 0.019 | 0.081 |
| TN-R | 1.39 ± 0.47 | 1.03 ± 0.39 | 0.90 ± 0.47 | 0.77 | 0.71 | 0.93 | 0.032 | 0.199 | 0.056 | 0.137 |
| Inflammation Panel—Plasma | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Protein | NPX | Fold Change | AD│Con | Con │MCI│AD | ||||||
| Con | MCI | AD | MCI│Con | AD│Con | AD│MCI | p-Value | FDR | p-Value | FDR | |
| IL-2RB | 1.29 ± 0.28 | 1.04 ± 0.18 | 1.03 ± 0.26 | 0.83 | 0.83 | 1.00 | 0.041 | 0.442 | 0.036 | 0.799 |
| OSM | 2.85 ± 0.56 | 3.39 ± 0.72 | 4.08 ± 1.08 | 1.52 | 2.84 | 1.87 | 0.005 | 0.150 | 0.009 | 0.338 |
| TGF-α | 3.76 ± 0.22 | 4.09 ± 0.42 | 4.34 ± 0.37 | 1.30 | 1.53 | 1.18 | <0.000 | 0.020 | 0.003 | 0.208 |
| TRANCE | 5.09 ± 0.36 | 4.90 ± 0.43 | 4.49 ± 0.72 | 0.89 | 0.71 | 0.80 | 0.030 | 0.477 | 0.048 | 0.631 |
| CXCL1 | 7.45 ± 0.75 | 7.05 ± 0.43 | 6.84 ± 0.45 | 0.70 | 0.61 | 0.87 | 0.039 | 0.499 | 0.061 | 0.703 |
| CXCL9 | 7.63 ± 0.61 | 8.32 ± 1.09 | 8.36 ± 0.92 | 2.10 | 1.81 | 0.86 | 0.049 | 0.395 | 0.139 | 1.000 |
| GDNF | 2.35 ± 0.25 | 2.65 ± 0.34 | 2.38 ± 0.22 | 1.24 | 1.02 | 0.82 | 0.786 | 0.940 | 0.042 | 0.693 |
| HGF | 8.21 ± 0.35 | 8.50 ± 0.43 | 8.60 ± 0.39 | 1.24 | 1.32 | 1.06 | 0.029 | 0.607 | 0.081 | 0.717 |
| Inflammation Panel–EVs | ||||||||||
| 4E-BP1 | 1.66 ± 0.65 | 1.11 ± 0.32 | 1.12 ± 0.42 | 0.64 | 0.65 | 1.02 | 0.040 | 0.345 | 0.025 | 0.189 |
| ADA | 1.81 ± 0.72 | 1.19 ± 0.26 | 1.24 ± 0.26 | 0.59 | 0.61 | 1.03 | 0.029 | 0.364 | 0.010 | 0.396 |
| CCL11 | 0.75 (0.68–0.91) | 0.78 (0.69–1.00) | 1.03 (0.93–1.25) | 1.02 | 1.21 | 1.19 | 0.001 | 0.024 | 0.011 b | 0.185 |
| CD244 | 5.84 ± 0.96 | 4.96 ± 0.39 | 4.93 ± 0.69 | 0.47 | 0.49 | 1.05 | 0.026 | 0.435 | 0.012 | 0.224 |
| CD40 | 10.15 ± 1.13 | 9.17 ± 0.50 | 9.00 ± 0.93 | 0.42 | 0.43 | 1.02 | 0.024 | 0.590 | 0.016 | 0.201 |
| TGF-α | 0.41 ± 0.26 | 0.14 ± 0.28 | 0.13 ± 0.28 | 0.83 | 0.83 | 1.00 | 0.034 | 0.338 | 0.049 | 0.149 |
| CD5 | 5.62 ± 1.19 | 4.45 ± 0.68 | 4.88 ± 0.57 | 0.36 | 0.47 | 1.31 | 0.091 | 0.372 | 0.016 | 0.151 |
| CXCL1 | 3.48 ± 1.03 | 3.03 ± 0.47 | 2.64 ± 0.71 | 0.63 | 0.53 | 0.84 | 0.048 | 0.307 | 0.069 | 0.196 |
| IL-18R1 | 5.26 ± 1.04 | 4.38 ± 0.60 | 4.73 ± 0.43 | 0.49 | 0.59 | 1.22 | 0.155 | 0.386 | 0.041 | 0.176 |
| SCF | 1.89 (1.55–2.60) | 1.94 (1.81–2.22) | 1.48 (1.30–1.87) | 1.03 | 0.75 | 0.73 | 0.071 | 0.328 | 0.034 b | 0.160 |
| TNFRSF9 | 1.42 (1.02–1.69) | 0.91 (0.77–1.16) | 1.20 (0.98–1.48) | 0.70 | 0.86 | 1.22 | 0.739 a | 0.945 | 0.033 b | 0.151 |
| uPA | 5.06 (4.60–5.53) | 4.22 (3.95–4.61) | 4.37 (4.21–4.54) | 0.56 | 0.63 | 1.12 | 0.052 a | 0.347 | 0.042 b | 0.166 |
| Protein(s) | AUC | Sensitivity (%) | Specificity (%) | 95% CI | p-Value |
|---|---|---|---|---|---|
| Plasma | |||||
| TGF-α | 0.93 | 80 | 100 | 0.82–1.00 | 0.001 |
| TGF-α │ CCL20 | 0.96 | 100 | 80 | 0.88–1.00 | 0.001 |
| MANF │ OSM | 0.96 | 90 | 100 | 0.88–1.00 | 0.001 |
| PLXNB3 │ TGF-α | 0.95 | 90 | 90 | 0.86–1.00 | 0.001 |
| TGF-α │ FGF-19 | 0.95 | 80 | 100 | 0.86–1.00 | 0.001 |
| OSM │ CCL20 | 0.94 | 80 | 90 | 0.84–1.00 | 0.001 |
| GFR-α-1 │ TGF-α | 0.94 | 100 | 80 | 0.84–1.00 | 0.001 |
| TGF-α │ DNER | 0.93 | 90 | 80 | 0.82–1.00 | 0.001 |
| TN-R │ TGF-α | 0.93 | 90 | 90 | 0.82–1.00 | 0.001 |
| OSM│ CXCL5 | 0.93 | 90 | 90 | 0.82–1.00 | 0.001 |
| PLXNB3 │ RSPO1 | 0.93 | 90 | 90 | 0.82–1.00 | 0.001 |
| uPA │ TGF-α | 0.92 | 90 | 80 | 0.80–1.00 | 0.001 |
| TGF-α │ CD40 | 0.92 | 90 | 80 | 0.80–1.00 | 0.001 |
| SKR3 │ TGF-α | 0.91 | 70 | 100 | 0.79–1.00 | 0.002 |
| TGF-α │ CCL23 | 0.91 | 80 | 90 | 0.79–1.00 | 0.002 |
| N-CDase │ TGF-α | 0.91 | 90 | 80 | 0.78–1.00 | 0.002 |
| CNTN5 │ TGF-α | 0.91 | 90 | 90 | 0.78–1.00 | 0.002 |
| Siglec-9 │ RSPO1 | 0.89 | 90 | 80 | 0.73–1.00 | 0.003 |
| Siglec-9 │ TGF-α | 0.89 | 100 | 80 | 0.73–1.00 | 0.003 |
| N-CDase │ HGF | 0.88 | 80 | 90 | 0.73–1.00 | 0.004 |
| EVs | |||||
| CCL11 | 0.88 | 90 | 80 | 0.73–1.00 | 0.004 |
| CLEC1B │ CCL11 | 0.95 | 100 | 80 | 0.86–1.00 | 0.001 |
| CCL11 │ CD40 | 0.95 | 100 | 80 | 0.86–1.00 | 0.001 |
| MCP-4 │ CCL11 | 0.93 | 100 | 90 | 0.79–1.00 | 0.001 |
| CD244 │ CCL11 | 0.92 | 100 | 80 | 0.79–1.00 | 0.001 |
| SKR3 │ CCL11 | 0.92 | 100 | 90 | 0.77–1.00 | 0.001 |
| JAM-B │ CCL11 | 0.92 | 100 | 90 | 0.77–1.00 | 0.001 |
| SIGLEC1 │ CCL11 | 0.91 | 90 | 80 | 0.78–1.00 | 0.002 |
| OPG │ CCL11 | 0.91 | 90 | 80 | 0.78–1.00 | 0.002 |
| uPA │ CCL11 | 0.91 | 90 | 80 | 0.78–1.00 | 0.002 |
| CCL11 │ MCP-2 | 0.91 | 100 | 70 | 0.78–1.00 | 0.002 |
| CLM-1 │ CCL11 | 0.91 | 100 | 80 | 0.78–1.00 | 0.002 |
| CCL11 │ CD5 | 0.91 | 90 | 90 | 0.76–1.00 | 0.002 |
| Siglec-9 │ CCL11 | 0.90 | 90 | 80 | 0.76–1.00 | 0.002 |
| CLM-6 │ CCL11 | 0.90 | 90 | 90 | 0.75–1.00 | 0.002 |
| SMOC2 │ OPG | 0.89 | 100 | 80 | 0.72–1.00 | 0.003 |
| IL18 │ 4E-BP1 | 0.88 | 90 | 80 | 0.73–1.00 | 0.004 |
| CCL11 │ PD-L1 | 0.88 | 80 | 90 | 0.73–1.00 | 0.004 |
| PLXNB3 │ CCL11 | 0.88 | 90 | 80 | 0.72–1.00 | 0.004 |
| CCL11 │ ADA | 0.88 | 90 | 80 | 0.71–1.00 | 0.004 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ellegaard Nielsen, J.; Sofie Pedersen, K.; Vestergård, K.; Georgiana Maltesen, R.; Christiansen, G.; Lundbye-Christensen, S.; Moos, T.; Risom Kristensen, S.; Pedersen, S. Novel Blood-Derived Extracellular Vesicle-Based Biomarkers in Alzheimer’s Disease Identified by Proximity Extension Assay. Biomedicines 2020, 8, 199. https://doi.org/10.3390/biomedicines8070199
Ellegaard Nielsen J, Sofie Pedersen K, Vestergård K, Georgiana Maltesen R, Christiansen G, Lundbye-Christensen S, Moos T, Risom Kristensen S, Pedersen S. Novel Blood-Derived Extracellular Vesicle-Based Biomarkers in Alzheimer’s Disease Identified by Proximity Extension Assay. Biomedicines. 2020; 8(7):199. https://doi.org/10.3390/biomedicines8070199
Chicago/Turabian StyleEllegaard Nielsen, Jonas, Kamilla Sofie Pedersen, Karsten Vestergård, Raluca Georgiana Maltesen, Gunna Christiansen, Søren Lundbye-Christensen, Torben Moos, Søren Risom Kristensen, and Shona Pedersen. 2020. "Novel Blood-Derived Extracellular Vesicle-Based Biomarkers in Alzheimer’s Disease Identified by Proximity Extension Assay" Biomedicines 8, no. 7: 199. https://doi.org/10.3390/biomedicines8070199
APA StyleEllegaard Nielsen, J., Sofie Pedersen, K., Vestergård, K., Georgiana Maltesen, R., Christiansen, G., Lundbye-Christensen, S., Moos, T., Risom Kristensen, S., & Pedersen, S. (2020). Novel Blood-Derived Extracellular Vesicle-Based Biomarkers in Alzheimer’s Disease Identified by Proximity Extension Assay. Biomedicines, 8(7), 199. https://doi.org/10.3390/biomedicines8070199