The Link between Chronic Stress and Accelerated Aging

 , and
, and 
{kind=link}
{kind=link}
Abstract
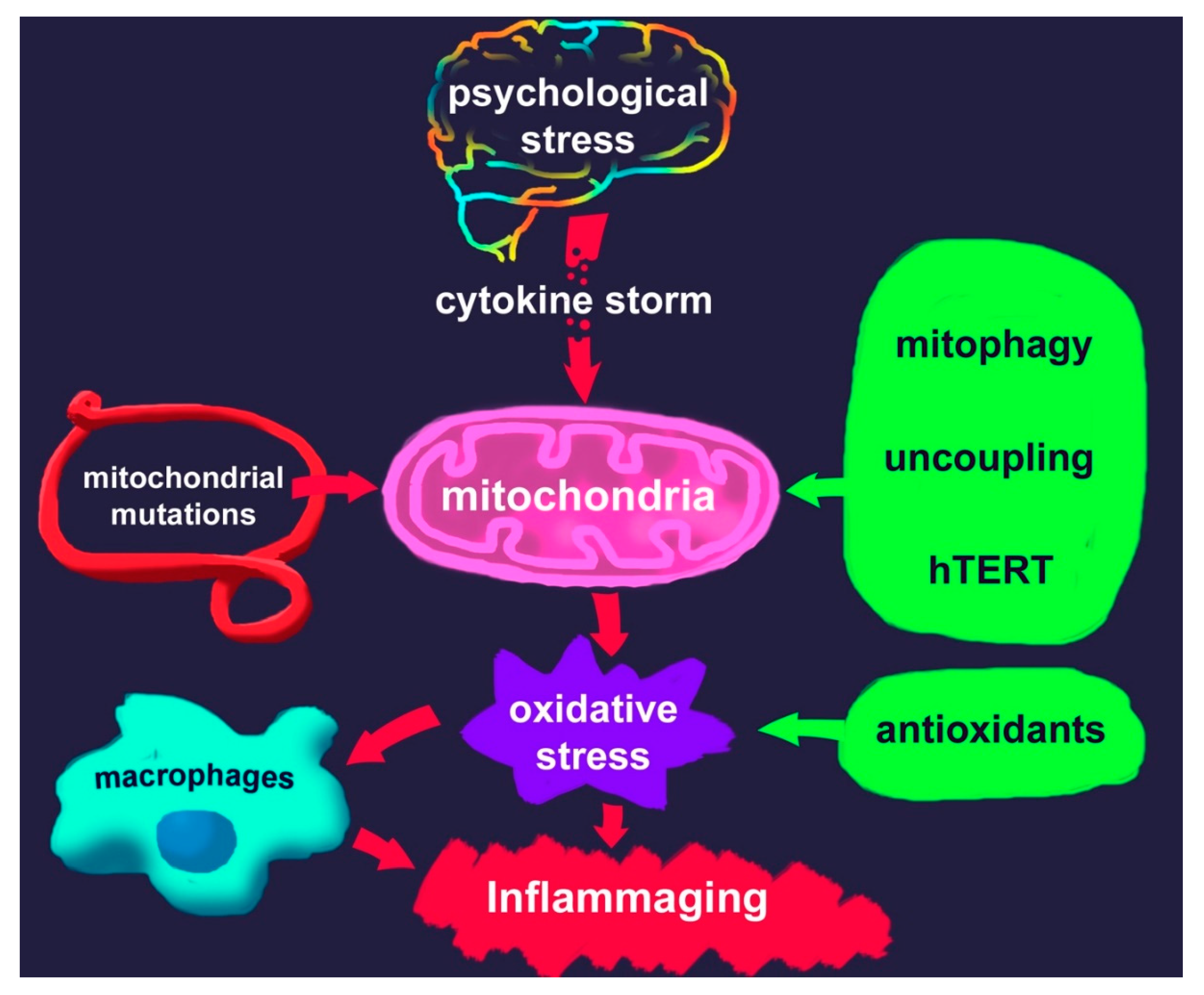
1. Psychological Stress and Aging
2. Cell Senescence
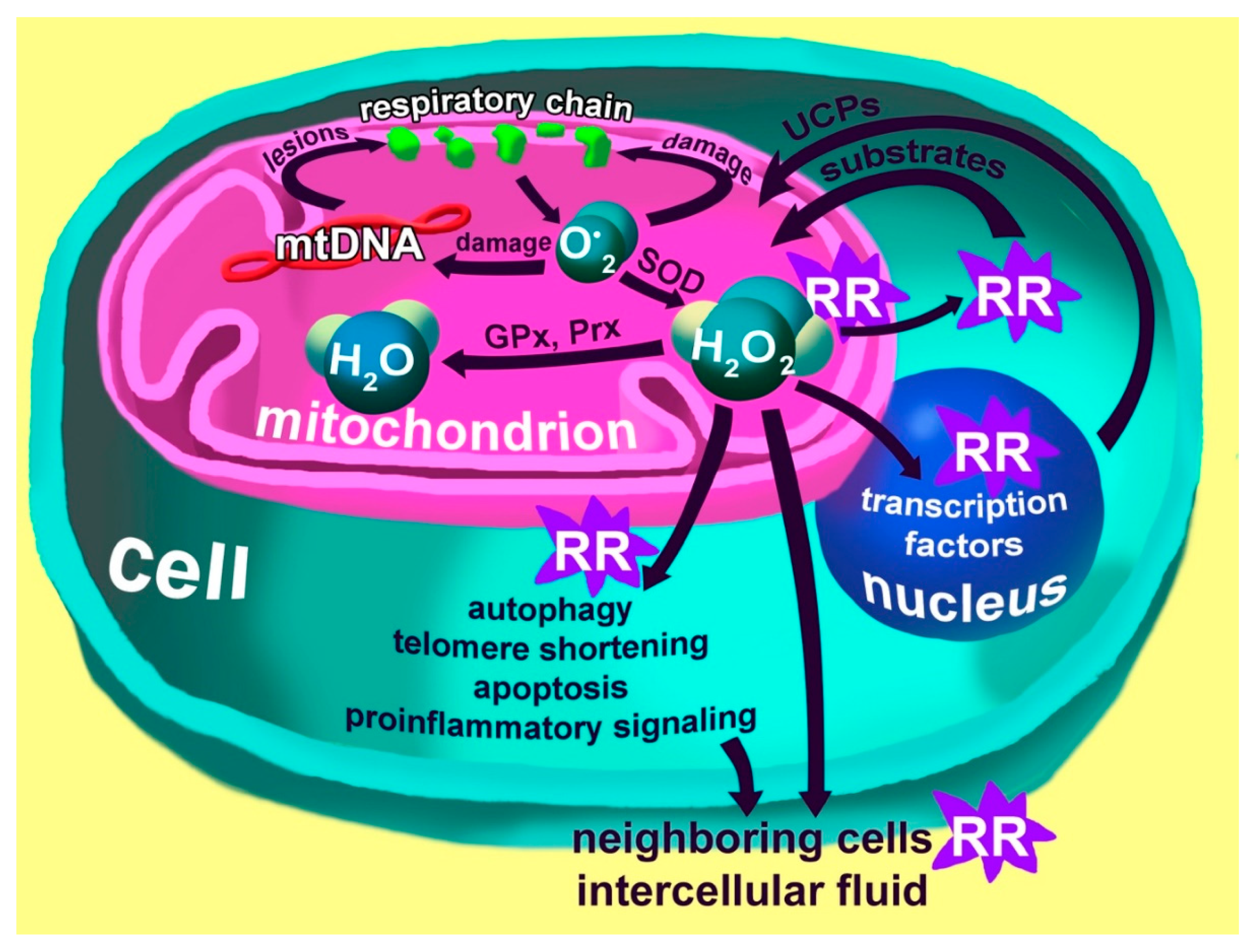
3. Mitochondria and ROS Production
4. The Special Role of Mitochondria and Macrophages in the Development of Chronic Inflammation
5. Oxidative Stress and the Pro-Inflammatory Phenotype of Immune Cells
5.1. Innate Immunity
5.2. Adaptive Immunity
6. Inflammaging
7. Potential Approaches to Reduce Oxidative Stress in Stress Disorders
7.1. Potential Anti-Inflammatory Treatment Strategies
7.2. Activators of Mitophagy
7.3. Uncouplers
7.4. hTERT Activators
7.5. Antioxidants
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef]
- O’Donovan, A.; Tomiyama, A.J.; Lin, J.; Puterman, E.; Adler, N.E.; Kemeny, M.; Wolkowitz, O.M.; Blackburn, E.H.; Epel, E.S. Stress appraisals and cellular aging: A key role for anticipatory threat in the relationship between psychological stress and telomere length. Brain Behav. Immun. 2012, 26, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Toft, H.; Bramness, J.G.; Lien, L.; Abebe, D.S.; Wampold, B.E.; Tilden, T.; Hestad, K.; Neupane, S.P. PTSD patients show increasing cytokine levels during treatment despite reduced psychological distress. Neuropsychiatr. Dis. Treat. 2018, 14, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Hori, H.; Kim, Y. Inflammation and post-traumatic stress disorder. Psychiatry Clin. Neurosci. 2019, 73, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Basterzi, A.D.; Aydemir, C.; Kisa, C.; Aksaray, S.; Tuzer, V.; Yazici, K.; Göka, E. IL-6 levels decrease with SSRI treatment in patients with major depression. Hum. Psychopharmacol. 2005, 20, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Salim, S. Oxidative stress: A potential link between emotional wellbeing and immune response. Curr. Opin. Pharmacol. 2016, 29, 70–76. [Google Scholar] [CrossRef]
- El Assar, M.; Angulo, J.; Carnicero, J.A.; Walter, S.; García-García, F.J.; López-Hernández, E.; Sánchez-Puelles, J.M.; Rodríguez-Mañas, L. Frailty Is Associated With Lower Expression of Genes Involved in Cellular Response to Stress: Results From the Toledo Study for Healthy Aging. J. Am. Med. Dir. Assoc. 2017, 18. [Google Scholar] [CrossRef]
- Erusalimsky, J.D. Oxidative stress, telomeres and cellular senescence: What non-drug interventions might break the link? [published online ahead of print, 2020 Feb 13]. Free Radic. Biol. Med. 2020. [Google Scholar] [CrossRef]
- Epel, E.S.; Blackburn, E.H.; Lin, J.; Dhabhar, F.S.; Adler, N.E.; Morrow, J.D.; Cawthon, R.M. Accelerated telomere shortening in response to life stress. Proc. Natl. Acad. Sci. USA 2004, 101, 17312–17315. [Google Scholar] [CrossRef]
- Watfa, G.; Dragonas, C.; Brosche, T.; Dittrich, R.; Sieber, C.C.; Alecu, C.; Benetos, A.; Nzietchueng, R. Study of telomere length and different markers of oxidative stress in patients with Parkinson’s disease. J. Nutr. Health Aging 2011, 15, 277–281. [Google Scholar] [CrossRef]
- Salpea, K.D.; Talmud, P.J.; Cooper, J.A.; Maubaret, C.G.; Stephens, J.W.; Abelak, K.; Humphries, S.E. Association of telomere length with type 2 diabetes, oxidative stress and UCP2 gene variation. Atherosclerosis 2010, 209, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Starr, J.M.; Shiels, P.G.; Harris, S.E.; Pattie, A.; Pearce, M.S.; Relton, C.L.; Deary, I.J. Oxidative stress, telomere length and biomarkers of physical aging in a cohort aged 79 years from the 1932 Scottish Mental Survey. Mech. Ageing Dev. 2008, 129, 745–751. [Google Scholar] [CrossRef]
- Margaritis, M.; Sanna, F.; Lazaros, G.; Akoumianakis, I.; Patel, S.; Antonopoulos, A.S.; Duke, C.; Herdman, L.; Psarros, C.; Oikonomou, E.K.; et al. Predictive value of telomere length on outcome following acute myocardial infarction: Evidence for contrasting effects of vascular vs. blood oxidative stress. Eur. Heart J. 2017, 38, 3094–3104. [Google Scholar] [CrossRef] [PubMed]
- Achuthan, S.; Santhoshkumar, T.R.; Prabhakar, J.; Nair, S.A.; Pillai, M.R. Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J. Biol. Chem. 2011, 286, 37813–37829. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Olovnikov, A.M. The role of incomplete terminal repair of chromosomal DNA in the aging of neurons and postmitotic organisms. Biol. Bull. 1995, 504. [Google Scholar]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, J.E.; Revesz, D.; Wolkowitz, O.M.; Penninx, B.W.J.H. Cellular aging in depression: Permanent imprint or reversible process? Bioessays 2014, 36, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W.; Alex, P.; Lin, A.P.; Wolf, E.J.; Miller, D.R. Oxidative Stress, Inflammation, and Neuroprogression in Chronic PTSD. Harv. Rev. Psychiatry 2018, 26, 57–69. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Roy, J.; Galano, J.M.; Durand, T.; Le Guennec, J.Y.; Lee, J.C. Physiological role of reactive oxygen species as promoters of natural defenses. FASEB J. 2017, 31, 3729–3745. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; De Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Tylee, D.S.; Chandler, S.D.; Nievergelt, C.M.; Liu, X.; Pazol, J.; Woelk, C.H.; Lohr, J.B.; Kremen, W.S.; Baker, D.G.; Glatt, S.J.; et al. Resiliency Study Investigators. Blood-based gene-expression biomarkers of post-traumatic stress disorder among deployed marines: A pilot study. Psychoneuroendocrinology 2015, 51, 472–494. [Google Scholar] [CrossRef]
- Logue, M.W.; Smith, A.K.; Baldwin, C.; Wolf, E.J.; Guffanti, G.; Ratanatharathorn, A.; Stone, A.; Schichman, S.A.; Humphries, D.; Binder, E.B.; et al. An analysis of gene expression in PTSD implicates genes involved in the glucocorticoid receptor pathway and neural responses to stress. Psychoneuroendocrinology 2015, 57, 1–13. [Google Scholar] [CrossRef]
- Rose, G.; Crocco, P.; De Rango, F.; Montesanto, A.; Passarino, G. Further support to the uncoupling-to survive theory: The genetic variation of human UCPgenes is associated with longevity. PLoS ONE 2011, 6, e29650. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Albertini, M.C.; Orciani, M.; Ceka, A.; Cricca, M.; Procopio, A.D.; Bonafè, M. DNA damage response (DDR) and senescence: Shuttled inflamma-miRNAs on the stage of inflamm-aging. Oncotarget 2015, 6, 35509–35521. [Google Scholar] [CrossRef] [PubMed]
- Baëhl, S.; Garneau, H.; Le Page, A.; Lorrain, D.; Viens, I.; Svotelis, A.; Lord, J.M.; Phillips, A.C.; Cabana, F.; Larbi, A.; et al. Altered neutrophil functions in elderly patients during a 6-month follow-up period after a hip fracture. Exp. Gerontol. 2015, 65, 58–68. [Google Scholar] [CrossRef]
- Linton, P.J.; Thoman, M.L. Immunosenescence in monocytes, macrophages, and dendritic cells: Lessons learned from the lung and heart. Immunol. Lett. 2014, 162, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.C.; Goldstein, D.R.; Montgomery, R.R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013, 13, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, S.; Weyand, C.M.; Goronzy, J.J. Mechanisms of immunosenescence: Lessons from models of accelerated immune aging. Ann. N. Y. Acad. Sci. 2012, 1247, 69–82. [Google Scholar] [CrossRef]
- Gounder, S.S.; Abdullah, B.J.J.; Radzuanb, N.E.I.B.M.; Zain, F.D.B.M.; Sait, N.B.M.; Chua, C.; Subramani, B. Effect of Aging on NK Cell Population and Their Proliferation at Ex Vivo Culture Condition. Anal. Cell. Pathol. 2018, 2018, 7871814. [Google Scholar] [CrossRef]
- Nilsson, B.O.; Ernerudh, J.; Johansson, B.; Evrin, P.E.; Löfgren, S.; Ferguson, F.G.; Wikby, A. Morbidity does not influence the T-cell immune risk phenotype in the elderly: Findings in the Swedish NONA Immune Study using sample selection protocols. Mech. Ageing Dev. 2003, 124, 469–476. [Google Scholar] [CrossRef]
- Olsson, J.; Wikby, A.; Johansson, B.; Löfgren, S.; Nilsson, B.O.; Ferguson, F.G. Age-related change in peripheral blood T-lymphocyte subpopulations and cytomegalovirus infection in the very old: The Swedish longitudinal OCTO immune study. Mech. Ageing Dev. 2000, 121, 187–201. [Google Scholar] [CrossRef]
- Tabibian-Keissar, H.; Hazanov, L.; Schiby, G.; Rosenthal, N.; Rakovsky, A.; Michaeli, M.; Shahaf, G.L.; Pickman, Y.; Rosenblatt, K.; Melamed, D.; et al. Aging affects B-cell antigen receptor repertoire diversity in primary and secondary lymphoid tissues. Eur. J. Immunol. 2016, 46, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Sauce, D. Naive T cells: The crux of cellular immune aging? Exp. Gerontol. 2014, 54, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Larbi, A.; Fulop, T. From “truly naïve” to “exhausted senescent” T cells: When markers predict functionality. Cytom. A 2014, 85, 25–35. [Google Scholar] [CrossRef]
- Dock, J.N.; Effros, R.B. Role of CD8 T Cell Replicative Senescence in Human Aging and in HIV-mediated Immunosenescence. Aging Dis. 2011, 2, 382–397. [Google Scholar]
- Fessler, J.; Ficjan, A.; Duftner, C.; Dejaco, C. The impact of aging on regulatory T-cells. Front. Immunol. 2013, 4, 231. [Google Scholar] [CrossRef]
- De la Fuente, M.; Miquel, J. An update of the oxidation-inflammation theory of aging: The involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 2009, 15, 3003–3026. [Google Scholar] [CrossRef]
- Fülöp, T.; Larbi, A.; Witkowski, J.M. Human Inflammaging. Gerontology 2019, 65, 495–504. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Weyand, C.M. Successful and Maladaptive T Cell Aging. Immunity 2017, 46, 364–378. [Google Scholar] [CrossRef]
- Feduccia, A.A.; Jerome, L.; Yazar-Klosinski, B.; Emerson, A.; Mithoefer, M.M.; Doblin, R. Breakthrough for trauma treatment: Safety and efficacy of MDMA-assisted psychotherapy compared to Paroxetine and Sertraline. Front. Psychiatry 2019, 10, 650. [Google Scholar] [CrossRef]
- Lee, B.; Sur, B.; Yeom, M.; Shim, I.; Lee, H.; Hahm, D.H. Effects of systemic administration of ibuprofen on stress response in a rat model of post-traumatic stress disorder. Korean J. Physiol. Pharmacol. 2016, 20, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R.; Golier, J.A.; Kaufman, S. Circadian rhythm of salivary cortisol in Holocaust survivors with and without PTSD. Am. J. Psychiatry 2005, 162, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Khoury, N.M.; Marvar, P.J.; Gillespie, C.F.; Wingo, A.; Schwartz, A.; Bradley, B.; Kramer, M.; Ressler, K.J. The renin-angiotensin pathway in posttraumatic stress disorder: Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are associated with fewer traumatic stress symptoms. J. Clin. Psychiatry 2012, 73, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Narita-Ohtaki, R.; Hori, H.; Itoh, M.; Lin, M.; Niwa, M.; Ino, K.; Imai, R.; Ogawa, S.; Sekiguchi, A.; Matsui, M.; et al. Cognitive function in Japanese women with posttraumatic stress disorder: Association with exercise habits. J. Affect. Disord. 2018, 236, 306–312. [Google Scholar] [CrossRef]
- Uemura, M.; Ohira, T.; Yasumura, S.; Otsuru, A.; Maeda, M.; Harigane, M.; Horikoshi, N.; Suzuki, Y.; Yabe, H.; Takahashi, H.; et al. Association between psychological distress and dietary intake among evacuees after the Great East Japan Earthquake in a cross-sectional study: The Fukushima Health Management Survey. BMJ Open 2016, 6, e011534. [Google Scholar] [CrossRef]
- Bower, J.E.; Irwin, M.R. Mind-body therapies and control of inflammatory biology: A descriptive review. Brain Behav. Immun. 2016, 51, 1–11. [Google Scholar] [CrossRef]
- Michopoulos, V.; Beurel, E.; Gould, F.; Dhabhar, F.S.; Schultebraucks, K.; Galatzer-Levy, I.; Rothbaum, B.O.; Ressler, K.J.; Nemeroff, C.B. Association of Prospective Risk for Chronic PTSD Symptoms with Low TNFα and IFNγ Concentrations in the Immediate Aftermath of Trauma Exposure. Am. J. Psychiatry 2020, 177, 58–65. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef]
- Sebori, R.; Kuno, A.; Hosoda, R.; Hayashi, T.; Horio, Y. Resveratrol Decreases Oxidative Stress by Restoring Mitophagy and Improves the Pathophysiology of Dystrophin-Deficient mdx Mice. Oxid. Med. Cell Longev. 2018, 2018, 9179270. [Google Scholar] [CrossRef] [PubMed]
- Vernucci, E.; Tomino, C.; Molinari, F.; Limongi, D.; Aventaggiato, M.; Sansone, L.; Tafani, M.; Russo, M.A. Mitophagy and Oxidative Stress in Cancer and Aging: Focus on Sirtuins and Nanomaterials. Oxid. Med. Cell Longev. 2019, 2019, 6387357. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, K.; Huang, C.; Lin, D.; Zhou, Y.; Wu, Y.; Tian, N.; Fan, P.; Pan, X.; Xu, D.; et al. SIRT3 Activation by Dihydromyricetin Suppresses Chondrocytes Degeneration via Maintaining Mitochondrial Homeostasis. Int. J. Biol. Sci. 2018, 14, 1873–1882. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Nisar, M.; Huang, C.; Pan, X.; Lin, D.; Zheng, G.; Jin, H.; Chen, D.; Tian, N.; Huang, Q.; et al. Small molecule natural compound agonist of SIRT3 as a therapeutic target for the treatment of intervertebral disc degeneration. Exp. Mol. Med. 2018, 50, 146. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Brand, M.D. Mitochondrial matrix reactive oxygen species production is very sensitive to mild uncoupling. Biochem. Soc. Trans. 2003, 31, 1300–1301. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chan, S.L.; De Souza-Pinto, N.C.; Slevin, J.R.; Wersto, R.P.; Zhan, M.; Mustafa, K.; De Cabo, R.; Mattson, M.P. Mitochondrial UCP4 mediates an adaptive shift in energy metabolism and increases the resistance of neurons to metabolic and oxidative stress. Neuromol. Med. 2006, 8, 389–414. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Kazak, L.; Jedrychowski, M.P.; Lu, G.Z.; Erickson, B.K.; Szpyt, J.; Pierce, K.A.; Laznik-Bogoslavski, D.; Vetrivelan, R.; Clish, C.B.; et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016, 532, 112–116. [Google Scholar] [CrossRef]
- Tao, H.; Zhang, Y.; Zeng, X.; Shulman, G.I.; Jin, S. Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat. Med. 2014, 20, 1263–1269. [Google Scholar] [CrossRef]
- Geisler, J.G. 2,4 Dinitrophenol as Medicine. Cells 2019, 8, 280. [Google Scholar] [CrossRef]
- Caldeira da Silva, C.C.; Cerqueira, F.M.; Barbosa, L.F.; Medeiros, M.H.; Kowaltowski, A.J. Mild mitochondrial uncoupling in mice affects energy metabolism, redox balance and longevity. Aging Cell 2008, 7, 552–560. [Google Scholar] [CrossRef]
- Padalko, V.I. Uncoupler of oxidative phosphorylation prolongs the lifespan of Drosophila. Biochemistry 2005, 70, 986–989. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Huang, J.; Wang, G. Mitochondria, Telomeres and Telomerase Subunits. Front. Cell Dev. Biol. 2019, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Jäger, K.; Walter, M. Therapeutic Targeting of Telomerase. Genes 2016, 7, 39. [Google Scholar] [CrossRef]
- Fauce, S.R.; Jamieson, B.D.; Chin, A.C.; Mitsuyasu, R.T.; Parish, S.T.; Ng, H.L.; Kitchen, C.M.; Yang, O.O.; Harley, C.B.; Effros, R.B. Telomerase-based pharmacologic enhancement of antiviral function of human CD8+ T lymphocytes. J. Immunol. 2008, 181, 7400–7406. [Google Scholar] [CrossRef] [PubMed]
- Bernardes de Jesus, B.; Schneeberger, K.; Vera, E.; Tejera, A.; Harley, C.B.; Blasco, M.A. The telomerase activator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence. Aging Cell 2011, 10, 604–621. [Google Scholar] [CrossRef] [PubMed]
- Sprouse, A.A.; Steding, C.E.; Herbert, B.S. Pharmaceutical regulation of telomerase and its clinical potential. J. Cell. Mol. Med. 2012, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Heiss, E.H.; Schilder, Y.D.; Dirsch, V.M. Chronic treatment with resveratrol induces redox stress- and ataxia telangiectasia-mutated (ATM)-dependent senescence in p53-positive cancer cells. J. Biol. Chem. 2007, 282, 26759–26766. [Google Scholar] [CrossRef] [PubMed]
- De la Lastra, C.A.; Villegas, I. Resveratrol as an antioxidant and pro-oxidant agent: Mechanisms and clinical implications. Biochem. Soc. Trans. 2007, 35, 1156–1160. [Google Scholar] [CrossRef]
- Singh, A.; Sati, S.; Mishra, R. Resveratrol: Antioxidant-prooxidant. Int. J. Technol. Res. Sci. 2016, 1, 106–112. [Google Scholar]
- Wang, F.; Chang, G.; Geng, X. NGF and TERT co-transfected BMSCs improve the restoration of cognitive impairment in vascular dementia rats. PLoS ONE 2014, 9, e98774. [Google Scholar] [CrossRef]
- Saretzki, G.; Murphy, M.P.; Von Zglinicki, T. MitoQ counteracts telomere shortening and elongates lifespan of fibroblasts under mild oxidative stress. Aging Cell 2003, 2, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Liao, N.; Shi, Y.; Zhang, C.; Zheng, Y.; Wang, Y.; Zhao, B.; Zeng, Y.; Liu, X.; Liu, J. Antioxidants inhibit cell senescence and preserve stemness of adipose tissue-derived stem cells by reducing ROS generation during long-term in vitro expansion. Stem Cell Res. Ther. 2019, 10, 306. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Egorov, M.V.; Krasilshchikova, M.S.; Lyamzaev, K.G.; Manskikh, V.N.; Moshkin, M.P.; Novikov, E.A.; Popovich, I.G.; Rogovin, K.A.; Shabalina, I.G.; et al. Effects of the mitochondria-targeted antioxidant SkQ1 on lifespan of rodents. Aging 2011, 3, 1110–1119. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yegorov, Y.E.; Poznyak, A.V.; Nikiforov, N.G.; Sobenin, I.A.; Orekhov, A.N. The Link between Chronic Stress and Accelerated Aging. Biomedicines 2020, 8, 198. https://doi.org/10.3390/biomedicines8070198
Yegorov YE, Poznyak AV, Nikiforov NG, Sobenin IA, Orekhov AN. The Link between Chronic Stress and Accelerated Aging. Biomedicines. 2020; 8(7):198. https://doi.org/10.3390/biomedicines8070198
Chicago/Turabian StyleYegorov, Yegor E., Anastasia V. Poznyak, Nikita G. Nikiforov, Igor A. Sobenin, and Alexander N. Orekhov. 2020. "The Link between Chronic Stress and Accelerated Aging" Biomedicines 8, no. 7: 198. https://doi.org/10.3390/biomedicines8070198
APA StyleYegorov, Y. E., Poznyak, A. V., Nikiforov, N. G., Sobenin, I. A., & Orekhov, A. N. (2020). The Link between Chronic Stress and Accelerated Aging. Biomedicines, 8(7), 198. https://doi.org/10.3390/biomedicines8070198