Meta-Analysis of Hypoxic Transcriptomes from Public Databases



Abstract
1. Introduction
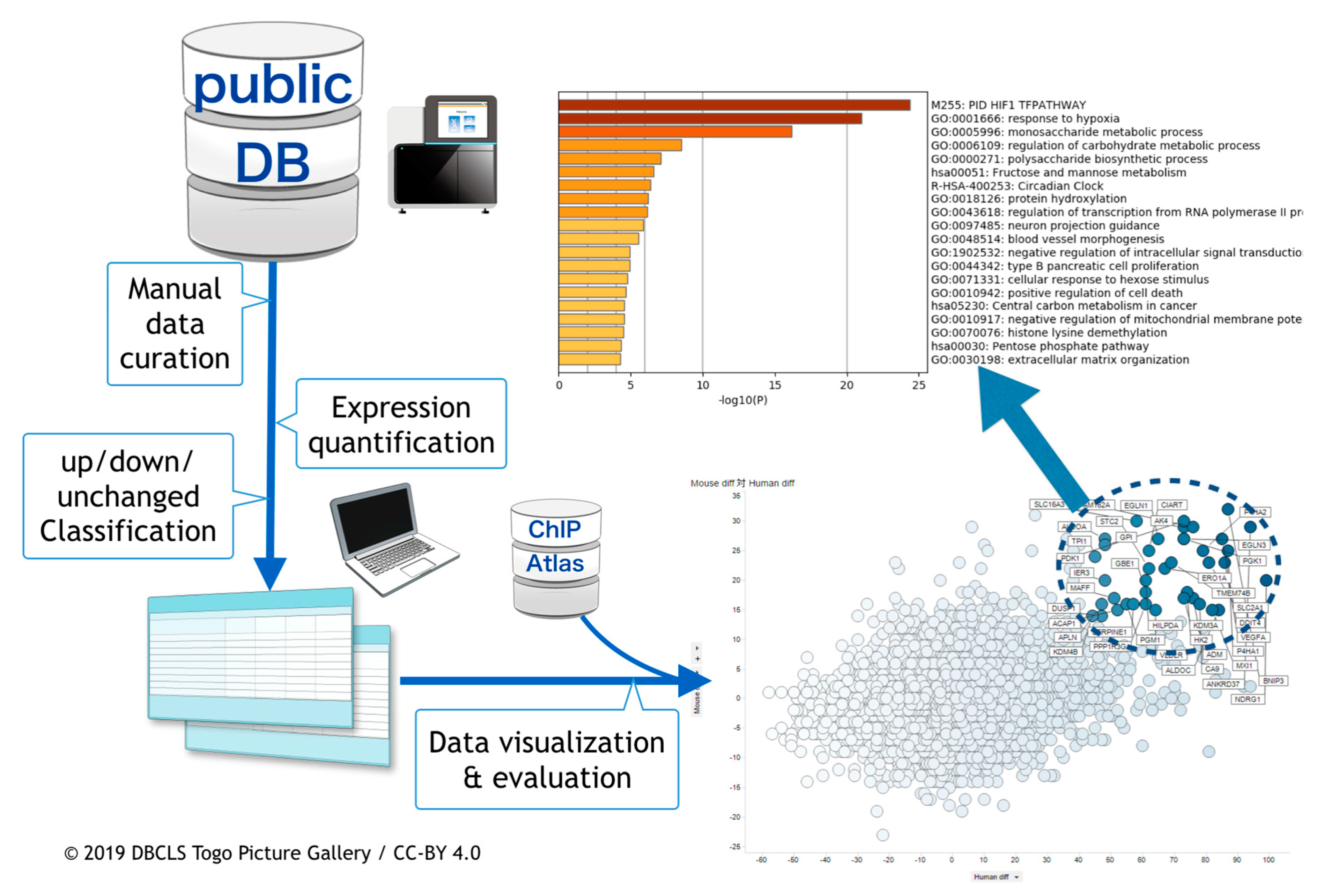
2. Materials and Methods
2.1. Curation of Public Gene Expression Data
2.2. Gene Expression Quantification
2.3. Meta-Analysis of ChIP-Seq Data
2.4. Visualization and the Integrated Functional Analysis of Genes
3. Results
3.1. Curation of Hypoxic Transriptome Data in Public Databases
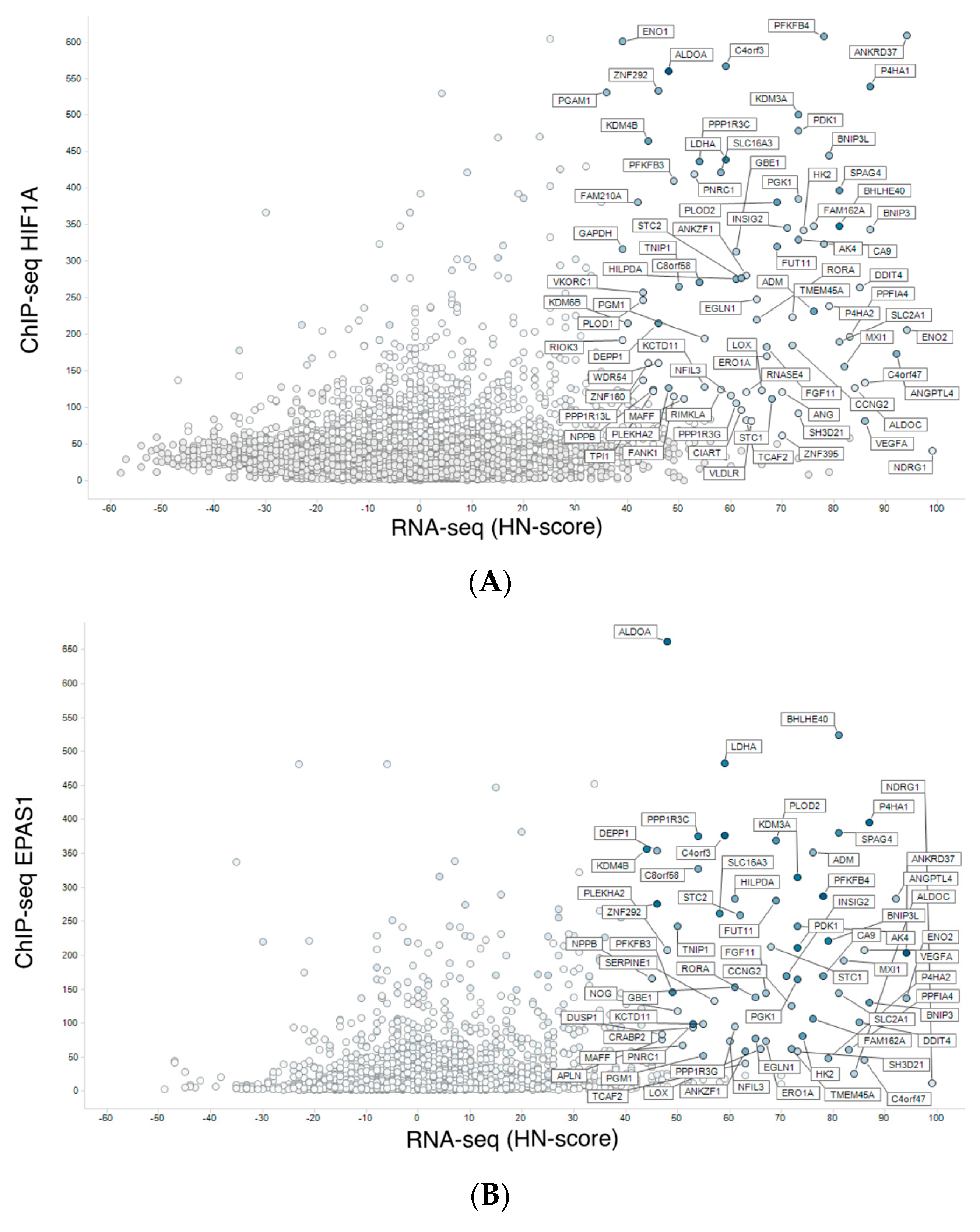
3.2. Meta-Analysis of Hypoxia-Responsive Genes
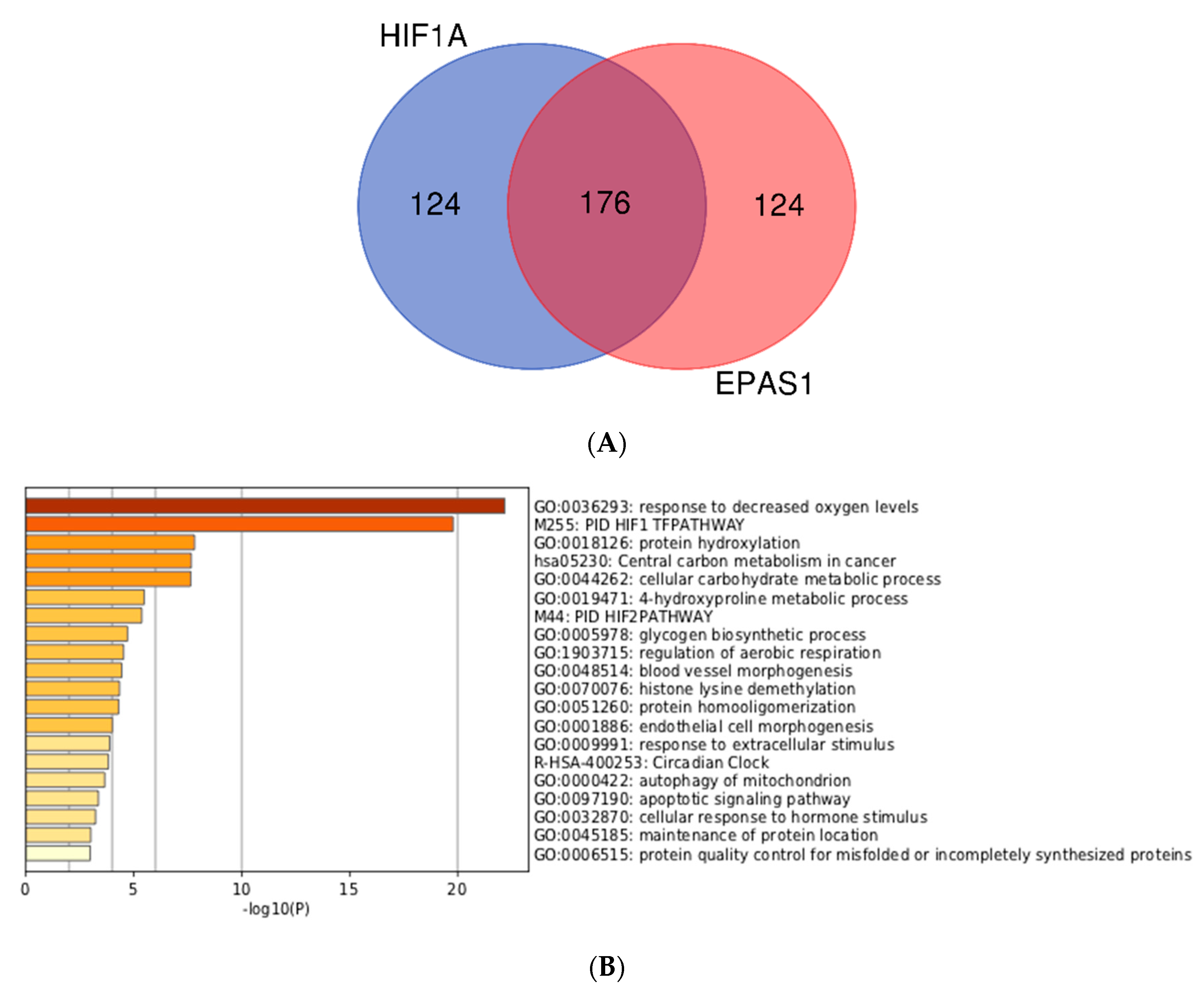
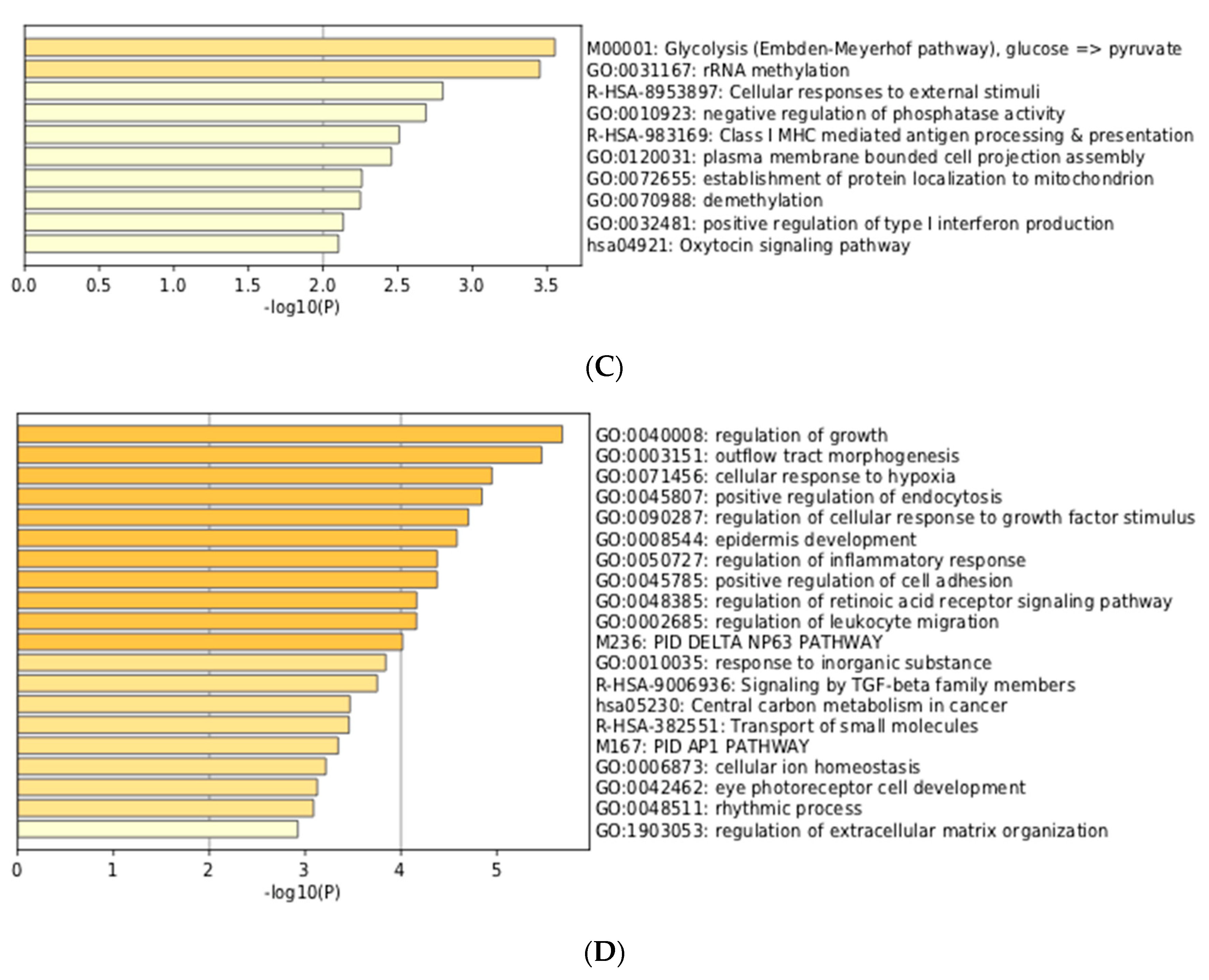
3.3. Integration of Meta-Analyzed ChIP-Seq Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2018, 47, D711–D715. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K. An intimate crosstalk between iron homeostasis and oxygen metabolism regulated by the hypoxia-inducible factors (HIFs). Free Radic. Biol. Med. 2019, 133, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.; Simon, M. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.R.; Huang, S.; Nagy, M.S.; Yadagiri, V.K.; Fields, C.; Mukherjee, D.; Bons, B.; Dedhia, P.H.; Chin, A.M.; Tsai, Y.-H.; et al. Bacterial colonization stimulates a complex physiological response in the immature human intestinal epithelium. elife 2017, 6, e29132. [Google Scholar] [CrossRef] [PubMed]
- Bono, H. All of gene expression (AOE): An integrated index for public gene expression databases. BioRxiv 2019, 626754. [Google Scholar] [CrossRef]
- Kodama, Y.; Mashima, J.; Kosuge, T.; Ogasawara, O. DDBJ update: The Genomic Expression Archive (GEA) for functional genomics data. Nucleic Acids Res. 2019, 47, D69–D73. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Shumway, M.; Leinonen, R.; International Nucleotide Sequence Database Collaboration. The Sequence Read Archive: Explosive growth of sequencing data. Nucleic Acids Res. 2012, 40, D54–D56. [Google Scholar] [CrossRef] [PubMed]
- Bono, H. Meta-analysis of hypoxic transcriptomes from public databases. BioRxiv 2018, 267310. [Google Scholar] [CrossRef]
- Hiraoka, Y.; Yamada, K.; Kawasaki, Y.; Hirose, H.; Matsumoto, Y.; Ishikawa, K.; Yasumizu, Y. Ikra: Rnaseq Pipeline Centered on Salmon. 2019. Available online: https://doi.org/10.5281/zenodo.3352573 (accessed on 9 January 2020).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F. Trim Galore. 2015. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 9 January 2020).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef] [PubMed]
- Oki, S.; Ohta, T.; Shioi, G.; Hatanaka, H.; Ogasawara, O.; Okuda, Y.; Kawaji, H.; Nakaki, R.; Sese, J.; Meno, C. ChIP-Atlas: A data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018, 19, e46255. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Bono, H.; Hiyama, K.; Kawamoto, T.; Kato, Y.; Nakanishi, T.; Nishiyama, M.; Hiyama, E.; Hirohashi, N.; Sueoka, E.; et al. Differentiated embryo chondrocyte plays a crucial role in DNA damage response via transcriptional regulation under hypoxic conditions. PLoS ONE 2018, 13, e0192136. [Google Scholar] [CrossRef] [PubMed]
- Benita, Y.; Kikuchi, H.; Smith, A.D.; Zhang, M.Q.; Chung, D.C.; Xavier, R.J. An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009, 37, 4587–4602. [Google Scholar] [CrossRef]
- Hu, C.J.; Wang, L.Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef]
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragoussis, J.; Ratcliffe, P.J. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 2009, 284, 16767–16775. [Google Scholar] [CrossRef]
- Ortiz-Barahona, A.; Villar, D.; Pescador, N.; Amigo, J.; del Peso, L. Genome-wide identification of hypoxia-inducible factor binding sites and target genes by a probabilistic model integrating transcription-profiling data and in silico binding site prediction. Nucleic Acids Res. 2010, 38, 2332–2345. [Google Scholar] [CrossRef] [PubMed]
- Smythies, J.A.; Sun, M.; Masson, N.; Salama, R.; Simpson, P.D.; Murray, E.; Neumann, V.; Cockman, M.E.; Choudhry, H.; Ratcliffe, P.J.; et al. Inherent DNA-binding specificities of the HIF-1α and HIF-2α transcription factors in chromatin. EMBO Rep. 2019, 20, e46401. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Gene | Human Up | Human Down | Human HN-Score | Mouse Gene | Mouse Up | Mouse Down | Mouse HN-Score | Total HN-Score |
|---|---|---|---|---|---|---|---|---|
| ANKRD37 | 101 | 7 | 94 | Ankrd37 | 35 | 6 | 29 | 123 |
| NDRG1 | 104 | 5 | 99 | Ndrg1 | 22 | 2 | 20 | 119 |
| BNIP3 | 92 | 5 | 87 | Bnip3 | 33 | 1 | 32 | 119 |
| P4HA1 | 92 | 5 | 87 | P4ha1 | 27 | 2 | 25 | 112 |
| DDIT4 | 94 | 9 | 85 | Ddit4 | 29 | 2 | 27 | 112 |
| VEGFA | 92 | 6 | 86 | Vegfa | 23 | 0 | 23 | 109 |
| FAM162A | 82 | 6 | 76 | Fam162a | 30 | 1 | 29 | 105 |
| SLC2A1 | 88 | 7 | 81 | Slc2a1 | 28 | 5 | 23 | 104 |
| P4HA2 | 83 | 4 | 79 | P4ha2 | 27 | 2 | 25 | 104 |
| PDK1 | 80 | 7 | 73 | Pdk1 | 32 | 2 | 30 | 103 |
| AK4 | 82 | 9 | 73 | Ak4 | 31 | 2 | 29 | 102 |
| PGK1 | 78 | 5 | 73 | Pgk1 | 30 | 3 | 27 | 100 |
| EGLN3 | 82 | 9 | 73 | Egln3 | 29 | 2 | 27 | 100 |
| ALDOC | 93 | 9 | 84 | Aldoc | 17 | 2 | 15 | 99 |
| MXI1 | 88 | 6 | 82 | Mxi1 | 16 | 1 | 15 | 97 |
| ENO2 | 99 | 5 | 94 | Eno2 | 13 | 11 | 2 | 96 |
| CA9 | 88 | 10 | 78 | Car9 | 21 | 5 | 16 | 94 |
| ANGPTL4 | 98 | 6 | 92 | Angptl4 | 8 | 6 | 2 | 94 |
| ADM | 83 | 7 | 76 | Adm | 20 | 3 | 17 | 93 |
| TMEM74B | 76 | 7 | 69 | Tmem74b | 24 | 1 | 23 | 92 |
| HK2 | 82 | 8 | 74 | Hk2 | 21 | 3 | 18 | 92 |
| EGLN1 | 69 | 4 | 65 | Egln1 | 27 | 0 | 27 | 92 |
| BNIP3L | 84 | 5 | 79 | Bnip3l | 12 | 0 | 12 | 91 |
| KDM3A | 78 | 5 | 73 | Kdm3a | 18 | 1 | 17 | 90 |
| C4orf47 | 91 | 5 | 86 | 1700029J07Rik | 7 | 3 | 4 | 90 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bono, H.; Hirota, K. Meta-Analysis of Hypoxic Transcriptomes from Public Databases. Biomedicines 2020, 8, 10. https://doi.org/10.3390/biomedicines8010010
Bono H, Hirota K. Meta-Analysis of Hypoxic Transcriptomes from Public Databases. Biomedicines. 2020; 8(1):10. https://doi.org/10.3390/biomedicines8010010
Chicago/Turabian StyleBono, Hidemasa, and Kiichi Hirota. 2020. "Meta-Analysis of Hypoxic Transcriptomes from Public Databases" Biomedicines 8, no. 1: 10. https://doi.org/10.3390/biomedicines8010010
APA StyleBono, H., & Hirota, K. (2020). Meta-Analysis of Hypoxic Transcriptomes from Public Databases. Biomedicines, 8(1), 10. https://doi.org/10.3390/biomedicines8010010