Diagnosis and Management of Progressive Multiple Sclerosis


Abstract
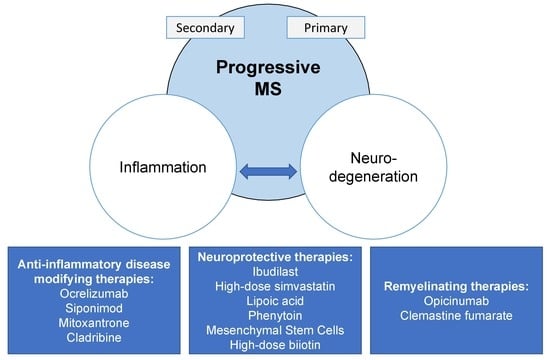
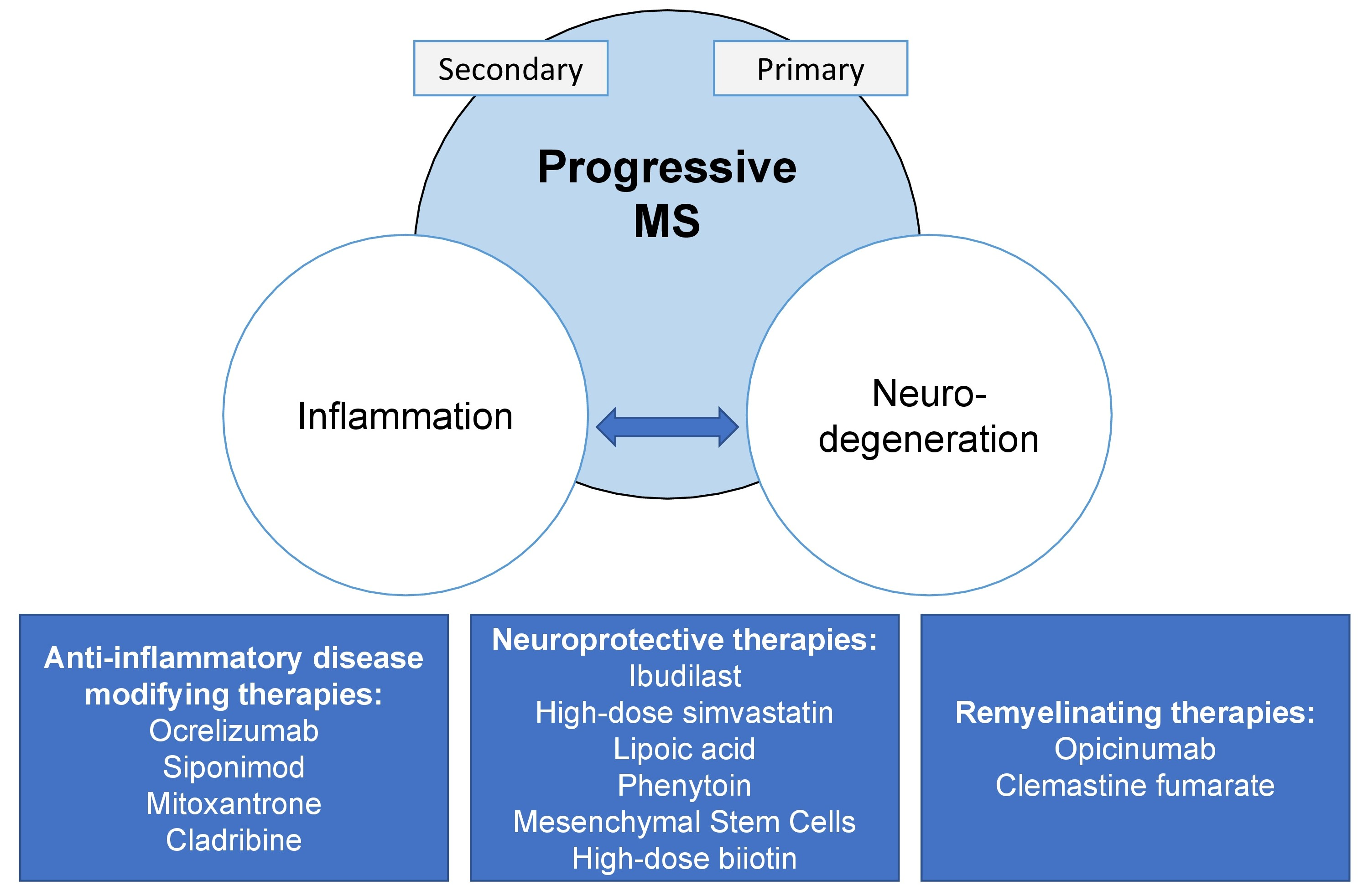
1. Introduction
2. Pathogenesis
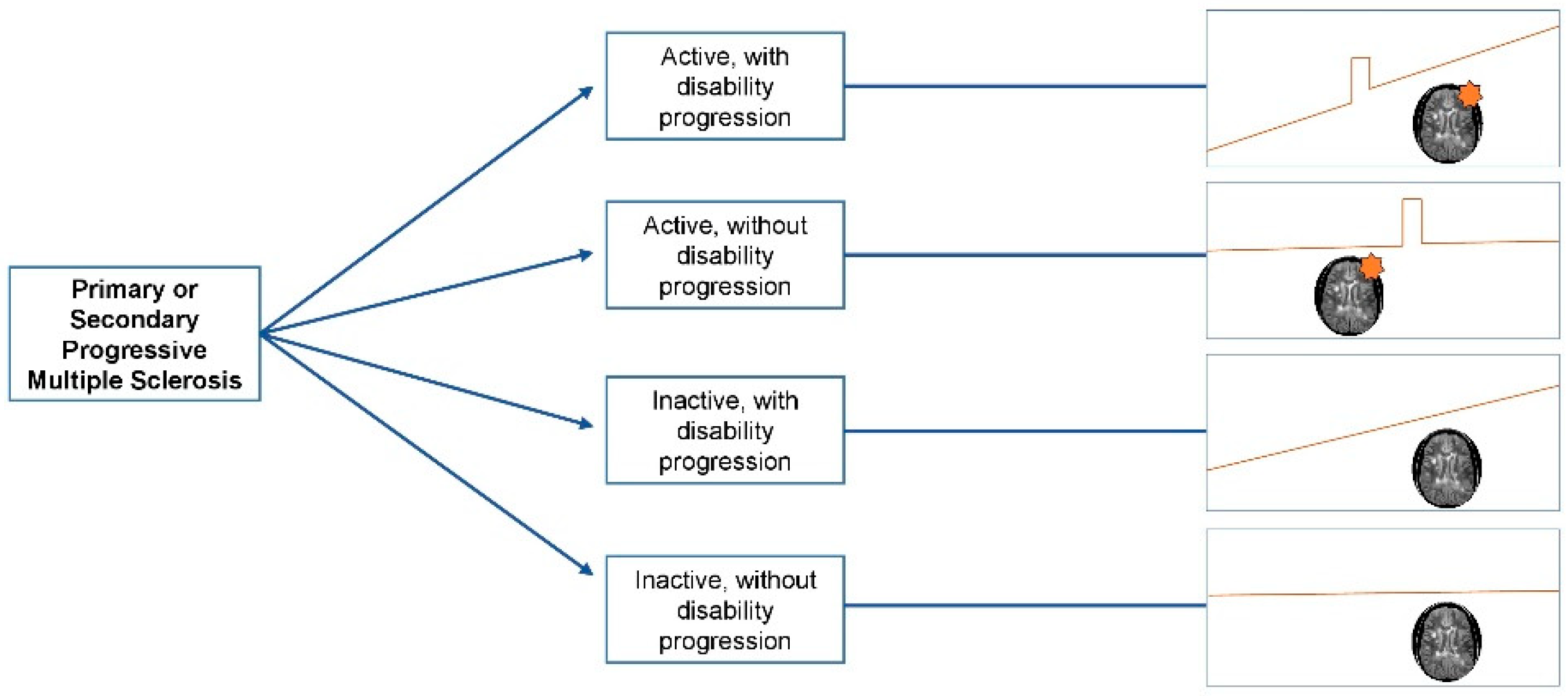
3. Diagnostic Criteria and Disease Course Definitions
4. Disability Outcome Measures
5. Measuring Disease Progression
6. Treatment
6.1. Anti-Inflammatory Disease-Modifying Therapies
6.1.1. Approved Therapies
6.1.2. Therapies with Negative or Weak Effect in Progressive MS
6.2. Remyelination and Neuroprotection in Progressive MS
6.3. Symptomatic Management
7. Challenges in Progressive MS Treatment and Research
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wallin, M.T.; Culpepper, W.J.; Campbell, J.D.; Nelson, M.L.; Langer-Gould, A.; Marrie, R.A.; Cutter, G.R.; Kaye, W.E.; Wagner, L.; Tremlett, H.; et al. The prevalence of MS in the United States: A population-based estimate using health claims data. Neurology 2019, 92, e1019–e1024. [Google Scholar] [CrossRef] [PubMed]
- Browne, P.; Chandraratna, D.; Angood, C.; Tremlett, H.; Baker, C.; Taylor, B.V.; Thompson, A.J. Atlas of Multiple Sclerosis 2013: A growing global problem with widespread inequity. Neurology 2014, 83, 1022–1024. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mörk, S.; Bö, L. Axonal Transection in the Lesions of Multiple Sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Kornek, B.; Storch, M.K.; Weissert, R.; Wallstroem, W.; Stefferl, A.; Olsson, T.; Linington, C.; Schmidbauer, M.; Lassmann, H. Multiple sclerosis and chronic autoimmune encephalomyelitis: A comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 2000, 157, 267–276. [Google Scholar] [CrossRef]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Bruck, W.; et al. Clinical and Pathological Insights into the Dynamic Nature of the White Matter Multiple Sclerosis Plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef]
- De Stefano, N.; Giorgio, A.; Battaglini, M.; Rovaris, M.; Sormani, M.P.; Barkhof, F.; Korteweg, T.; Enzinger, C.; Fazekas, F.; Calabrese, M.; et al. Assessing brain atrophy rates in a large population of untreated multiple sclerosis subtypes. Neurology 2010, 74, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, C.F.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Kutzelnigg, A.; Parisi, J.E.; Stadelmann, C.; Brück, W.; Lassmann, H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005, 128, 2705–2712. [Google Scholar]
- Amato, M.P.; Portaccio, E.; Goretti, B.; Zipoli, V.; Battaglini, M.; Bartolozzi, M.L.; Stromillo, M.L.; Guidi, L.; Siracusa, G.; Sorbi, S.; et al. Association of Neocortical Volume Changes with Cognitive Deterioration in Relapsing-Remitting Multiple Sclerosis. Arch. Neurol. 2007, 64, 1157–1161. [Google Scholar] [CrossRef]
- Calabrese, M.; Agosta, F.; Rinaldi, F.; Mattisi, I.; Grossi, P.; Favaretto, A.; Atzori, M.; Bernardi, V.; Barachino, L.; Rinaldi, L.; et al. Cortical Lesions and Atrophy Associated with Cognitive Impairment in Relapsing-Remitting Multiple Sclerosis. Arch. Neurol. 2009, 66, 1144–1150. [Google Scholar] [CrossRef]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial changes within axons in multiple sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Stys, P.K. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009, 8, 280–291. [Google Scholar] [CrossRef]
- Ontaneda, D.; Thompson, A.J.; Fox, R.J.; ACohen, J. Progressive multiple sclerosis: Prospects for disease therapy, repair, and restoration of function. Lancet 2017, 389, 1357–1366. [Google Scholar] [CrossRef]
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011, 134, 2755–2771. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007, 130, 1089–1104. [Google Scholar] [CrossRef]
- Brown, J.W.L.; Coles, A.; Horakova, D.; Havrdova, E.; Izquierdo, G.; Prat, A.; Girard, M.; Duquette, P.; Trojano, M.; Lugaresi, A.; et al. Association of Initial Disease-Modifying Therapy with Later Conversion to Secondary Progressive Multiple Sclerosis. JAMA 2019, 321, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, R.; Quaglini, S.; Tavazzi, E.; Amato, M.P.; Paolicelli, D.; Zipoli, V.; Romani, A.; Tortorella, C.; Portaccio, E.; D’Onghia, M.; et al. Immunomodulatory therapies delay disease progression in multiple sclerosis. Mult. Scler. J. 2016, 22, 1732–1740. [Google Scholar] [CrossRef]
- Trojano, M.; Pellegrini, F.; Paolicelli, D.; Fuiani, A.; Zimatore, G.B.; Tortorella, C.; Simone, I.L.; Patti, F.; Ghezzi, A.; Zipoli, V.; et al. Real-life impact of early interferon beta therapy in relapsing multiple sclerosis. Ann. Neurol. 2009, 66, 513–520. [Google Scholar] [CrossRef]
- Trojano, M.; Pellegrini, F.; Fuiani, A.; Paolicelli, D.; Zipoli, V.; Zimatore, G.B.; Monte, E.D.; Portaccio, C.; Lepore, V.; Livrea, P.; et al. New natural history of interferon-beta-treated relapsing multiple sclerosis. Ann. Neurol. 2007, 61, 300–306. [Google Scholar] [CrossRef]
- Haider, L.; Simeonidou, C.; Steinberger, G.; Hametner, S.; Grigoriadis, N.; Deretzi, G.; Kovacs, G.G.; Kutzelnigg, A.; Lassmann, H.; Frischer, J.M. Multiple sclerosis deep grey matter: The relation between demyelination, neurodegeneration, inflammation and iron. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1386–1395. [Google Scholar] [CrossRef]
- Hametner, S.; Wimmer, I.; Haider, L.; Pfeifenbring, S.; Brück, W.; Lassmann, H. Iron and neurodegeneration in the multiple sclerosis brain. Ann. Neurol. 2013, 74, 848–861. [Google Scholar] [CrossRef]
- Harding, K.E.; Liang, K.; Cossburn, M.D.; Ingram, G.; Hirst, C.; Pickersgill, T.; Te Water, N.; Johann, W.; Ben-Shlomo, M.; Robertson, Y.; et al. Long-term outcome of paediatric-onset multiple sclerosis: A population-based study. J Neurol. Neurosurg. Psychiatry 2013, 84, 141–147. [Google Scholar] [CrossRef]
- Leray, E.; Yaouanq, J.; Le Page, E.; Coustans, M.; Laplaud, D.; Oger, J.; Edan, G. Evidence for a two-stage disability progression in multiple sclerosis. Brain 2010, 133, 1900–1913. [Google Scholar] [CrossRef]
- Confavreux, C.; Moreau, T.; Vukusic, S.; Adeleine, P. Relapses and Progression of Disability in Multiple Sclerosis. N. Engl. J. Med. 2000, 343, 1430–1438. [Google Scholar] [CrossRef]
- Beiki, O.; Frumento, P.; Bottai, M.; Manouchehrinia, A.; Hillert, J. Changes in the Risk of Reaching Multiple Sclerosis Disability Milestones in Recent Decades: A Nationwide Population-Based Cohort Study in Sweden. JAMA Neurol. 2019, 1–7. [Google Scholar] [CrossRef]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.K.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 Revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Lublin, F.; Stephen, C.; Reingold, P.; Cohen, J.A.; Cutter, G.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef]
- Whitaker, J.N.; McFarland, H.F.; Rudge, P.; Reingold, R.S. Outcomes assessment in multiple sclerosis trials. Mult. Scler. 1995, 1, 37–47. [Google Scholar] [CrossRef]
- Noseworthy, J.H.; Vandervoort, M.K.; Wong, C.J.; Ebers, G.C. Interrater variability with the Expanded Disability Status Scale (EDSS) and Functional Systems (FS) in a multiple sclerosis clinical trial. Neurology 1990, 40, 971. [Google Scholar] [CrossRef]
- Goodkin, D.E.; Cookfair, D.; Wende, K.; Bourdette, D.; Pullicino, P.; Scherokman, B.; Whitham, R. Inter- and intrarater scoring agreement using grades 1.0 to 3.5 of the Kurtzke Expanded Disability Status Scale (EDSS). Neurology 1992, 42, 859. [Google Scholar] [CrossRef]
- Kragt, J.J.; Nielsen, J.M.; Van Der Linden, F.A.; Uitdehaag, B.M.; Polman, C.H. How similar are commonly combined criteria for EDSS progression in multiple sclerosis? Mult. Scler. J. 2006, 12, 782–786. [Google Scholar] [CrossRef]
- Ontaneda, D.; ACohen, J.; Amato, M.P. Clinical outcome measures for progressive MS trials. Mult. Scler. J. 2017, 23, 1627–1635. [Google Scholar] [CrossRef]
- Cutter, G.R.; Baier, M.L.; Rudick, R.A.; Cookfair, D.L.; Fischer, J.S.; Petkau, J.; Syndulko, K.; Weinshenker, B.G.; Antel, J.P.; Confavreux, C.; et al. Development of a multiple sclerosis functional composite as a clinical trial outcome measure. Brain 1999, 122, 871–882. [Google Scholar] [CrossRef]
- Fischer, J.; Rudick, R.; Cutter, G.; Reingold, S. The Multiple Sclerosis Functional Composite measure (MSFC): An integrated approach to MS clinical outcome assessment. Mult. Scler. J. 1999, 5, 244–250. [Google Scholar] [CrossRef]
- Cohen, J.A.; Cutter, G.R.; Fischer, J.S.; Goodman, A.D.; Heidenreich, F.R.; Jak, A.J.; Kniker, J.E.; Kooijmans, M.F.; Lull, J.M.; Sandrock, A.W.; et al. Use of the Multiple Sclerosis Functional Composite as an Outcome Measure in a Phase 3 Clinical Trial. Arch. Neurol. 2001, 58, 961–967. [Google Scholar] [CrossRef]
- Polman, C.H.; Rudick, R.A. The Multiple Sclerosis Functional Composite: A clinically meaningful measure of disability. Neurology 2010, 74, 8–15. [Google Scholar] [CrossRef]
- Kalkers, N.; Bergers, L.; De Groot, V.; Lazeron, R.; Van Walderveen, M.; Uitdehaag, B.; Polman, C.; Barkhof, F. Concurrent validity of the MS Functional Composite using MRI as a biological disease marker. Neurology 2001, 56, 215–219. [Google Scholar] [CrossRef]
- Miller, D.M.; Rudick, R.A.; Cutter, G.; Baier, M.; Fischer, J.S. Clinical Significance of the Multiple Sclerosis Functional Composite. Arch. Neurol. 2000, 57, 1319–1324. [Google Scholar] [CrossRef]
- Cohen, J.A.; Cutter, G.R.; Fischer, J.S.; Goodman, A.; Heidenreich, F.R.; Kooijmans, M.F.; Sandrock, A.W.; Rudick, R.A.; Simon, J.H.; Simonian, N.A.; et al. Benefit of interferon -1a on MSFC progression in secondary progressive multiple sclerosis. Neurology 2002, 59, 679–687. [Google Scholar] [CrossRef]
- Baier, M.L.; Cutter, G.R.; Rudick, R.A.; Miller, D.; Cohen, J.A.; Weinstock-Guttman, B.; Mass, M.; Balcer, L.J. Low-contrast letter acuity testing captures visual dysfunction in patients with multiple sclerosis. Neurology 2005, 64, 992–995. [Google Scholar] [CrossRef]
- Balcer, L.J.; Baier, M.L.; Cohen, J.A.; Kooijmans, M.F.; Sandrock, A.W.; Nano-Schiavi, M.L.; Pfohl, D.C.; Mills, M.; Bowen, J.; Ford, C.; et al. Contrast letter acuity as a visual component for the Multiple Sclerosis Functional Composite. Neurology 2003, 61, 1367–1373. [Google Scholar] [CrossRef]
- Freeman, J.; Hobart, J.; Thompson, A. Kurtzke scales revisited: The application of psychometric methods to clinical intuition. Brain 2000, 123, 1027–1040. [Google Scholar]
- Rasova, K.; Martinkova, P.; Vyskotova, J.; Sedova, M. Assessment set for evaluation of clinical outcomes in multiple sclerosis: Psychometric properties. Patient Relat. Outcome Meas. 2012, 3, 59–70. [Google Scholar] [CrossRef]
- ACohen, J.; Coles, A.J.; Arnold, D.L.; Confavreux, C.; Fox, E.J.; Hartung, H.-P.; Havrdova, E.K.; Selmaj, K.W.; Weiner, H.L.; Fisher, E.; et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: A randomised controlled phase 3 trial. Lancet 2012, 380, 1819–1828. [Google Scholar]
- Polman, C.; Hohlfeld, R.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; Burtin, P.; Kappos, L.; Radue, E.-W.; O’Connor, P.; Calabresi, P.; et al. A Placebo-Controlled Trial of Oral Fingolimod in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar]
- Meyer-Moock, S.; Feng, Y.-S.; Maeurer, M.; Dippel, F.-W.; Kohlmann, T. Systematic literature review and validity evaluation of the Expanded Disability Status Scale (EDSS) and the Multiple Sclerosis Functional Composite (MSFC) in patients with multiple sclerosis. BMC Neurol. 2014, 14, 58. [Google Scholar] [CrossRef]
- Rudick, R.A.; Miller, D.; Bethoux, F.; Rao, S.M.; Lee, J.-C.; Stough, D.; Reece, C.; Schindler, D.; Mamone, B.; Alberts, J. The Multiple Sclerosis Performance Test (MSPT): An iPad-Based Disability Assessment Tool. J. Vis. Exp. 2014, e51318. [Google Scholar] [CrossRef]
- Macaron, G.; Moss, B.P.; Li, H.; Baldassari, L.; Rao, S.; Schindler, D.; Alberts, J.; Weber, M.; Ayers, M.; Bethoux, F.; et al. Technology-enabled assessments to enhance multiple sclerosis clinical care and research. Neurol. Clin. Pract. 2019, in press. [Google Scholar]
- Motl, R.W.; A Cohen, J.; Benedict, R.; Phillips, G.; LaRocca, N.; Hudson, L.D.; Rudick, R. Multiple Sclerosis Outcome Assessments Consortium Validity of the timed 25-foot walk as an ambulatory performance outcome measure for multiple sclerosis. Mult. Scler. J. 2017, 23, 704–710. [Google Scholar] [CrossRef]
- Feng, J.; Qu, J.; Felix, C.; McGinley, M.; Nakamura, K.; Macaron, G.; Moss, B.; Li, H.; Jones, S.; Rao, S.; et al. Quantitative MRI and patient-reported outcomes validate clinically meaningful changes on tests of ambulation and hand function. ACTRIMS 2019, P050. [Google Scholar]
- Feys, P.; Lamers, I.; Francis, G.; Benedict, R.; Phillips, G.; LaRocca, N.; Hudson, L.D.; Rudick, R. Multiple Sclerosis Outcome Assessments Consortium the Nine-Hole Peg Test as a manual dexterity performance measure for multiple sclerosis. Mult. Scler. J. 2017, 23, 711–720. [Google Scholar] [CrossRef]
- Benedict, R.H.; DeLuca, J.; Phillips, G.; LaRocca, N.; Hudson, L.D.; Rudick, R. Multiple Sclerosis Outcome Assessments Consortium Validity of the Symbol Digit Modalities Test as a cognition performance outcome measure for multiple sclerosis. Mult. Scler. J. 2017, 23, 721–733. [Google Scholar] [CrossRef]
- Macaron, G.; Moss, B.; Baldassari, L.E.; Conway, D.; McGinley, M.; Alshehri, E.; Feng, J.; Bermel, R.; Boissy, A.; Cohen, J.A.; et al. Cross-sectional predictive value of clinically meaningful change in processing speed on self-reported cognition and MRI metrics. ACTRIMS 2019, P034. [Google Scholar]
- Balcer, L.J.; Raynowska, J.; Nolan, R.; Galetta, S.L.; Kapoor, R.; Benedict, R.; Phillips, G.; LaRocca, N.; Hudson, L.; Rudick, R.; et al. Validity of low-contrast letter acuity as a visual performance outcome measure for multiple sclerosis. Mult. Scler. J. 2017, 23, 734–747. [Google Scholar] [CrossRef]
- Oh, J.; Ontaneda, D.; Azevedo, C.; Klawiter, E.; Absinta, M.; Arnold, D.; Bakshi, R.; Calabresi, P.; Crainiceanu, C.; Dewey, B.; et al. Imaging outcome measures of neuroprotection repair in MS: A consensus statement from NAIMS. Neurology 2019, 92, 519–533. [Google Scholar] [CrossRef]
- Ontaneda, D.; Fox, R.J.; Chataway, J. Clinical trials in progressive multiple sclerosis: Lessons learned and future perspectives. Lancet Neurol. 2015, 14, 208–223. [Google Scholar] [CrossRef]
- Mahajan, K.R.; Ontaneda, D. The Role of Advanced Magnetic Resonance Imaging Techniques in Multiple Sclerosis Clinical Trials. Neurotherapeutics 2017, 14, 905–923. [Google Scholar] [CrossRef]
- Hardmeier, M.; Leocani, L.; Fuhr, P. A new role for evoked potentials in MS? Repurposing evoked potentials as biomarkers for clinical trials in MS. Mult. Scler. J. 2017, 23, 1309–1319. [Google Scholar] [CrossRef]
- Saidha, S.; Al-Louzi, O.; Ratchford, J.N.; Bhargava, P.; Oh, J.; Newsome, S.D.; Prince, J.L.; Pham, D.; Roy, S.; Van Zijl, P.; et al. Optical Coherence Tomography Reflects Brain Atrophy in Multiple Sclerosis: A Four-Year Study. Ann. Neurol. 2015, 78, 801–813. [Google Scholar] [CrossRef]
- Novakova, L.; Zetterberg, H.; Sundström, P.; Axelsson, M.; Khademi, M.; Gunnarsson, M.; Malmeström, C.; Svenningsson, A.; Olsson, T.; Piehl, F.; et al. Monitoring disease activity in multiple sclerosis using serum neurofilament light protein. Neurology 2017, 89, 2230–2237. [Google Scholar] [CrossRef]
- Damasceno, A.; Dias-Carneiro, R.P.C.; Moraes, A.S.; Boldrinib, V.O.; Quintilianob, R.P.S.; de Paula Galdino da Silvab, V.A.; Fariasb, A.S.; Brandãob, C.O.; Damascenoa, B.P.; dos Santos, L.M.B.; et al. Clinical MRI correlates of CSF neurofilament light chain levels in relapsing progressive multiple sclerosis. Mult. Scler. Relat. Disord. 2019, 30, 149–153. [Google Scholar] [CrossRef]
- Martin, S.J.; McGlasson, S.; Hunt, D.; Overell, J. Cerebrospinal fluid neurofilament light chain in multiple sclerosis and its subtypes: A meta-analysis of case-control studies. J. Neurol. Neurosurg. Psychiatry 2019. [Google Scholar] [CrossRef]
- Pawlitzki, M.; Schreiber, S.; Bittner, D.; Kreipe, J.; Leypoldt, F.; Rupprecht, K.; Carare, R.O.; Meuth, S.G.; Vielhaber, S.; Körtvélyessy, P. CSF Neurofilament Light Chain Levels in Primary Progressive MS: Signs of Axonal Neurodegeneration. Front. Neurol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Barro, C.; Naegelin, Y.; Schädelin, S.; Giardiello, A.; Zecca, C.; Blennow, K.; Zetterberg, H.; Leppert, D.; Gobbi, C.; Kuhle, J.; et al. Serum Neurofilament light: A biomarker of neuronal damage in multiple sclerosis. Ann. Neurol. 2017, 81, 857–870. [Google Scholar]
- Barro, C.; Benkert, P.; Disanto, G.; Tsagkas, C.; Amann, M.; Naegelin, Y.; Leppert, D.; Gobbi, C.; Granziera, C.; Yaldizli, O.; et al. Serum neurofilament as a predictor of disease worsening and brain and spinal cord atrophy in multiple sclerosis. Brain 2018, 141, 2382–2391. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; De Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef]
- Fox, E.; Markowitz, C.; Applebee, A.; Montalban, X.; Wolinsky, J.S.; Belachew, S.; Damian, F.; Han, J.; Musch, B.; Giovannoni, G. Effect of ocrelizumab on upper limb function in patients with primary progressive multiple sclerosis (PPMS) in the oratorio study (ENCORE). J. Neurol. Neurosurg. Psychiatry 2018, 89, A14. [Google Scholar] [CrossRef]
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 2009, 66, 460–471. [Google Scholar] [CrossRef]
- Naegelin, Y.; Naegelin, P.; Von Felten, S.; Lorscheider, J.; Sonder, J.; Uitdehaag, B.M.J.; Scotti, B.; Zecca, C.; Gobbi, C.; Kappos, L.; et al. Association of Rituximab Treatment with Disability Progression Among Patients With Secondary Progressive Multiple Sclerosis. JAMA Neurol. 2019, 76, 274. [Google Scholar] [CrossRef]
- Gelfand, J.M.; Cree, B.A.C.; Hauser, S.L. Ocrelizumab and Other CD20+ B-Cell-Depleting Therapies in Multiple Sclerosis. Neurotherapeutics 2017, 14, 835–841. [Google Scholar] [CrossRef]
- Klein, C.; Lammens, A.; Schäfer, W.; Georges, G.; Schwaiger, M.; Mössner, E.; Hopfner, K.-P.; Umana, P.; Niederfellner, G. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. mAbs 2013, 5, 22–33. [Google Scholar] [CrossRef]
- Feng, J.J.; Ontaneda, D. Treating primary-progressive multiple sclerosis: Potential of ocrelizumab and review of B-cell therapies. Degener. Neurol. Neuromuscul. Dis. 2017, 7, 31–45. [Google Scholar] [CrossRef]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Jackson, S.J.; Giovannoni, G.; Baker, D. Fingolimod modulates microglial activation to augment markers of remyelination. J. Neuroinflamm. 2011, 8, 76. [Google Scholar] [CrossRef]
- Lublin, F.; Miller, D.H.; Freedman, M.S.; Cree, B.A.C.; Wolinsky, J.S.; Weiner, H.; Lubetzki, C.; Hartung, H.-P.; Montalban, X.; Uitdehaag, B.M.J.; et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet 2016, 387, 1075–1084. [Google Scholar] [CrossRef]
- Martinelli Boneschi, F.; Vacchi, L.; Rovaris, M.; Capra, R.; Comi, G. Mitoxantrone for multiple sclerosis. Cochrane Database Syst. Rev. 2013, 5, CD002127. [Google Scholar] [CrossRef]
- Hartung, H.P.; Gonsette, R.; König, N.; Kwiecinski, H.; Guseo, A.; Morrissey, S.; Krapf, S.; Zwingers, T. Mitoxantrone in Multiple Sclerosis Study Group. Mitoxantrone in progressive multiple sclerosis: A placebo-controlled, double-blind, randomised, multicentre trial. Lancet 2002, 28, 2018–2025. [Google Scholar] [CrossRef]
- Krapf, H.; Morrissey, S.P.; Zenker, O.; Zwingers, T.; Gonsette, R.; Hartung, H.-P. Effect of mitoxantrone on MRI in progressive MS: Results of the MIMS trial. Neurology 2005, 65, 690–695. [Google Scholar] [CrossRef]
- Grey Née Cotte, S.; Salmen Née Stroet, A.; Von Ahsen, N.; Starck, M.; Winkelmann, A.; Zettl, U.K.; Comabella, M.; Montalban, X.; Zipp, F.; Fleischer, V.; et al. Lack of efficacy of mitoxantrone in primary progressive Multiple Sclerosis irrespective of pharmacogenetic factors: A multi-center, retrospective analysis. J. Neuroimmunol. 2015, 278, 277–279. [Google Scholar] [CrossRef]
- Pelfrey, C.M.; Cotleur, A.C.; Zamor, N.; Lee, J.C.; Robert, R.J. Immunological studies of mitoxantrone in primary progressive multiple sclerosis. J. Neuroimmunol. 2006, 175, 192–199. [Google Scholar] [CrossRef]
- Cotte, S.; Von Ahsen, N.; Kruse, N.; Huber, B.; Winkelmann, A.; Zettl, U.K.; Starck, M.; König, N.; Téllez, N.; Dorr, J.; et al. ABC-transporter gene-polymorphisms are potential pharmacogenetic markers for mitoxantrone response in multiple sclerosis. Brain 2009, 132, 2517–2530. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Press Announcement−FDA Approves New Oral Drug to Treat Multiple Sclerosis. Office of the Commissioner. Available online: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm634469.htm (accessed on 2 April 2019).
- Giovannoni, G.; Comi, G.; Cook, S.; Rammohan, K.; Rieckmann, P.; Sørensen, P.S.; Vermersch, P.; Chang, P.; Hamlett, A.; Musch, B.; et al. A Placebo-Controlled Trial of Oral Cladribine for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2010, 362, 416–426. [Google Scholar] [CrossRef]
- Montalban, X.; Leist, T.P.; Cohen, B.A.; Moses, H.; Campbell, J.; Hicking, C.; Dangond, F. Cladribine tablets added to IFN-β in active relapsing multiple sclerosis. Neurol. Neuroimmunol. NeuroInflamm. 2018, 5, 477. [Google Scholar] [CrossRef]
- Leist, T.P.; Comi, G.; Cree, B.A.C.; Coyle, P.K.; Freedman, M.S.; Hartung, H.-P.; Vermersch, P.; Casset-Semanaz, F.; Scaramozza, M. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): A phase 3 randomised trial. Lancet Neurol. 2014, 13, 257–267. [Google Scholar] [CrossRef]
- Sipe, J. Cladribine in treatment of chronic progressive multiple sclerosis. Lancet 1994, 344, 9–13. [Google Scholar] [CrossRef]
- Rice, G.P.A.; Filippi, M.; Comi, G. Cladribine and progressive MS: Clinical and MRI outcomes of a multicenter controlled trial. Neurology 2000, 54, 1145–1155. [Google Scholar] [CrossRef]
- Filippi, M.; Rovaris, M.; Iannucci, G.; Mennea, S.; Sormani, M.P.; Comi, G. Whole brain volume changes in patients with progressive multiple sclerosis treated with cladribine. Neurology 2000, 55, 1714–1718. [Google Scholar] [CrossRef]
- Polman, C.; Pozzilli, C.; Thompson, A.; Beckmann, K.; Dahlke, F. Final analysis of the European multicenter trial on IFNbeta-1b in secondary-progressive multiple sclerosis. Neurology 2001, 57, 1969–1975. [Google Scholar]
- The North American Study Group. Interferon beta-1b in secondary progressive MS: Results from a 3-year controlled study. Neurology 2004, 63, 1788–1795. [Google Scholar] [CrossRef]
- Kappos, L.; Weinstock-Guttman, B.; Pozzilli, C.; Thompson, A.J.; Dahlke, F.; Beckmann, K.; Polman, C.; McFarland, H.; European (EU-SPMS) Interferon beta-1b in Secondary Progressive Multiple Sclerosis Trial Steering Committee and Independent Advisory Board; North American (NA-SPMS) Interferon beta-1b in Secondary Progressive Multiple Sclerosis Trial Steering Committee and Independent Advisory Board. Interferon beta-1b in secondary progressive multiple sclerosis. A combined analysis of the two trials. Neurology 2004, 63, 1779–1787. [Google Scholar] [CrossRef]
- Secondary Progressive Efficacy Clinical Trial of Recombinant Interferon-Beta-1a in MS (SPECTRIMS) Study Group. Randomized controlled trial of interferon- beta-1a in secondary progressive MS: Clinical results. Neurology 2001, 56, 1496–1504. [Google Scholar] [CrossRef]
- Andersen, O.; Elovaara, I.; Färkkilä, M.; Hansen, H.J.; Mellgren, S.; Myhr, K.-M.; Sandberg-Wollheim, M.; Soelberg, S. Multicentre, randomised, double blind, placebo controlled, phase III study of weekly, low dose, subcutaneous interferon beta-1a in secondary progressive multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2004, 75, 706–710. [Google Scholar] [CrossRef]
- Leary, S.M.; Miller, D.H.; Stevenson, V.L.; Brex, P.A.; Chard, D.T.; Thompson, A.J. Interferon beta-1a in primary progressive MS: An exploratory, randomized, controlled trial. Neurology 2003, 60, 44–51. [Google Scholar] [CrossRef]
- Montalban, X. Overview of European pilot study of interferon beta-Ib in primary progressive multiple sclerosis. Mult. Scler. 2004, 10, S62–S64. [Google Scholar]
- Montalban, X.; Garriga, J.S.; Tintoré, M.; Brieva, L.; Aymerich, F.; Rio, J.; Porcel, J.; Borràs, C.; Nos, C.; Rovira, À.; et al. A single-center, randomized, double-blind, placebo-controlled study of interferon beta-1b on primary progressive and transitional multiple sclerosis. Mult. Scler. J. 2009, 15, 1195–1205. [Google Scholar] [CrossRef]
- Wolinsky, J.S.; Narayana, P.A.; O’Connor, P.; Coyle, P.K.; Ford, C.; Johnson, K.; Miller, A.; Pardo, L.; Kadosh, S.; Ladkani, D.; et al. Glatiramer acetate in primary progressive multiple sclerosis: Results of a multinational, multicenter, double-blind, placebo-controlled trial. Ann. Neurol. 2007, 61, 14–24. [Google Scholar] [CrossRef]
- Wolinsky, J.S.; Shochat, T.; Weiss, S.; Ladkani, D. Glatiramer acetate treatment in PPMS: Why males appear to respond favorably. J. Neurol. Sci. 2009, 286, 92–98. [Google Scholar] [CrossRef]
- Ho, P.-R.; Campbell, N.; Chang, I.; Deykin, A.; Forrestal, F.; Lucas, N.; Yu, B.; Arnold, D.L.; Hartung, H.-P.; Miller, A.; et al. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): A phase 3, randomised, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurol. 2018, 17, 405–415. [Google Scholar]
- Graves, D.; Vernino, S. Immunotherapies in Neurologic Disorders. Med. Clin. N. Am. 2012, 96, 497–523. [Google Scholar] [CrossRef]
- Salzer, J.; Svenningsson, R.; Alping, P.; Novakova, L.; Bjorck, A.; Fink, K.; Islam-Jakobsson, P.; Malmestrom, C.; Axelsson, M.; Vagberg, M. Rituximab in multiple sclerosis: A retrospective observational study on safety and efficacy. Neurology 2016, 87, 2074–2081. [Google Scholar] [CrossRef]
- Komori, M.; Lin, Y.C.; Cortese, I.; Blake, A.; Ohayon, J.; Cherup, J.; Maric, D.; Kosa, P.; Wu, T.; Bielekova, B. Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis. Ann. Clin. Transl. Neurol. 2016, 3, 166–179. [Google Scholar] [CrossRef]
- Bhargava, P.; Wicken, C.; Smith, M.D.; Strowd, R.E.; Cortese, I.; Reich, D.S.; Calabresi, P.A.; Mowry, E.M. Trial of intrathecal rituximab in progressive multiple sclerosis patients with evidence of leptomeningeal contrast enhancement. Mult. Scler. Relat. Disord. 2019, 30, 136–140. [Google Scholar] [CrossRef]
- Bergman, J.; Burman, J.; Gilthorpe, J.D.; Zetterberg, H.; Jiltsova, E.; Bergenheim, T.; Svenningsson, A. Intrathecal treatment trial of rituximab in progressive MS: An open-label phase 1b study. Neurology 2018, 91, e1893–e1901. [Google Scholar] [CrossRef]
- Topping, J.; Dobson, R.; Lapin, S.; Maslyanskiy, A.; Kropshofer, H.; David, L.; Giovannoni, G.; Evdoshenko, E. The effects of intrathecal rituximab on biomarkers in multiple sclerosis. Mult. Scler. Relat. Disord. 2016, 6, 49–53. [Google Scholar] [CrossRef]
- British and Dutch Multiple Sclerosis Azathioprine Trial Group. Double-masked trial of azathioprine in multiple sclerosis. Lancet 1988, 2, 179–183. [Google Scholar]
- Kappos, L.; Pätzold, U.; Dommasch, D.; Poser, S.; Haas, J.; Krauseneck, P.; Malin, J.-P.; Fierz, W.; Graffenried, B.U.; Gugerli, U.S. Cyclosporine versus azathioprine in the long-term treatment of multiple sclerosis? results of the german multicenter study. Ann. Neurol. 1988, 23, 56–63. [Google Scholar] [CrossRef]
- Uccelli, A.; Capello, E.; Fenoglio, D.; Incagliato, M.; Valbonesi, M.; Mancardi, G.L. Intravenous immunoglobulin, plasmalymphocytapheresis and azathioprine in chronic progressive multiple sclerosis. Neurol. Sci. 1994, 15, 49–53. [Google Scholar] [CrossRef]
- Perini, P.; Calabrese, M.; Tiberio, M.; Ranzato, F.; Battistin, L.; Gallo, P. Mitoxantrone versus cyclophosphamide in secondary-progressive multiple sclerosis: A comparative study. J. Neurol. 2006, 253, 1034–1040. [Google Scholar] [CrossRef]
- Zephir, H.; De Seze, J.; Duhamel, A.; Debouverie, M.; Hautecoeur, P.; Lebrun, C.; Malíková, I.; Pelletier, J.; Sénéchal, O.; Vermersch, P. Treatment of progressive forms of multiple sclerosis by cyclophosphamide: A cohort study of 490 patients. J. Neurol. Sci. 2004, 218, 73–77. [Google Scholar] [CrossRef]
- Brochet, B.; Deloire, M.S.A.; Perez, P.; Loock, T.; Baschet, L.; Debouverie, M.; Pittion, S.; Ouallet, J.-C.; Clavelou, P.; De Seze, J.; et al. Double-Blind Controlled Randomized Trial of Cyclophosphamide versus Methylprednisolone in Secondary Progressive Multiple Sclerosis. PLoS ONE 2017, 12, e0168834. [Google Scholar] [CrossRef]
- Weiner, H.L.; Mackin, G.A.; Orav, E.J.; Hafler, D.A.; Dawson, D.M.; Lapierre, Y.; Herndon, R.; Lehrich, J.R.; Hauser, S.L.; Turel, A.; et al. Intermittent cyclophosphamide pulse therapy in progressive multiple sclerosis: Final report of the Northeast Cooperative Multiple Sclerosis Treatment Group. Neurology 1993, 43, 910. [Google Scholar] [CrossRef]
- Hommes, O.R.; Sørensen, P.S.; Fazekas, F.; Enriquez, M.M.; Koelmel, H.W.; Fernández, Ó.; Pozzilli, C.; O’Connor, P. Intravenous immunoglobulin in secondary progressive multiple sclerosis: Randomised placebo-controlled trial. Lancet 2004, 364, 1149–1156. [Google Scholar] [CrossRef]
- Goodkin, D.; Rudick, R.; Medendorp, S.V.; Daughtry, M.; Van Dyke, C. Low-dose oral methotrexate in chronic progressive multiple sclerosis: Analyses of serial MRIs. Neurology 1996, 47, 1153–1157. [Google Scholar] [CrossRef]
- Goodkin, D.E.; Rudick, R.A.; Medendorp, S.V.; Daughtry, M.M.; Schwetz, K.M.; Fischer, J.; Van Dyke, C. Low-dose (7.5 mg) oral methotrexate reduces the rate of progression in chronic progressive multiple sclerosis. Ann. Neurol. 1995, 37, 30–40. [Google Scholar] [CrossRef]
- Lugaresi, A.; Caporale, C.; Farina, D.; Marzoli, F.; Bonanni, L.; Muraro, P.A.; De Luca, G.; Iarlori, C.; Gambi, D. Low-dose oral methotrexate treatment in chronic progressive multiple sclerosis. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2001, 22, 209–210. [Google Scholar] [CrossRef]
- The Multiple Sclerosis Study Group. Efficacy and toxicity of cyclosporine in chronic progressive multiple sclerosis: A randomized, double-blinded, placebo-controlled clinical trial. Ann Neurol. 1990, 27, 591–605. [Google Scholar] [CrossRef]
- Fakih, R.; Matiello, M.; Chitnis, T.; Stankiewicz, J.M. Efficacy and safety of mycophenolate mofetil in progressive multiple sclerosis patients. J. Neurol. 2018, 265, 2688–2694. [Google Scholar] [CrossRef]
- Barkhof, F.; Giovannoni, G.; Hartung, H.P.; Cree, B.; Uccelli, A.; Sormani, M.P.; Krieger, S.; Uitdehaag, B.; Vollmer, T.; Montalban, X. ARPEGGIO: A randomized, placebo-controlled study to evaluate oral laquinimod in patients with primary progressive multiple sclerosis (PPMS) (P7.210). Neurology 2015, 84, P7.210. [Google Scholar]
- Freedman, M.S.; Bar-Or, A.; Oger, J.; Traboulsee, A.; Patry, D.; Young, C.; Olsson, T.; Li, D.; Hartung, H.P.; Krantz, M.; et al. A phase III study evaluating the efficacy safety of MBP8298 in secondary progressive multiple sclerosis. Neurology 2011, 77, 1551–1560. [Google Scholar] [CrossRef]
- Deshmukh, V.A.; Tardif, V.; Lyssiotis, C.A.; Green, C.C.; Kerman, B.; Kim, H.J.; Padmanabhan, K.; Swoboda, J.G.; Ahmad, I.; Kondo, T.; et al. A regenerative approach to the treatment of multiple sclerosis. Nature 2013, 502, 327–332. [Google Scholar] [CrossRef]
- Mei, F.; Fancy, S.P.J.; Shen, Y.A.; Niu, J.; Zhao, C.; Presley, B.; Miao, E.; Lee, S.; Mayoral, S.R.; Redmond, S.A.; et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med. 2014, 20, 954–960. [Google Scholar] [CrossRef]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): A randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef]
- Jepson, S.; Vought, B.; Gross, C.H.; Gan, L.; Austen, D.; Frantz, J.D.; Zwahlen, J.; Lowe, D.; Markland, W.; Krauss, R. LINGO-1, a Transmembrane Signaling Protein, Inhibits Oligodendrocyte Differentiation and Myelination through Intercellular Self-interactions. J. Boil. Chem. 2012, 287, 22184–22195. [Google Scholar] [CrossRef]
- Mi, S.; Miller, R.H.; Lee, X.; Scott, M.L.; Shulag-Morskaya, S.; Shao, Z.; Chang, J.; Thill, G.; Levesque, M.; Zhang, M.; et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat. Neurosci. 2005, 8, 745–751. [Google Scholar] [CrossRef]
- Cadavid, D.; Balcer, L.; Galetta, S.; Aktas, O.; Ziemssen, T.; Vanopdenbosch, L.; Frederiksen, J.; Skeen, M.; Jaffe, G.J.; Butzkueven, H.; et al. Safety and efficacy of opicinumab in acute optic neuritis (RENEW): A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2017, 16, 189–199. [Google Scholar] [CrossRef]
- Mellion, M.; Edwards, K.R.; Hupperts, R.; Drulović, J.; Montalban, X.; Hartung, H.P.; Brochet, B.; Calabresi, P.A.; Rudick, R.; Ibrahim, A. Efficacy Results from the Phase 2b SYNERGY Study: Treatment of Disabling Multiple Sclerosis with the Anti-LINGO-1 Monoclonal Antibody Opicinumab (S33.004). Neurology 2017, 88, S33.004. [Google Scholar]
- .McCroskery, P.; Selmaj, K.; Fernandez, O.; Grimaldi, L.; Silber, E.; Pardo, G.; Freedman, M.S.; Zhang, Y.; Xu, L.; Cadavid, D. Safety and Tolerability of Opicinumab in Relapsing Multiple Sclerosis. Neurology 2017, 88, P5.369. [Google Scholar]
- Cho, Y.; Crichlow, G.V.; Vermeire, J.J.; Leng, L.; Du, X.; Hodsdon, M.E.; Bucala, R.; Cappello, M.; Gross, M.; Gaeta, F.; et al. Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proc. Natl. Acad. Sci. USA 2010, 107, 11313–11318. [Google Scholar] [CrossRef]
- Fox, R.J.; Coffey, C.S.; Conwit, R.; Cudkowicz, M.E.; Gleason, T.; Goodman, A.; Klawiter, E.C.; Matsuda, K.; McGovern, M.; Naismith, R.T.; et al. Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N. Engl. J. Med. 2018, 379, 846–855. [Google Scholar] [CrossRef]
- Greenwood, J.; Steinman, L.; Zamvil, S.S. Statin therapy and autoimmune disease: From protein prenylation to immunomodulation. Nat. Rev. Immunol. 2006, 6, 358–370. [Google Scholar] [CrossRef]
- Chataway, J.; Schuerer, N.; Alsanousi, A.; Chan, D.; MacManus, D.; Hunter, K.; Anderson, V.; Bangham, C.R.M.; Clegg, S.; Nielsen, C.; et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): A randomised, placebo-controlled, phase 2 trial. Lancet 2014, 383, 2213–2221. [Google Scholar] [CrossRef]
- Rocamonde, B.; Paradells, S.; Barcia, J.; Barcia, C.; García-Verdugo, J.M.; Miranda, M.; Gómez, F.R.; Soria, J. Neuroprotection of lipoic acid treatment promotes angiogenesis and reduces the glial scar formation after brain injury. Neuroscience 2012, 224, 102–115. [Google Scholar] [CrossRef]
- Rochette, L.; Ghibu, S.; Richard, C.; Zeller, M.; Cottin, Y.; Vergely, C. Direct and indirect antioxidant properties of alpha-lipoic acid and therapeutic potential. Mol. Nutr. Food Res. 2013, 57, 114–125. [Google Scholar] [CrossRef]
- Spain, R.; Powers, K.; Murchison, C.; Heriza, E.; Winges, K.; Yadav, V.; Cameron, M.; Kim, E.; Horak, F.; Simon, J.; et al. Lipoic acid in secondary progressive multiple sclerosis: A randomized controlled pilot trial. Neur. Neuroimmunol. Neuroinflamm. 2017, 4, e374. [Google Scholar] [CrossRef]
- Lo, A.C.; Saab, C.Y.; Black, J.A.; Waxman, S.G. Phenytoin Protects Spinal Cord Axons and Preserves Axonal Conduction and Neurological Function in a Model of Neuroinflammation in Vivo. J. Neurophysiol. 2003, 90, 3566–3571. [Google Scholar] [CrossRef]
- Raftopoulos, R.; Hickman, S.J.; Toosy, A.; Sharrack, B.; Mallik, S.; Paling, D.; Altmann, D.R.; Yiannakas, M.C.; Malladi, P.; Sheridan, R.; et al. Phenytoin for neuroprotection in patients with acute optic neuritis: A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 259–269. [Google Scholar] [CrossRef]
- Cohen, J.A. Mesenchymal Stem Cell Transplantation in Multiple Sclerosis. J. Neurol. Sci. 2013, 333, 43–49. [Google Scholar] [CrossRef]
- Bonab, M.M.; Yazdanbakhsh, S.; Lotfi, J.; Alimoghaddom, K.; Talebian, F.; Hooshmand, F.; Ghavamzadeh, A.; Nikbin, B. Does mesenchymal stem cell therapy help multiple sclerosis patients? Report of a pilot study. Iran. J. Immunol. IJI 2007, 4, 50–57. [Google Scholar]
- Bonab, M.M.; Sahraian, M.A.; Aghsaie, A.; Karvigh, S.A.; Hosseinian, S.M.; Nikbin, B.; Lotfi, J.; Khorramnia, S.; Motamed, M.R.; Togha, M.; et al. Autologous mesenchymal stem cell therapy in progressive multiple sclerosis: An open label study. Curr. Stem Cell Res. Ther. 2012, 7, 407–414. [Google Scholar] [CrossRef]
- Yamout, B.; Hourani, R.; Salti, H.; Barada, W.; El-Hajj, T.; Al-Kutoubi, A.; Herlopian, A.; Baz, E.K.; Mahfouz, R.; Khalil-Hamdan, R.; et al. Bone marrow mesenchymal stem cell transplantation in patients with multiple sclerosis: A pilot study. J. Neuroimmunol. 2010, 227, 185–189. [Google Scholar] [CrossRef]
- Lublin, F.D.; Bowen, J.D.; Huddlestone, J.; Kremenchutzky, M.; Carpenter, A.; Corboy, J.R.; Freedman, M.S.; Krupp, L.; Paulo, C.; Hariri, R.J.; et al. Human placenta-derived cells (PDA-001) for the treatment of adults with multiple sclerosis: A randomized, placebo-controlled, multiple-dose study. Mult. Scler. Relat. Disord. 2014, 3, 696–704. [Google Scholar] [CrossRef]
- Karussis, D.; Karageorgiou, C.; Vaknin-Dembinsky, A.; Gowda-Kurkalli, B.; Gomori, J.M.; Kassis, I.; Bulte, J.W.M.; Petrou, P.; Ben-Hur, T.; Abramsky, O.; et al. Safety and Immunological Effects of Mesenchymal Stem Cell Transplantation in Patients with Multiple Sclerosis and Amyotrophic Lateral Sclerosis. Arch. Neurol. 2010, 67, 1187–1194. [Google Scholar] [CrossRef]
- Harris, V.K.; Vyshkina, T.; Sadiq, S.A. Clinical safety of intrathecal administration of mesenchymal stromal cell–derived neural progenitors in multiple sclerosis. Cytotherapy 2016, 18, 1476–1482. [Google Scholar] [CrossRef]
- Harris, V.K.; Stark, J.; Vyshkina, T.; Blackshear, L.; Joo, G.; Stefanova, V.; Sara, G.; Sadiq, S.A. Phase I Trial of Intrathecal Mesenchymal Stem Cell-derived Neural Progenitors in Progressive Multiple Sclerosis. EBioMedicine 2018, 29, 23–30. [Google Scholar] [CrossRef]
- Li, J.-F.; Zhang, D.-J.; Geng, T.; Chen, L.; Huang, H.; Yin, H.-L.; Zhang, Y.-Z.; Lou, J.-Y.; Cao, B.; Wang, Y.-L. The Potential of Human Umbilical Cord-Derived Mesenchymal Stem Cells as a Novel Cellular Therapy for Multiple Sclerosis. Cell Transplant. 2014, 23, 113–122. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, H.; Hua, B.; Wang, H.; Wang, J.; Han, Z.; Sun, L. Allogeneic mesenchymal stem cells transplantation in treatment of multiple sclerosis. Mult. Scler. J. 2009, 15, 644–646. [Google Scholar] [CrossRef]
- Cohen, J.A.; Atkins, H.; Banwell, B.; Bar-Or, A.; Bebo, B.; Bowen, J.; Burt, R.; Calabresi, P.; Cohen, J.; Comi, G.; et al. Cell-based therapeutic strategies for multiple sclerosis. Brain 2017, 140, 2776–2796. [Google Scholar]
- Uccelli, A.; Brundin, L.; Clanet, M.; Fernandez, O.; Nabavi, S.M.; Muraro, P.A.; Oliveri, R.S.; Radue, E.W.; Sellner, J.; Sorensen, P.S.; et al. MEsenchymal StEm cells for Multiple Sclerosis (MESEMS): A randomized, double blind, cross-over phase I/II clinical trial with autologous mesenchymal stem cells for the therapy of multiple sclerosis. Trials 2019, 20, 263. [Google Scholar] [CrossRef]
- Sedel, F.; Bernard, D.; Mock, D.M.; Tourbah, A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology 2016, 110, 644–653. [Google Scholar] [CrossRef]
- Sedel, F.; Papeix, C.; Bellanger, A.; Touitou, V.; Lebrun-Frenay, C.; Galanaud, D.; Gout, O.; Lyon-Caen, O.; Tourbah, A. High doses of biotin in chronic progressive multiple sclerosis: A pilot study. Mult. Scler. Relat. Disord. 2015, 4, 159–169. [Google Scholar] [CrossRef]
- Tourbah, A.; Lebrun-Frenay, C.; Edan, G.; Clanet, M.; Papeix, C.; Vukusic, S.; De Sèze, J.; Debouverie, M.; Gout, O.; Clavelou, P.; et al. MD1003 (high-dose biotin) for the treatment of progressive multiple sclerosis: A randomised, double-blind, placebo-controlled study. Mult. Scler. J. 2016, 22, 1719–1731. [Google Scholar] [CrossRef]
- Birnbaum, G.; Stulc, J. High dose biotin as treatment for progressive multiple sclerosis. Mult. Scler. Relat. Disord. 2017, 18, 141–143. [Google Scholar] [CrossRef]
- Baldassari, L.E.; Feng, J.; Clayton, B.L.; Oh, S.-H.; Sakaie, K.; Tesar, P.J.; Wang, Y.; Cohen, J.A. Developing therapeutic strategies to promote myelin repair in multiple sclerosis. Expert Rev. Neurother. 2019, 1–17. [Google Scholar] [CrossRef]
- Najm, F.J.; Madhavan, M.; Zaremba, A.; Shick, E.; Karl, R.T.; Factor, D.C.; Miller, T.E.; Nevin, Z.S.; Kantor, C.; Sargent, A.; et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature 2015, 522, 216–220. [Google Scholar] [CrossRef]
- Cohen, J.A.; Baldassari, L.E.; Atkins, H.L.; Bowen, J.D.; Bredeson, C.; Carpenter, P.A.; Corboy, J.R.; Freedman, M.S.; Griffith, L.M.; Lowsky, R.; et al. Autologous Hematopoietic Cell Transplantation for Treatment-Refractory Relapsing Multiple Sclerosis: Position Statement from the American Society for Blood and Marrow Transplantation. Boil. Blood Marrow Transplant. 2019, 25, 845–854. [Google Scholar] [CrossRef]
- Marrie, R.A.; Hanwell, H. General health issues in multiple sclerosis: Comorbidities, secondary conditions, and health behaviors. Continuum 2013, 19, 1046–1057. [Google Scholar] [CrossRef]
- Ontaneda, D. Inadequate outcome measures are the biggest impediment to successful clinical trials in progressive MS-Commentary. Mult. Scler. 2017, 23, 508–509. [Google Scholar] [CrossRef]
- Giovannoni, G.; Airas, L.; Bove, R.; Boyko, A.; Cutter, G.; Hobart, J.; Kuhle, J.; Oh, J.; Tur, C.; Garas, M.; et al. Ocrelizumab Treatment Effect on Upper Limb Function in PPMS Patients with Disability: Subgroup Results of the ORATORIO Study to Inform the ORATORIO-HAND Study Design; ECTRIMS Online Library: London, UK, 2018; p. 619. [Google Scholar]
- Sumowski, J.F.; Benedict, R.; Enzinger, C.; Filippi, M.; Geurts, J.J.; Hamalainen, P.; Hulst, H.; Inglese, M.; Leavitt, V.M.; Rocca, M.A.; et al. Cognition in multiple sclerosis State of the field and priorities for the future. Neurology 2018, 90, 278–288. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Therapy | Potential Mechanism of Action | Trials | Primary Endpoint | Results |
|---|---|---|---|---|
| Remyelination strategies | ||||
| Clemastine fumarate | First-generation anti-histamine, promotes remyelination and oligodendrocyte differentiation via anti-muscarinic effect [123,124] | ReBUILD [125] | Shortening of P100 latency delay on visual-evoked potentials at 150 days | Improvement in P100 latency of 1.7 ms/eye (95% CI 0.5 to 2.9, p = 0.0048) with clemastine |
| Opicinumab | Anti-LINGO-1 antibody, promotes remyelination and oligodendrocyte differentiation via blocking of inhibitory adhesion molecule [126,127] | RENEW [128] (in acute unilateral optic neuritis) | 24-week change in optic nerve conduction latency using full-field visual evoked potential | Non-significant trend towards improvement in the intention-to-treat analysis, modest but significant benefit at week 32 in the per-protocol analysis |
| SYNERGY [129,130] (in RRMS and SPMS with active disease) | Percentage of participants with ≥3 month confirmed improvement of composite endpoint (EDSS, T25FW, 9HPT, PASAT) over 72 weeks | Benefit seen in those receiving the 30 mg/kg dose. | ||
| Neuroprotection strategies | ||||
| Ibudilast | Phosphodiesterase-inhibitor, inhibits macrophage migration inhibitory factor, and toll-like receptor 4 [131] | SPRINT-MS [132] | Progression of whole brain atrophy over 96 weeks | 48% slowing in the rate of atrophy progression with ibudilast compared to placebo |
| Simvastatin | HMG-CoA reductase inhibitor, inhibit MHCII-restricted antigen presentation, shifts cytokine production from a pro- to an anti-inflammatory response, decreases T-cell proliferation [133] | MS-STAT [134] | Progression of whole-brain atrophy, change in EDSS and total MS Impact Scale-29 at 24 months | Decrease in annualized rate of whole brain atrophy compared to placebo, benefit on EDSS and MS Impact Scale-29 as well |
| Lipoic acid | Endogenous antioxidant, various potential mechanisms, including free radical scavenging, oxidative damage repair, downregulation of inflammatory cytokines, T-cell migration in CNS inhibition [135,136] | Spain et al. [137]. | Annual percent change of brain volume | 68% reduction in the rate of brain atrophy compared to placebo over 24 months |
| Phenytoin | Selective sodium-channel inhibitor, reverses sodium influx, which drives calcium influx via reverse operation of the sodium/calcium exchanger after axonal injury [138] | Raftopoulos et al. [139]. | RNFL thickness in the affected eye | 30% reduction in the extent of RNFL loss with phenytoin compared with placebo at 6 months |
| Mesenchymal stem cells * | Pluripotent non-hematopoietic precursor cells (isolated from bone marrow or adipose tissue), release of soluble trophic factors that promote intrinsic tissue repair mechanisms [140] | Multiple small clinical trials and open label studies using variable route of administration and dosing regimens [141,142,143,144,145,146,147,148,149] | Variable endpoints depending on trial | Good safety and tolerability, efficacy not yet established [150] |
| Phase II randomized, double-blind trial, MESEMS (NCT01854957) [151] | Safety, reduction in the total number of contrast-gadolinium enhancing lesions | Ongoing | ||
| Open-label study, MSC-NTF Cells (NCT03799718) | Safety, T25FW change from baseline, changes in neurotrophic factors | Ongoing | ||
| High-dose biotin (MD 1003) * | Essential co-factor for five carboxylases involved in fatty acid synthesis and energy production, promotes remyelination, and reduces axonal hypoxia [152] | Sedel et al. (pilot study) [153] | Shortening of P100 latency on visual-evoked potentials | Improvement or normalization of P100 latency |
| Tourbah et al. (randomized, double-blind placebo-controlled trial) [154] | Proportion of patients with disability reversal on EDSS or T25FW at month 9, confirmed at month 12 | 2.6% of treated patients achieved the primary endpoint versus none of the placebo-treated patients (p = 0.005) | ||
| Birnbaum et al. (open-label study of compound medication, not MD 1003) [155] | EDSS worsening or improvement while on treatment (3 to 12 months) | No benefits observed | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macaron, G.; Ontaneda, D. Diagnosis and Management of Progressive Multiple Sclerosis. Biomedicines 2019, 7, 56. https://doi.org/10.3390/biomedicines7030056
Macaron G, Ontaneda D. Diagnosis and Management of Progressive Multiple Sclerosis. Biomedicines. 2019; 7(3):56. https://doi.org/10.3390/biomedicines7030056
Chicago/Turabian StyleMacaron, Gabrielle, and Daniel Ontaneda. 2019. "Diagnosis and Management of Progressive Multiple Sclerosis" Biomedicines 7, no. 3: 56. https://doi.org/10.3390/biomedicines7030056
APA StyleMacaron, G., & Ontaneda, D. (2019). Diagnosis and Management of Progressive Multiple Sclerosis. Biomedicines, 7(3), 56. https://doi.org/10.3390/biomedicines7030056