Stem Cell-Based Therapies for Multiple Sclerosis: Current Perspectives

Abstract
1. Introduction
2. Immunoablation Followed by Hematopoietic Stem Cell Transplantation
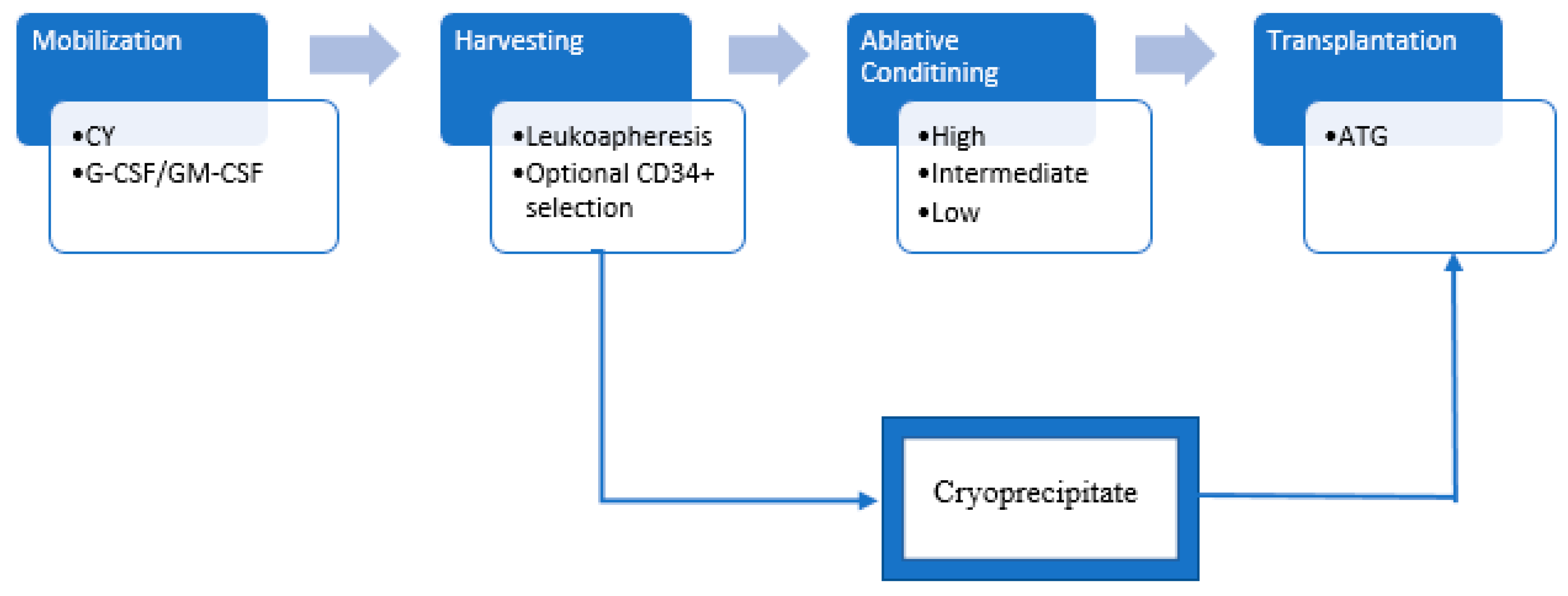
2.1. The Procedure
2.2. Immune Mechanisms
2.3. Current Clinical Knowledge
3. Mesenchymal Stem Cells
3.1. Preclinical Animal Studies with Mesenchymal Stem Cells
3.2. Clinical Trials Using Mesenchymal Stem Cells for the Treatment of Multiple Sclerosis
4. Other Stem Cells under Research
4.1. Neuronal Stem Cells (NSC)
4.2. Human Embryonic Stem Cells (hESC)
4.3. Induced Pluripotent Stem Cells (iPSC)
5. Conclusions
Funding
Conflicts of Interest
References
- Frischer, J.M.; Steiner, J.P.; Dal-Bianco, A.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Reingold, S.C.; Steiner, J.P.; Polman, C.H.; Cohen, J.A.; Freedman, M.S.; Kappos, L.; Thompson, A.J.; Wolinsky, J.S. The challenge of follow-on biologics for treatment of multiple sclerosis. Neurology 2009, 73, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Carter, J.L. Multiple sclerosis: Current and emerging disease-modifying therapies and treatment strategies. Mayo Clin. Proc. 2014, 89, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Freedman, M.S.; Selchen, D.; Arnold, D.L.; Prat, A.; Banwell, B.; Yeung, M.; Morgenthau, D.; Lapierre, Y. Treatment optimization in MS: Canadian MS Working Group updated recommendations. Can. J. Neurol. Sci. 2013, 40, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Sormani, M.P.; Li, D.K.; Bruzzi, P.; Stubinski, B.; Cornelisse, P.; Rocak, S.; De Stefano, N. Combined MRI lesions and relapses as a surrogate for disability in multiple sclerosis. Neurology 2011, 77, 1684–1690. [Google Scholar] [CrossRef] [PubMed]
- Stem Cells in MS. Available online: https://www.nationalmssociety.org/Research/Research-News-Progress/Stem-Cells-in-MS (accessed on 11 January 2019).
- Delemarre, E.M.; van den Broek, T.; Mijnheer, G.; Meerding, J.; Wehrens, E.J.; Olek, S.; Boes, M.; van Herwijnen, M.J.; Broere, F.; van Royen, A.; et al. Autologous stem cell transplantation aids autoimmune patients by functional renewal and TCR diversification of regulatory T cells. Blood 2016, 12, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Freedman, M.S.; Bar-Or, A.; Atkins, H.L.; Karussis, D.; Frassoni, F.; Lazarus, H.; Scolding, N.; Slavin, S.; Le Blanc, K.; Uccelli, A.; et al. The therapeutic potential of mesenchymal stem cell transplantation as a treatment for multiple sclerosis: Consensus report of the International MSCT Study Group. Mult. Scler. 2010, 16, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Cho, S.R. Neurorestoration induced by mesenchymal stem cells. Yonsei Med. J. 2012, 53, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Picard-Riera, N.; Decker, L.; Delarasse, C.; Goude, K.; Nait-Oumesmar, B.; Liblau, R.; Pham-Dinh, D.; Baron-Van Evercooren, A. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc. Natl. Acad. Sci. USA 2002, 99, 13211–13216. [Google Scholar] [CrossRef] [PubMed]
- Thiruvalluvan, A.; Czepiel, M.; Kap, Y.A.; Mantingh-Otter, I.; Vainchtein, I.; Kuipers, J.; Bijlard, M.; Baron, W.; Giepmans, B.; Brück, W.; et al. Survival and functionality of human induced pluripotent stem cell-derived oligodendrocytes in a nonhuman primate model for multiple sclerosis. Stem Cells Transl. Med. 2016, 5, 1550–1561. [Google Scholar] [CrossRef] [PubMed]
- Aharonowiz, M.; Einstein, O.; Fainstein, N.; Lassmann, H.; Reubinoff, B.; Ben-Hur, T. Neuroprotective effect of transplanted human embryonic stem cell-derived neural precursors in an animal model of multiple sclerosis. PLoS ONE 2008, 3, e3145. [Google Scholar] [CrossRef] [PubMed]
- Fassas, A.; Anagnostopoulos, A.; Kazis, A.; Kapinas, K.; Sakellari, I.; Kimiskidis, V.; Tsompanakou, A. Peripheral blood stem cell transplantation in the treatment of progressive multiple sclerosis: First results of a pilot study. Bone Marrow Transplant. 1997, 20, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Snowden, J.A.; Patton, W.N.; O’Donnell, J.L.; Hannah, E.E.; Hart, D.N. Prolonged remission of longstanding systemic lupus erythematous after autologous bone marrow transplant for non-Hodgkin’s lymphoma. Bone Marrow Transplant. 1997, 19, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Van Gelder, M.; van Bekkum, D.W. Treatment of relapsing experimental autoimmune encephalomyelitis in rats with allogeneic bone marrow transplantation from a resistant strain. Bone Marrow Transplant. 1995, 16, 343–351. [Google Scholar] [PubMed]
- Van Gelder, M.; Kinwel-Bohre, E.P.; van Bekkum, D.W. Treatment of experimental allergic encephalomyelitis in rats with total body irradiation and syngeneic BMT. Bone Marrow Transplant. 1993, 11, 233–241. [Google Scholar] [PubMed]
- Van Gelder, M.; van Bekkum, D.W. Effective treatment of relapsing experimental autoimmune encephalomyelitis with pseudoautologous bone marrow transplantation. Bone Marrow Transplant. 1996, 18, 1029–1034. [Google Scholar] [PubMed]
- Muraro, P.A.; Douek, D.C.; Packer, A.; Chung, K.; Guenaga, F.J.; Cassiani-Ingoni, R.; Campbell, C.; Memon, S.; Nagle, J.W.; Hakim, F.T.; et al. Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J. Exp. Med. 2005, 201, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Muraro, P.A.; Robins, H.; Malhotra, S.; Howell, M.; Phippard, D.; Desmarais, C.; de Paula Alves Sousa, A.; Griffith, L.M.; Lim, N.; Nash, R.A.; et al. T cell repertoire following autologous stem cell transplantation for multiple sclerosis. J. Clin. Investig. 2014, 124, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- Glastone, D.E.; Peyster, R.; Baron, E.; Friedman-Urevich, S.; Sibony, P.; Melville, P.; Gottesman, M. High-dose cyclophosphamide for moderate to severe refractory multiple sclerosis. Am. J. Ther. 2011, 18, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, C.; Kaplin, A.I.; Brodsky, R.A.; Drachman, D.B.; Jones, R.J.; Pham, D.L.; Richert, N.D.; Pardo, C.A.; Yousem, D.M.; Hammond, E.; et al. Reduction of disease activity and disability with high-dose cyclophosphamide in patients with aggressive multiple sclerosis. Arch. Neurol. 2008, 65, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Harrinson, D.M.; Gladstone, D.E.; Hammond, E.; Cheng, J.; Jones, R.J.; Brodsky, R.A.; Kerr, D.; McArthur, J.C.; Kaplin, A. Treatment of relapsing-remitting multiple sclerosis with high-dose cyclophosphamide infusion followed by glatimer acetate maintenance. Mult. Scler. J. 2012, 18, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Saccardi, R.; Freedman, M.S.; Sormani, M.P.; Atkins, H.; Farge, D.; Griffith, L.M.; Kraft, G.; Mancardi, G.L.; Nash, R.; Pasquini, M.; et al. A prospective, randomized, controlled trial of autologous haematopoietic stem cell transplantation for aggressive multiple sclerosis: A position paper. Mult Scler. J. 2012, 18, 825–834. [Google Scholar] [CrossRef]
- Mancardi, G.; Saccardi, R. Autologous haematopoietic stem-cell transplantation in multiple sclerosis. Lancet Neurol. 2008, 7, 626–636. [Google Scholar] [CrossRef]
- Openshaw, H.; Stuve, O.; Antel, J.P.; Nash, R.; Lund, B.T.; Weiner, L.P.; Kashyap, A.; McSweeney, P.; Forman, S. Multiple sclerosis flares associated with recombinant granulocyte colony-stimulating factor. Neurology 2000, 54, 2147–2150. [Google Scholar] [CrossRef]
- Snowden, J.A.; Saccardi, R.; Allez, M.; Ardizzone, S.; Arnold, R.; Cervera, R.; Denton, C.; Hawkey, C.; Labopin, M.; Mancardi, G.; et al. Haematopoietic SCT in severe autoimmune diseases: Updated guidelines of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant. 2012, 47, 770–790. [Google Scholar] [CrossRef]
- Saiz, A.; Blanco, Y.; Berenguer, J.; Gómez-Choco, M.; Carreras, E.; Arbizu, T.; Graus, F. Clinical outcome 6 years after autologous hematopoietic stem cell transplantation in multiple sclerosis. Neurologia 2008, 23, 405–407. [Google Scholar] [PubMed]
- Shevchenko, J.; Kuznestsov, A.N.; Ionova, T.I.; Melnichenko, V.Y.; Fedorenko, D.A.; Kurbatova, K.A.; Gorodokin, G.I.; Novik, A.A. Long-term outcomes of autologous hematopoietic stem cell transplantation with reduced-intensity conditioning in multiple sclerosis: physician’s and patient’s perspectives. Ann. Hematol. 2015, 94, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Bacigalupo, A.; Ballen, K.; Giralt, S.; Lazarus, H.; Ho, V.; Apperley, J.; Slavin, S.; Pasquini, M.; Sandmaier, B.M.; Barrett, J.; et al. Defining the intensity of conditioning regimens: Working definitions. Biol. Blood Marrow Transplant. 2009, 15, 1628–1633. [Google Scholar] [CrossRef] [PubMed]
- Gratwohl, A.; Passweg, J.; Bocelli-Tyndall, C.; Fassas, A.; van Laar, J.M.; Farge, D.; Andolina, M.; Arnold, R.; Carreras, E.; Finke, J.; et al. Autologous hematopoietic stem cell transplantation for autoimmune diseases. Bone Marrow Transplant. 2005, 35, 869–879. [Google Scholar] [CrossRef]
- Saccardi, R.; Kozak, T.; Bocelli-Tyndall, C.; Fassas, A.; Kazis, A.; Havrdova, E.; Carreras, E.; Saiz, A.; Löwenberg, B.; te Boekhorst, P.A.; et al. Autologous stem cell transplantation for progressive multiple sclerosis: Update of the European Group for Blood and Marrow Transplantation autoimmune diseases working party database. Mult. Scler. J. 2006, 12, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Samjin, J.P.; te Boekhorst, P.A.; Mondria, T.; van Doorn, P.A.; Flach, H.Z.; van der Meché, F.G.A.; Cornelissen, J.; Hop, W.C.; Löwenberg, B.; Hintzen, R.Q. Intense T cell depletion followed by autologous bone marrow transplantation for severe multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2006, 77, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Nash, R.A.; Dansey, R.; Storek, J.; Georges, G.E.; Bowen, J.D.; Holmberg, L.A.; Kraft, G.H.; Mayes, M.D.; McDonagh, K.T.; Chen, C.S.; et al. Epstein-Barr virus-associated posttransplantation lymphoproliferative disorder after high-dose immunosuppressive therapy and autologous CD34-selected hematopoietic stem cell transplantation for severe autoimmune diseases. Biol. Blood Marrow Transplant. 2003, 9, 583–591. [Google Scholar] [CrossRef]
- Mancardi, G.L.; Sormani, M.P.; Di Gioia, M.; Vuolo, L.; Gualandi, F.; Amato, M.P.; Capello, E.; Currò, D.; Uccelli, A.; Bertolotto, A.; et al. Autologous haematopoietic stem cell transplantation with an intermediate intensity conditioning regimen in multiple sclerosis: The Italian multi-centre experience. Mult. Scler. J. 2012, 18, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Curro’, D.; Vuolo, L.; Gualandi, F.; Bacigalupo, A.; Roccatagliata, L.; Capello, E.; Uccelli, A.; Saccardi, R.; Mancardi, G. Low intensity lympho-ablative regimen followed by autologous hematopoietic stem cell transplantation in severe forms of multiple sclerosis: A MRI-based clinical study. Mult. Scler. J. 2015, 11, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Burt, R.K.; Loh, Y.; Cohen, B.; Stefoski, D.; Balabanov, R.; Katsamakis, G.; Oyama, Y.; Russell, E.J.; Stern, J.; Muraro, P.; et al. Autologous non-myeloablative haemopoeitic stem cell transplantation in relapsing-remitting multiple sclerosis: A phase I/II study. Lancet Neurol. 2009, 201, 805–816. [Google Scholar] [CrossRef]
- Hammerschlak, N.; Rodrigues, M.; Moraes, D.A.; Oliveira, M.C.; Stracieri, A.B.; Pieroni, F.; Barros, G.M.; Madeira, M.I.; Simões, B.P.; Barreira, A.A.; et al. Brazilian experience with two conditioning regimens in patients with multiple sclerosis: BEAM/horse ATG and CY/rabbit ATG. Bone Marrow Transplant. 2006, 45, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Muraro, P.A.; Martin, R.; Mancardi, G.L.; Nicholas, R.; Sormani, M.P.; Saccardi, R. Autologous haematopoietic stem cell transplantation for treatment of multiple sclerosis. Nat. Rev. Neurol. 2017, 13, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Arruda, L.C.; de Azevedo, J.T.; de Oliveira, G.L.V.; Scortegagna, G.T.; Rogrigues, E.S.; Palma, P.V.B.; Brum, D.G.; Guerreiro, C.T.; Marques, V.D.; Barreira, A.A.; et al. Immunological correlates of favorable long-term clinical outcome in multiple sclerosis patients after autologous hematopoietic stem cell transplantation. Clin. Immunol. 2016, 169, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Fassas, A.; Kazis, A. High-dose immunosuppression and autologous hematopoietic stem cell rescue for severe multiple sclerosis. J. Hematother. Stem Cell Res. 2003, 12, 701–711. [Google Scholar] [CrossRef]
- Sun, W.; Uday, P.; Hutton, G.J.; Zang, Y.C.; Krance, R.; Carrum, G.; Land, G.A.; Heslop, H.; Brenner, M.; Zhang, J.Z. Characteristics of T-cell receptor repertoire and myelin-reactive T cells reconstituted from autologous haematopoietic stem-cell grafts in multiple sclerosis. Brain 2004, 127, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Darlington, P.J.; Touil, T.; Doucet, J.S.; Gaucher, D.; Zeidan, J.; Gauchat, D.; Corsini, R.; Kim, H.J.; Duddy, M.; Jalili, F.; et al. Diminished Th17 (not Th1) responses underlie multiple sclerosis disease abrogation after hematopoietic stem cell transplantation. Ann. Neurol. 2013, 73, 341–354. [Google Scholar] [CrossRef]
- Gosselin, D.; Rivest, S. Immune mechanisms underlying the beneficial effects of autologous hematopoietic stem cell transplantation in multiple sclerosis. Neurotherapeutics 2011, 8, 643–649. [Google Scholar] [CrossRef]
- Bowen, J.D.; Kraft, G.H.; Wundes, A.; Guan, Q.; Maravilla, K.R.; Gooley, T.A.; McSweeney, P.A.; Pavletic, S.Z.; Openshaw, H.; Storb, R.; et al. Autologous hematopoietic cell transplantation following high-dose immunosuppressive therapy for advanced multiple sclerosis: Long-term results. Bone Marrow Transplant. 2012, 47, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Muraro, P.A.; Pasquini, M.; Atkins, H.L.; Bowen, J.D.; Farge, D.; Fassas, D.; Freedman, M.S.; Georges, G.E.; Gualandi, F.; Hamerschlak, N.; et al. Long-term outcomes after autologous hematopoietic stem cell transplantation for multiple sclerosis. JAMA Neurol. 2017, 74, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Krasulová, E.; Trneny, M.; Kozak, T.; Vacková, B.; Pohlreich, D.; Kemlink, D.; Kobylka, P.; Kovárová, I.; Lhtáková, P.; Havrdov, E. High-dose immunoablation with autologous haematopoietic stem cell transplantation in aggressive multiple sclerosis: A single centre 10-year experience. Mult. Scler. J. 2010, 16, 685–693. [Google Scholar] [CrossRef]
- Fassas, A.; Kimiskidis, V.K.; Sakellari, I.; Kapinas, K.; Anagnostopoulos, A.; Tsimourtou, V.; Kazis, A. Long-term results of stem transplantation for MS: A single-center experience. Neurology 2011, 76, 1066–1070. [Google Scholar] [CrossRef] [PubMed]
- Cassanova, B.; Jarque, I.; Gascón, F.; Hernández-Boluda, J.C.; Pérez-Miralles, F.; de la Rubia, J.; Alcalá, C.; Sanz, J.; Mallada, J.; Cervelló, A.; et al. Autologous hematopoietic stem cell transplantation in relapsing-remitting multiple sclerosis: Comparison with secondary progressive multiple sclerosis. Neurol. Sci. 2017, 38, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Nash, R.A.; Hutton, G.J.; Racke, M.K.; Popat, U.; Devine, S.M.; Steinmiller, K.C.; Griffith, L.M.; Muraro, P.A.; Openshaw, H.; Sayre, P.H.; et al. High-dose immunosuppressive therapy and autologous HCT for relapsing-remitting MS. Neurology 2017, 88, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Nash, R.A.; Hutton, G.J.; Racke, M.K.; Popat, U.; Devine, S.M.; Griffith, L.M.; Muraro, P.A.; Openshaw, H.; Sayre, P.H.; Stüve, O.; et al. High-dose immunosuppressive therapy and autologous hematopoietic cell transplantation for relapsing-remitting multiple sclerosis (HALT-MS): A 3-year interim report. JAMA Neurol. 2015, 72, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Atkins, H.L.; Bowman, M.; Allan, D.; Anstee, G.; Arnold, D.L.; Bar-Or, A.; Bence-Bruckler, I.; Birch, P.; Bredeson, C.; Chen, J.; et al. Immunoablation and autologous haemopoietic stem-cell transplantation for aggressive multiple sclerosis: A multicentre single-group phase 2 trial. Lancet 2016, 388, 576–585. [Google Scholar] [CrossRef]
- Burt, R.K.; Balabanov, R.; Sharrack, B.; Morgan, A.; Quigley, K.; Yaung, K.; Helenowski, I.B.; Jovanovic, B.; Spahovic, D.; Arnautovic, I.; et al. Association of nonmyeloablative hematopoietic stem cell transplantation with neurological disability in patients with relapsing-remitting multiple sclerosis. JAMA 2015, 313, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Sormani, M.P.; Muraro, P.; Schiavetti, I.; Signori, A.; Laroni, A.; Saccardi, R.; Mancardi, G.L. Autologous hematopoietic stem cell transplantation in multiple sclerosis: A meta-analysis. Neurology 2017, 13, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Laroni, A.; Freedman, M.S. Mesenchymal stem cells for the treatment of multiple sclerosis and other neurological diseases. Lancet Neurol. 2011, 10, 649–656. [Google Scholar] [CrossRef]
- Rice, C.M.; Kemp, K.; Wilkins, A.; Scolding, N.J. Cell therapy for multiple sclerosis: An evolving concept with implications for other neurodegenerative diseases. Lancet 2013, 382, 1204–1213. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bowles, A.C.; Semon, J.A.; Scruggs, B.A.; Zhang, S.; Strong, A.L.; Gimble, J.M.; Bunnell, B.A. Transplantation of autologous adipose stem cells lacks therapeutic efficacy in the experimental autoimmune encephalomyelitis model. PLoS ONE 2014, 9, e85007. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chai, J.; Shen, C.; Han, Y.; Sun, T. Human umbilical cord-derived mesenchymal stem cells differentiate into epidermal-like cells using a novel co-culture technique. Cytotechnology 2014, 66, 699–708. [Google Scholar] [CrossRef]
- Bai, L.; Lennon, D.P.; Maier, K.; Caplan, A.L.; Miller, S.D.; Miller, R.H. Human bone marrow-derived mesenchymal stem cells induce Th2 polarized immune response and promote endogenous repair in animal models of multiple sclerosis. Glia 2009, 57, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- Zappia, E.; Casazza, S.; Pedemonte, E.; Benvenuto, F.; Bonanni, I.; Gerdoni, E.; Giunti, D.; Ceravolo, A.; Cazzanti, F.; Frassnoni, F.; et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood 2005, 106, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Chen, Y.; Cui, Y.; Lu, M.; Elias, S.B.; Mitchell, J.B.; Hammill, L.; Vanguri, P.; Chopp, M. Human bone marrow stromal cell treatment improves neurological functional recovery in EAE mice. Exp. Neurol. 2005, 195, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Gerdoni, E.; Gallo, B.; Casazza, S.; Musio, S.; Bonanni, I.; Pedemonte, E.; Mantegazza, R.; Frassoni, F.; Mancardi, G.; Pedotti, R.; et al. Mesenchymal stem cells effectively modulate pathogenic immune response in experimental autoimmune encephalomyelitis. Ann. Neurol. 2007, 61, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Kemp, K.; Hares, K.; Mallam, E.; Heesom, K.J.; Scolding, N.; Wilkins, A. Mesenchymal stem cell-secreted superoxide dismutase promotes cerebellar neuronal survival. J. Neurochem. 2010, 114, 1569–1580. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, A.; Kemp, K.; Ginty, M.; Hares, K.; Mallam, E.; Scolding, N. Human bone marrow derived mesenchymal stem cells secrete brain-derived neurotrophic factor which promotes neuronal survival in vitro. Stem Cell Res. 2009, 3, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, G.; Wekerle, H. EAE: An immunologist’s magic eye. Eur. Immunol. 2009, 39, 2031–2035. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Pavlovska, G.; Glover, C.P.; Uney, J.B.; Wraith, D.; Scolding, N.J. Human mesenchymal stem cells abrogate experimental allergic encephalomyelitis after intraperitoneal injection, and with sparse CNS infiltration. Neurosci. Lett. 2008, 448, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Kassis, I.; Grigoriadis, N.; Gowda-Kurkalli, B.; Mizrachi-Kol, R.; Ben-Hur, T.; Slavin, S.; Abramsky, O.; Karussis, D. Neuroprotection and immunomodulation with mesenchymal stem cells in chronic experimental autoimmune encephalitis. Arch. Neurol. 2008, 65, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Lennon, D.P.; Caplan, A.I.; DeChant, A.; Hecker, J.; Kranso, J.; Zaremba, A.; Miller, R.H. Hepatocyte growth factor mediates mesenchymal stem cell-induced recovery in multiple sclerosis models. Nat. Neurosci. 2012, 15, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Karussis, D.; Kassis, I. Use of stem cells for the treatment of multiple sclerosis. Expert Rev. Neurother. 2007, 7, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Rivera, F.J.; Couillard-Despres, D.; Pedre, X.; Ploetz, S.; Caioni, M.; Lois, C.; Bogdahn, U.; Aigner, L. Mesenchymal stem cells instruct oligodendrogenic fate decision on adult neural stem cells. Stem Cells 2006, 24, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Glenn, J.D.; Smith, M.D.; Calabresi, P.A.; Whartenby, K.A. Mesenchymal stem cells differentially modulate effector CD8+ T cell subsets and exacerbate experimental autoimmune encephalomyelitis. Stem Cells 2014, 32, 2744–2755. [Google Scholar] [CrossRef]
- Grigoriadis, N.; Lourbopoulos, A.; Lagoudaki, R.; Frischer, J.M.; Polyzoidou, E.; Touloumi, O.; Simeonidou, C.; Deretzi, G.; Kountouras, J.; Spandou, E. Variable behavior and complications of autologous bone marrow mesenchymal stem cells transplanted in experimental autoimmune encephalomyelitis. Exp. Neurol. 2011, 230, 78–89. [Google Scholar] [CrossRef] [PubMed]
- von Bahr, L.; Batsis, I.; Moll, G.; Häqq, M.; Szakos, A.; Sunberg, B.; Uzunel, M.; Ringden, O.; Le Blanc, K. Analysis of tissues following mesenchymal stromal cell therapy in humans indicate limited long-term engraftment and no ectopic tissue formation. Stem Cells 2012, 30, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Riordan, N.H.; Ichim, T.E.; Min, W.P.; Wang, H.; Solano, F.; Lara, F.; Alfaro, M.; Rodriguez, J.P.; Harman, R.J.; Patel, A.N.; et al. Non-expanded adipose stromal vascular fraction cell therapy for multiple sclerosis. J. Transl. Med. 2009, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Mohyeddin Bonab, M.; Yazdanbakhsh, S.; Loftfi, J.; Alimoghaddom, K.; Talebian, F.; Hooshmand, F.; Ghavamzadeh, A.; Nikbin, B. Does mesenchymal stem cell therapy help multiple sclerosis patients? Report of a pilot study. Iran. J. Immunol. 2007, 4, 50–57. [Google Scholar]
- Karussis, D.; Karageorgiou, C.; Vaknin-Dembinsky, A.; Gowda-Kurkalli, B.; Gomori, J.M.; Kassis, I.; Bulte, J.W.; Petrou, P.; Ben-Hur, T.; Abramsky, O.; et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch. Neurol. 2010, 67, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Yamout, B.; Hourani, R.; Salti, H.; Barada, W.; El-Hajj, T.; Al-Kutoubi, A.; Herlopian, A.; Baz, E.K.; Mahfouz, R.; Khalil-Hamdan, R.; et al. Bone marrow mesenchymal stem cell transplantation in patients with multiple sclerosis: A pilot study. J. Neuroimmunol. 2010, 227, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Bonab, M.M.; Sahraian, M.A.; Aghsaie, A.; Karvigh, S.A.; Hosseinian, S.M.; Nikbin, B.; Lotfi, J.; Khorramnia, S.; Motamed, M.R.; Togha, M.; et al. Autologous mesenchymal stem cell therapy in progressive multiple sclerosis: An open label study. Curr. Stem Cell Res. Ther. 2012, 7, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Llufriu, S.; Sepulveda, M.; Blanco, Y.; Marín, P.; Moreno, B.; Berenguer, J.; Gabilondo, I.; Martínez-Heras, E.; Sola-Valls, N.; Arnaiz, J.A.; et al. Randomized placebo-controlled phase II trial of autologous mesenchymal stem cells in multiple sclerosis. PLoS ONE 2014, 9, e113936. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.K.; Yan, Q.J.; Vyshkina, T.; Sahabi, S.; Liu, X.; Sadiq, S.A. Clinical and pathological effects of intrathecal injection of mesenchymal stem cell-derived neural progenitors in an experimental model of multiple sclerosis. J. Neurol. Sci. 2012, 313, 167–177. [Google Scholar] [CrossRef]
- Harris, V.K.; Faroqui, R.; Vyshkina, T.; Sadiq, S.A. Characterization of autologous mesenchymal stem cell-derived neural progenitors as a feasible source of stem cells for central nervous system applications in multiple sclerosis. Stem Cells Transl. Med. 2012, 1, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.K.; Stark, J.; Vyshkina, T.; Blackshear, L.; Joo, G.; Stefanova, V.; Sara, G.; Sadiq, S.A. Phase I trial of intrathecal mesenchymal stem cell-derived neural progenitors in progressive multiple sclerosis. eBioMedicine 2018, 29, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.K.; Vyshkina, T.; Sadiq, S.A. Clinical safety of intrathecal administration of mesenchymal stromal cell-derived neural progenitors in multiple sclerosis. Cytotherapy 2016, 18, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Dahbour, S.; Jamali, F.; Alhattab, D.; Al-Radaideh, A.; Ababneh, O.; Al-Ryalat, N.; Al-Bdour, M.; Hourani, B.; Msallam, M.; Rasheed, M.; et al. Mesenchymal stem cells and conditioned media in the treatment of multiple sclerosis patients: Clinical, ophthalmological and radiological assessments of safety and efficacy. CNS Neurosci. Ther. 2017, 2, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Dunne, C.; Hewson, J.; Wohl, C.; Wheatley, M.; Peterson, A.C.; Reynolds, B.A. Multipotent CNS stem cells are present in the adult mammalian spinal cord and ventricular neuroaxis. J. Neurosci. 1996, 16, 7599–7609. [Google Scholar] [CrossRef]
- Morshead, C.M.; Reynolds, B.A.; Craig, C.G.; McBurney, M.W.; Staines, W.A.; Morassutti, D.; Weiss, S.; van der Koou, D. Neural stem cells in the adult mammalian forebrain: A relatively quiescent subpopulation of subependymal cells. Neuron 1994, 13, 1071–1082. [Google Scholar] [CrossRef]
- Ming, G.L.; Song, H. Adult Neurogenesis in the Mammalian Brain: Significant Answers and Significant Questions. Neuron 2012, 70, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Bakemore, W.F.; Hans, S.K. The origin of remyelinating cells in the central nervous system. J. Neuroimm. 1999, 98, 69–76. [Google Scholar] [CrossRef]
- Armstrong, R.C.; Le, Q.T.; Flint, N.C.; Vana, A.C.; Zhou, Y.X. Endogenous cell repair of chronic demyelination. J. Neuropathol. Exp. Neurol. 2006, 65, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hur, T.; Einstein, O.; Mizrachi-Kol, R.; Ben-Menachem, O.; Reinhartz, E.; Karussis, D.; Abramsky, O. Transplanted multipotential neural precursor cells migrate into the inflamed white matter in response to experimental autoimmune encephalomyelitis. Glia 2003, 41, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Tourtellotte, W.W.; Rudick, R.A.; Trapp, B.D. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 2002, 346, 165–173. [Google Scholar] [CrossRef]
- Izrael, M.; Zhang, P.; Kaufman, R.; Shinder, V.; Ella, R.; Amit, M.; Itskovitz-Eldor, J.; Chebath, J.; Revel, M. Human oligodendrocytes derived from embryonic stem cells: Effect of noggin on phenotypic differentiation in vitro and on myelination in vivo. Mol. Cell Neurosci. 2007, 34, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Shroff, G. Human embryonic stem cells for the treatment of multiple sclerosis: A case report. Case Rep. Int. 2015, 4, 38–42. [Google Scholar] [CrossRef]
- Czepiel, M.; Balasubramaniyan, V.; Schaafsma, W.; Stancic, M.; Mikkers, H.; Hiusman, C.; Boddeke, E.; Copray, S. Differentiation of induced pluripotent stem cells into functional oligodendrocytes. Glia 2011, 59, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Laterza, C.; Merlini, A.; De Feo, D.; Ruffini, F.; Menon, R.; Onorati, M.; Fredickx, E.; Muzio, L.; Lombardo, A.; Comi, G.; et al. iPSC-derived neural precursors exert a neuroprotective role in immune-mediated demyelination via the secretion of LIF. Nat. Commun. 2013, 4, 2597. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhang, Z.N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent stem cells. Nature 2011, 474, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Okano, H.; Nakamura, M.; Yoshida, K.; Okada, Y.; Tsuji, O.; Nori, S.; Ikeda, E.; Yamanaka, S.; Miura, K. Steps toward safe cell therapy using induced pluripotent stem cells. Circ. Res. 2013, 112, 523–533. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Intensity | Regimen |
|---|---|
| High | High dose of busulfan combined with cyclophosphamide and ATG |
| Intermediate | BEAM (BCNU, etoposide, cytosine arabinoside, melphalan) +/− ATG |
| Low (nonmyeloablative) | Cyclophosphamide and ATG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuascut, F.X.; Hutton, G.J. Stem Cell-Based Therapies for Multiple Sclerosis: Current Perspectives. Biomedicines 2019, 7, 26. https://doi.org/10.3390/biomedicines7020026
Cuascut FX, Hutton GJ. Stem Cell-Based Therapies for Multiple Sclerosis: Current Perspectives. Biomedicines. 2019; 7(2):26. https://doi.org/10.3390/biomedicines7020026
Chicago/Turabian StyleCuascut, Fernando X., and George J. Hutton. 2019. "Stem Cell-Based Therapies for Multiple Sclerosis: Current Perspectives" Biomedicines 7, no. 2: 26. https://doi.org/10.3390/biomedicines7020026
APA StyleCuascut, F. X., & Hutton, G. J. (2019). Stem Cell-Based Therapies for Multiple Sclerosis: Current Perspectives. Biomedicines, 7(2), 26. https://doi.org/10.3390/biomedicines7020026