
Aptamer Technology: Adjunct Therapy for Malaria


Abstract
:1. Introduction
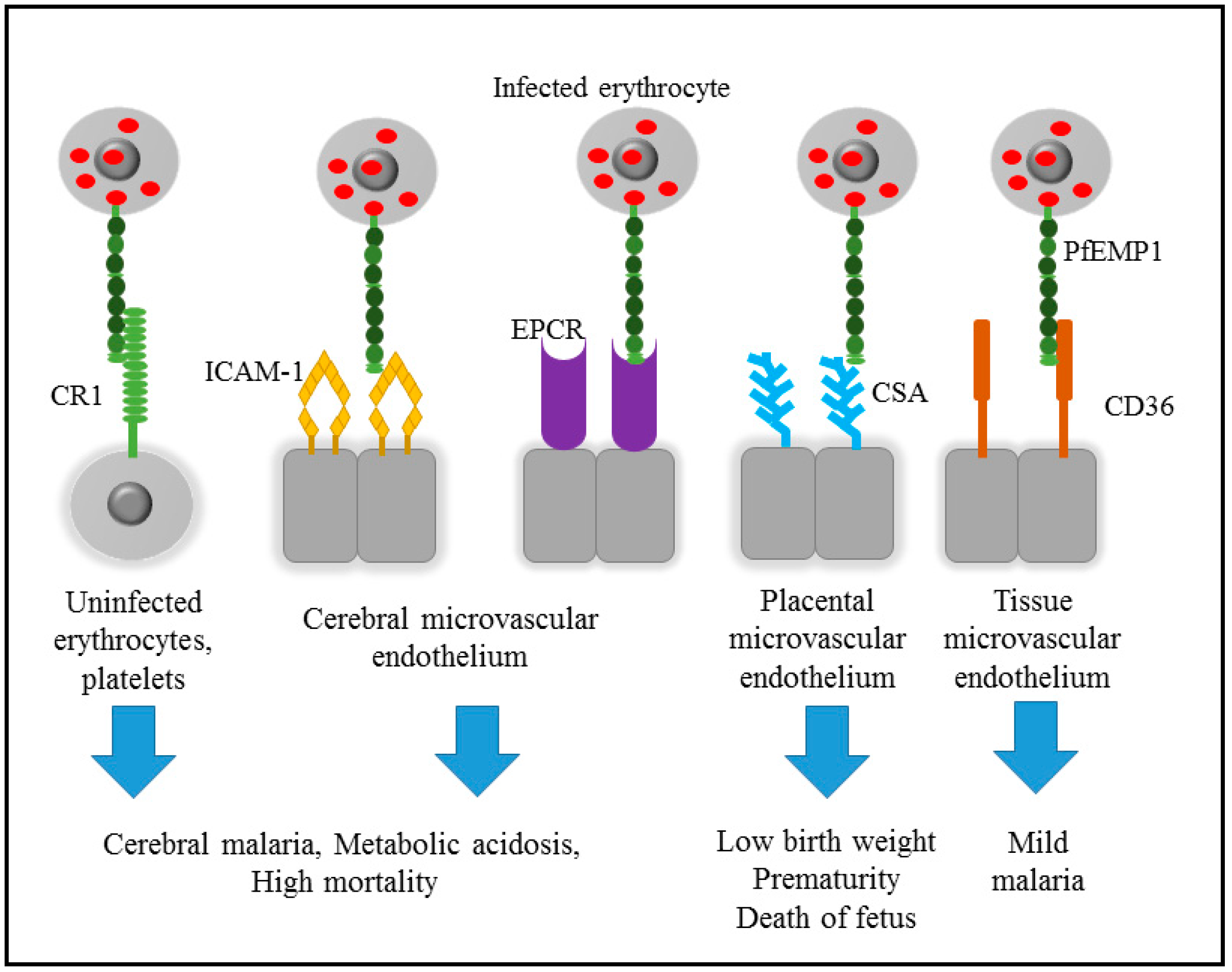
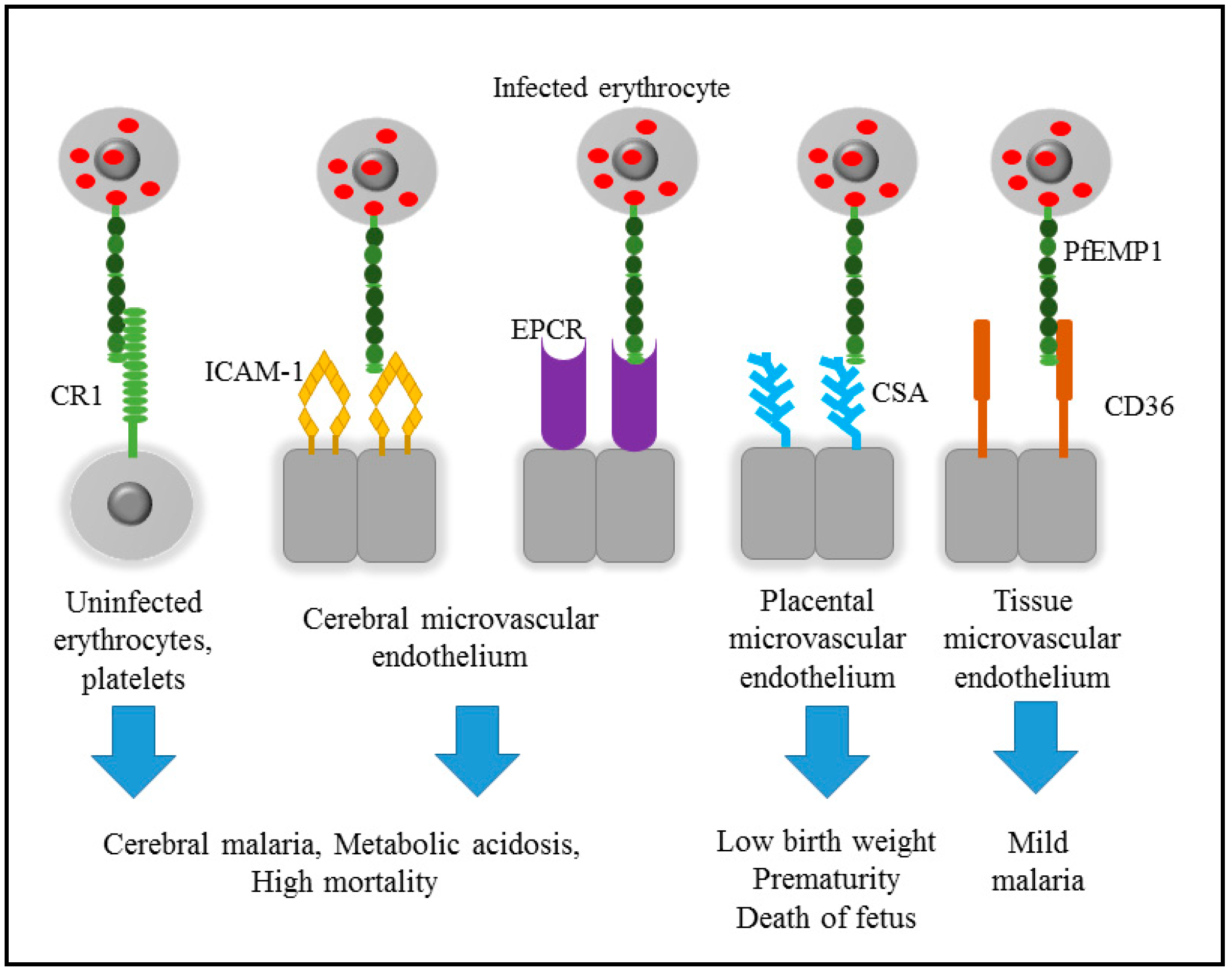
2. Pathogenicity of Malaria
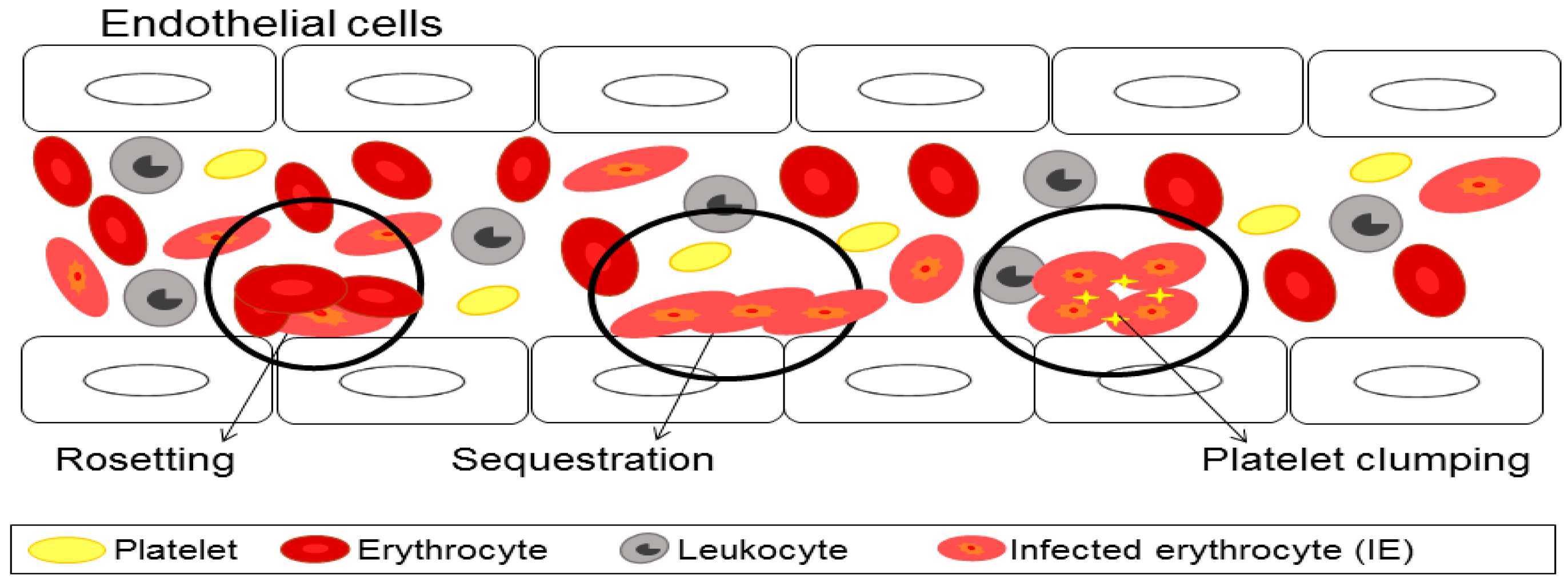
3. Cytoadherence of Infected Erythrocyte
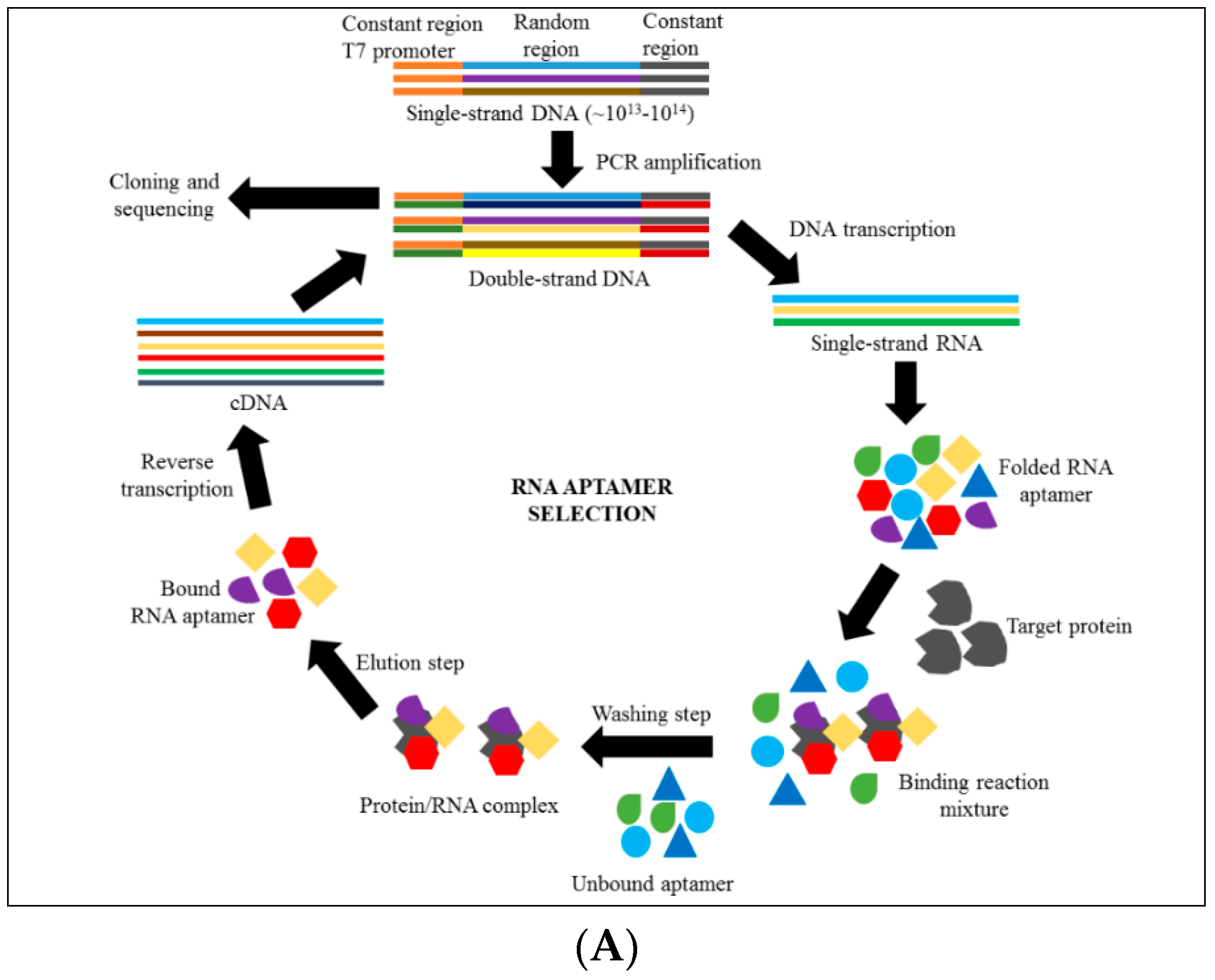
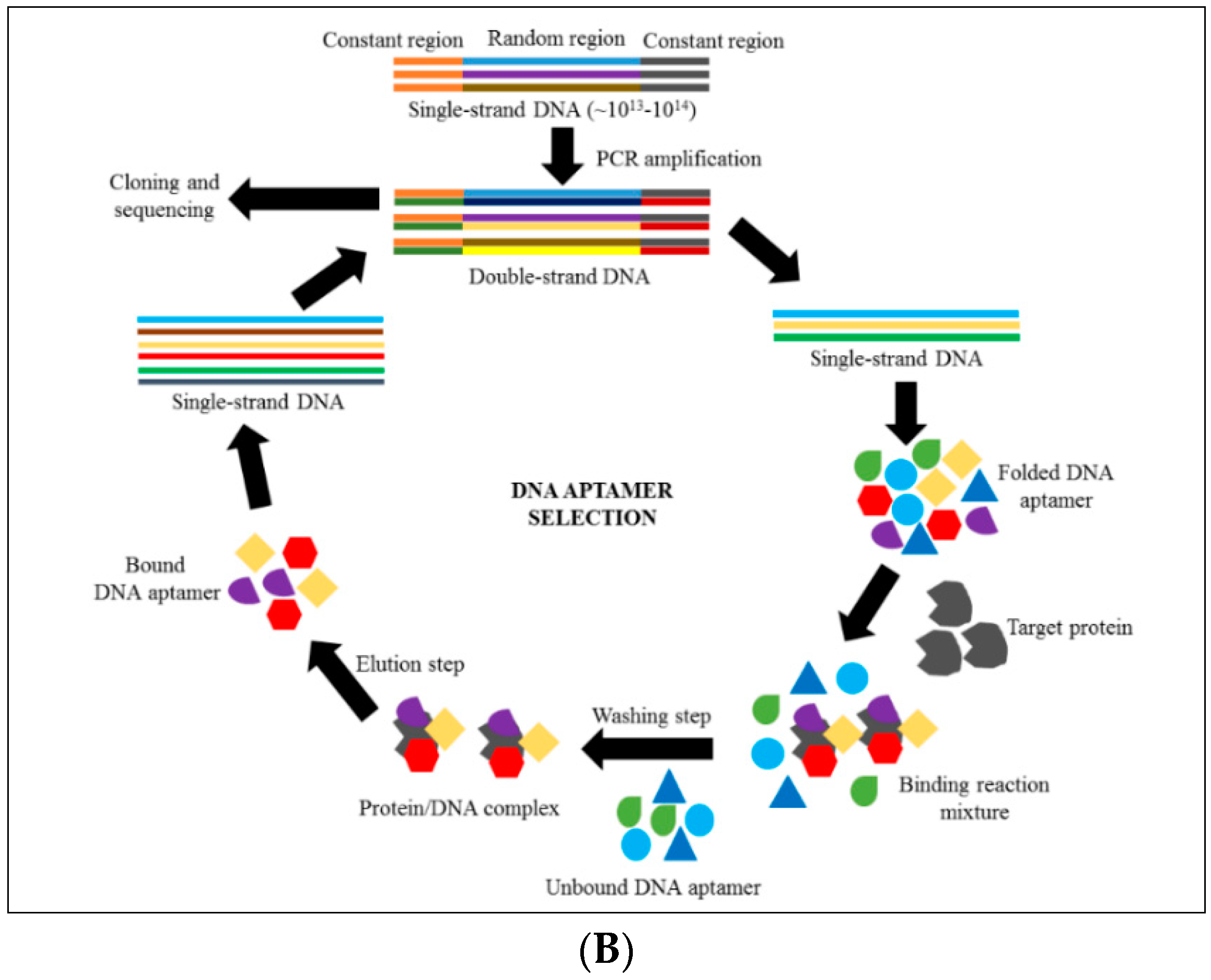
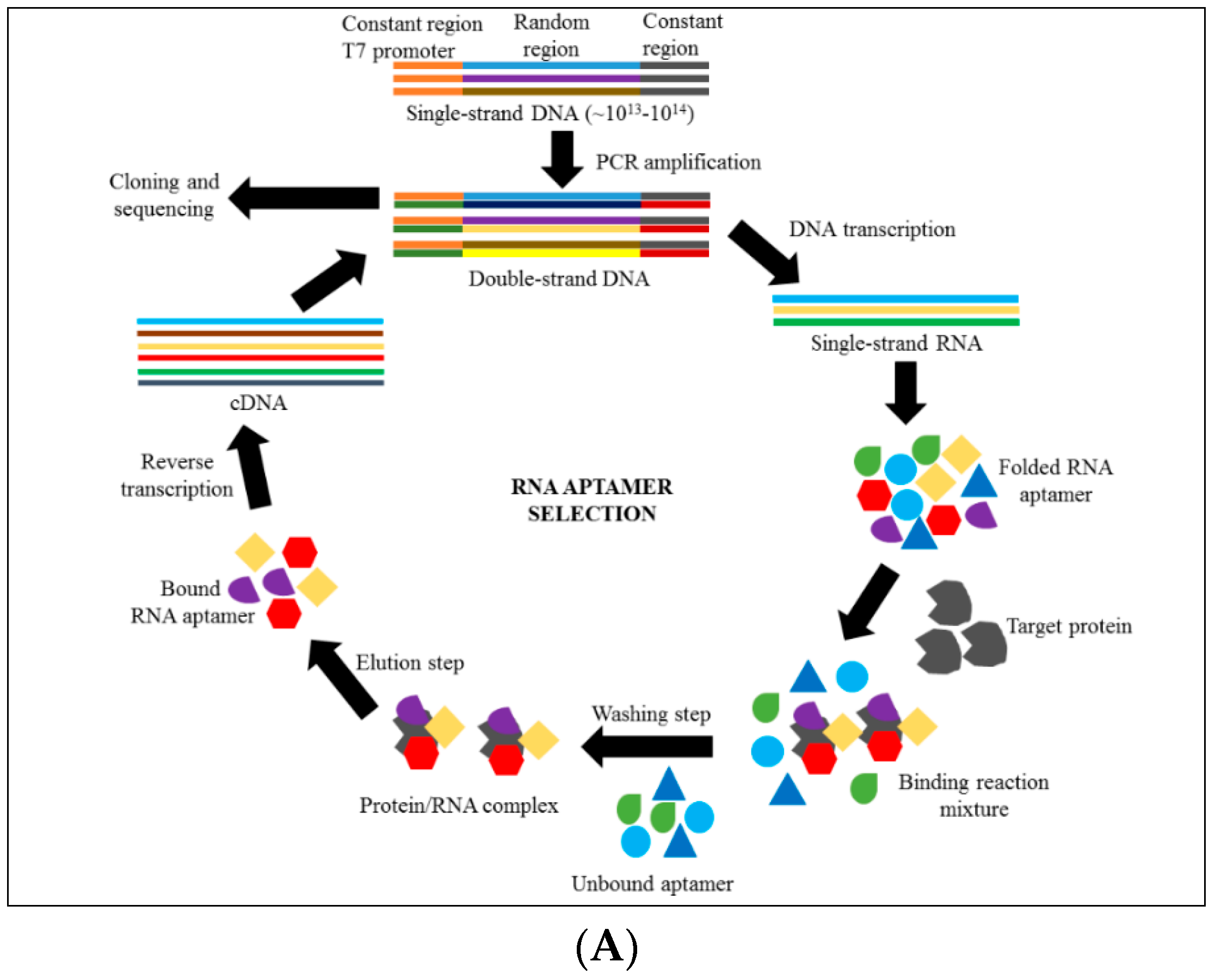
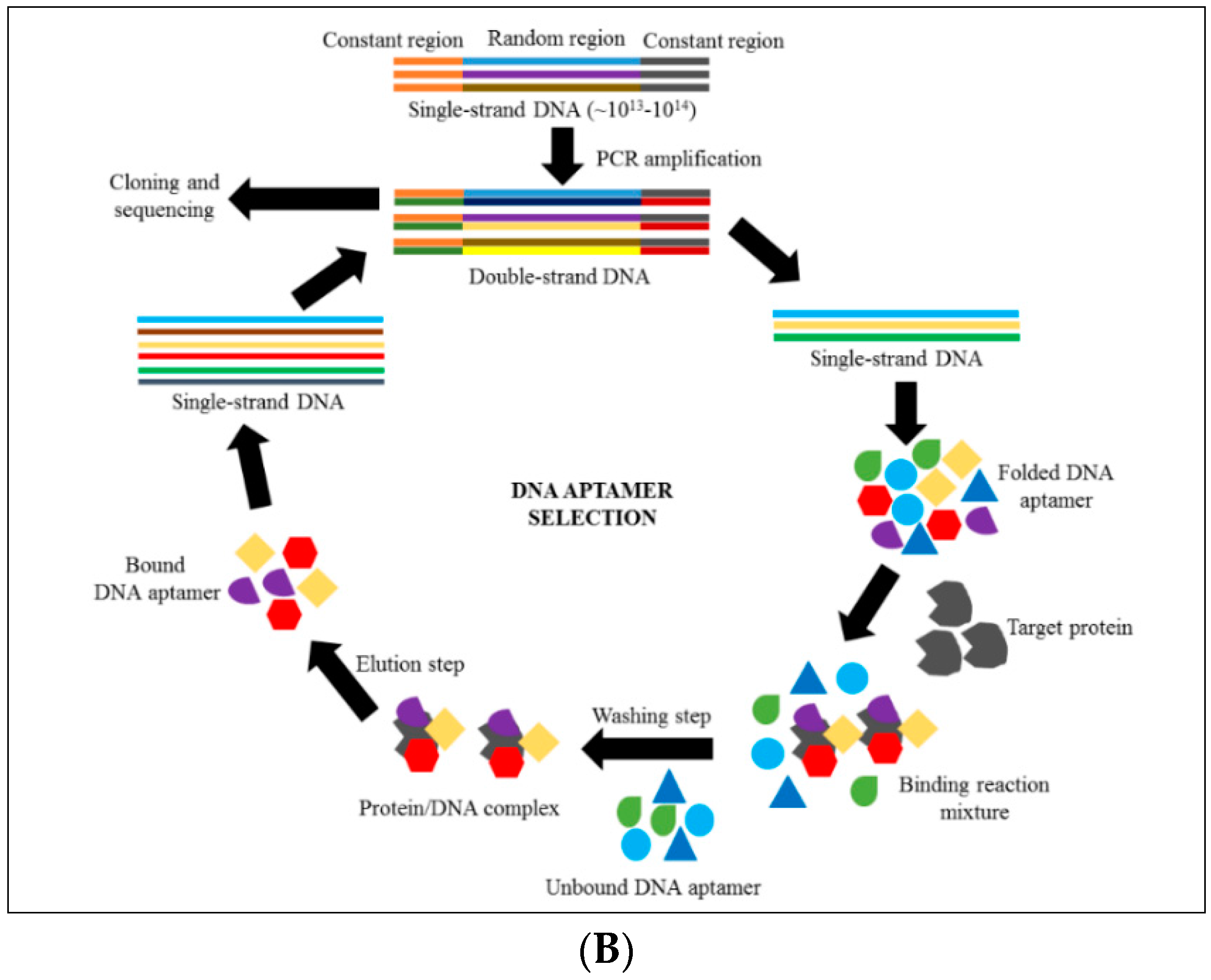
4. Introduction to Aptamer Technology
5. Aptamers as Therapeutic Agents Compared with Antibodies
6. Discovering Anti-Cytoadhrence through Aptamer Technology
7. Current Aptamer Development for Malaria Therapy
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Alemu, A.; Shiferaw, Y.; Addis, Z.; Mathewos, B.; Birhan, W. Effect of malaria on HIV/AIDS transmission and progression. Parasites Vectors 2013, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Laishram, D.D.; Sutton, P.L.; Nanda, N.; Sharma, V.L.; Sobti, R.C.; Carlton, J.M.; Joshi, H. The complexities of malaria disease manifestations with a focus on asymptomatic malaria. Malar. J. 2012, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). World Malaria Report; WHO: Geneva, Switzerland, 2015; p. 280. [Google Scholar]
- Winzeler, E.A. Malaria research in the post-genomic era. Nature 2008, 455, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Fatih, F.A.; Staines, H.M.; Siner, A.; Ahmed, M.A.; Woon, L.C.; Pasini, E.M.; Kocken, C.H.; Singh, B.; Cox-Singh, J.; Krishna, S. Susceptibility of human plasmodium knowlesi infections to anti-malarials. Malar. J. 2013, 12, 425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatih, F.A.; Siner, A.; Ahmed, A.; Woon, L.C.; Craig, A.G.; Singh, B.; Krishna, S.; Cox-Singh, J. Cytoadherence and virulence—The case of Plasmodium knowlesi malaria. Malar. J. 2012, 11, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snow, R.W.; Guerra, C.A.; Noor, A.M.; Myint, H.Y.; Hay, S.I. The global distribution of clinical episodes of plasmodium falciparum malaria. Nature 2005, 434, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Storm, J.; Craig, A.G. Pathogenesis of cerebral malaria—Inflammation and cytoadherence. Front. Cell. Infect. Microbiol. 2014, 4, 100. [Google Scholar] [CrossRef] [PubMed]
- Doolan, D.L.; Dobano, C.; Baird, J.K. Acquired immunity to malaria. Clin. Microbiol. Rev. 2009, 22, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Tangpukdee, N.; Duangdee, C.; Wilairatana, P.; Krudsood, S. Malaria diagnosis: A brief review. Korean J. Parasitol. 2009, 47, 93–102. [Google Scholar] [CrossRef] [PubMed]
- White, N.J. Drug resistance in malaria. Br. Med. Bull. 1998, 54, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.H.; Ackerman, H.C.; Su, X.Z.; Wellems, T.E. Malaria biology and disease pathogenesis: Insight for new treatments. Nat. Med. 2013, 19, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Antony, H.A.; Parija, S.C. Antimalarial drug resistance: An overview. Trop. Parasitol. 2016, 6, 30–41. [Google Scholar] [PubMed]
- Raza, A.; Ghanchi, N.; Khan, M.; Beg, M. Prevalence of drug resistance associated mutations in plasmodium vivax against sulphadoxine-pyrimethamine in Southern Pakistan. Malar. J. 2013, 12, 261. [Google Scholar] [CrossRef] [PubMed]
- Farooq, U.; Mahajan, R.C. Drug resistance in malaria. J. Vector Borne Dis. 2004, 41, 45–53. [Google Scholar] [PubMed]
- Tun, K.M.; Imwong, M.; Lwin, K.M.; Win, A.A.; Hlaing, T.M.; Hlaing, T.; Lin, K.; Kyaw, M.P.; Plewes, K.; Faiz, M.A.; et al. Spread of artemisinin-resistant plasmodium falciparum in myanmar: A cross-sectional survey of the k13 molecular marker. Lancet Infect. Dis. 2015, 15, 415–421. [Google Scholar] [CrossRef]
- Dondorp, A.M.; Yeung, S.; White, L.; Nguon, C.; Day, N.P.J.; Socheat, D.; von Seidlein, L. Artemisinin resistance: Current status and scenarios for containment. Nat. Rev. Microbiol. 2010, 8, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Tun, K.M.; Jeeyapant, A.; Imwong, M.; Thein, M.; Aung, S.S.M.; Hlaing, T.M.; Yuentrakul, P.; Promnarate, C.; Dhorda, M.; Woodrow, C.J.; et al. Parasite clearance rates in upper myanmar indicate a distinctive artemisinin resistance phenotype: A therapeutic efficacy study. Malar. J. 2016, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Carrara, V.I.; Lwin, K.M.; Phyo, A.P.; Ashley, E.; Wiladphaingern, J.; Sriprawat, K.; Rijken, M.; Boel, M.; McGready, R.; Proux, S.; et al. Malaria burden and artemisinin resistance in the mobile and migrant population on the thai–myanmar border, 1999–2011: An observational study. PLoS Med. 2013, 10, e1001398. [Google Scholar] [CrossRef] [PubMed]
- Starzengruber, P.; Swoboda, P.; Fuehrer, H.-P.; Khan, W.A.; Hofecker, V.; Siedl, A.; Fally, M.; Graf, O.; Teja-Isavadharm, P.; Haque, R.; et al. Current status of artemisinin-resistant falciparum malaria in south asia: A randomized controlled artesunate monotherapy trial in Bangladesh. PLoS ONE 2012, 7, e52236. [Google Scholar] [CrossRef] [PubMed]
- Rowe, J.A.; Claessens, A.; Corrigan, R.A.; Arman, M. Adhesion of plasmodium falciparum-infected erythrocytes to human cells: Molecular mechanisms and therapeutic implications. Expert Rev. Mol. Med. 2009, 11, e16. [Google Scholar] [CrossRef] [PubMed]
- Storm, J; Craig, A.G. Pathogenesis of cerebral malaria-inflammation and cytoadherence. Front. Cell. Infec. Microbil. 2014, 4, 100. [Google Scholar]
- Kraemer, S.M.; Smith, J.D. A family affair: Var genes, pfEMP1 binding, and malaria disease. Curr. Opin. Microbiol. 2006, 9, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.G.; Khairul, M.F.; Patil, P.R. Cytoadherence and severe malaria. Malays. J. Med. Sci. 2012, 19, 5–18. [Google Scholar] [PubMed]
- Chan, J.A.; Howell, K.B.; Reiling, L.; Ataide, R.; Mackintosh, C.L.; Fowkes, F.J.; Petter, M.; Chesson, J.M.; Langer, C.; Warimwe, G.M.; et al. Targets of antibodies against plasmodium falciparum-infected erythrocytes in malaria immunity. J. Clin. Investig. 2012, 122, 3227–3238. [Google Scholar] [CrossRef] [PubMed]
- Penman, B.; Gupta, S. Evolution of virulence in malaria. J. Biol. 2008, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, A.; Neculai, D.; Kain, K.C. CD36 and malaria: Friends or foes? A decade of data provides some answers. Trends Parasitol. 2014, 30, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Grau, G.E.; Craig, A.G. Cerebral malaria pathogenesis: Revisiting parasite and host contributions. Future Microbiol. 2012, 7, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Baruch, D.I.; Brady, C.; Murray, A.G.; Looareesuwan, S.; Kubes, P.; Ho, M. Recombinant pfemp1 peptide inhibits and reverses cytoadherence of clinicalplasmodium falciparum isolates in vivo. Blood Cancer J. 2003, 101, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q. The naturally acquired immunity in severe malaria and its implication for a PfEMP-1 based vaccine. Microbes Infect. 2007, 9, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Dhangadamajhi, G.; Kar, S.K.; Ranjit, M. The survival strategies of malaria parasite in the red blood cell and host cell polymorphisms. Malaria Res. Treat. 2010, 2010, 973094. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 mediates the innate host response to β-amyloid. J. Exp. Med. 2003, 197, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.P.; Lee, K.; Gillrie, M.R.; Roa, L.; Amrein, M.; Ho, M. CD36 reccruits α5β1 integrin to promote cytoadherence of P. Falciparum-infected erythrocytes. PLoS Pathog. 2013, 9, e1003590. [Google Scholar] [CrossRef] [PubMed]
- Turner, G.D.; Morrison, H.; Jones, M.; Davis, T.M.; Looareesuwan, S.; Buley, I.D.; Gatter, K.C.; Newbold, C.I.; Pukritayakamee, S.; Nagachinta, B.; et al. An immunohistochemical study of the pathology of fatal malaria. Evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration. Am. J. Pathol. 1994, 145, 1057–1069. [Google Scholar] [PubMed]
- Knowles, D.I.; Tolidjian, B.; Marboe, C.; Dagati, V.; Grimes, M.; Chess, L. Monoclonal anti-human monocyte antibodies okmi and OKM5 possess distinctive tissue distribution including differential reactivity with vascular endothelium. J. Immunol. 1984, 132, 2170–2173. [Google Scholar] [PubMed]
- Ho, M.; Davis, T.M.; Silamut, K.; Bunnag, D.; White, N.J. Rosette formation of plasmodium falciparum-infected erythrocytes from patients with acute malaria. Infect. Immun. 1991, 59, 2135–2139. [Google Scholar] [PubMed]
- Ochola, L.B.; Siddondo, B.R.; Ocholla, H.; Nkya, S.; Kimani, E.N.; Williams, T.N.; Makale, J.O.; Liljander, A.; Urban, B.C.; Bull, P.C.; et al. Specific receptor usage in plasmodium falciparum cytoadherence is associated with disease outcome. PLoS ONE 2011, 6, e14741. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, A.; Joergensen, L.; Barbati, Z.R.; Craig, A.; Hviid, L.; Jensen, A.T.R. Transfected HEK293 cells expressing functional recombinant intercellular adhesion molecule 1 (ICAM-1)—A receptor associated with severe plasmodium falciparum malaria. PLoS ONE 2013, 8, e69999. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.T.; Magistrado, P.; Sharp, S.; Joergensen, L.; Lavstsen, T.; Chiucchiuini, A.; Salanti, A.; Vestergaard, L.S.; Lusingu, J.P.; Hermsen, R.; et al. Plasmodium falciparum associated with severe childhood malaria preferentially expresses PfEMP1 encoded by group a var genes. J. Exp. Med. 2004, 199, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.S.; Turner, L.; Wang, C.W.; Petersen, J.E.; Kraft, M.; Lusingu, J.P.A.; Mmbando, B.; Marquard, A.M.; Bengtsson, D.B.; Hviid, L.; et al. Plasmodium falciparum expressing domain cassette 5 type PfEMP1 (DC5-PfEMP1) bind PECAM1. PLoS ONE 2013, 8, e69117. [Google Scholar] [CrossRef] [PubMed]
- Faille, D.; Combes, V.; Mitchell, A.J.; Fontaine, A.; Juhan-Vague, I.; Alessi, M.-C.; Chimini, G.; Fusaï, T.; Grau, G.E. Platelet microparticles: A new player in malaria parasite cytoadherence to human brain endothelium. FASEB J. 2009, 23, 3449–3458. [Google Scholar] [CrossRef] [PubMed]
- Avril, M.; Tripathi, A.K.; Brazier, A.J.; Andisi, C.; Janes, J.H.; Soma, V.L.; Sullivan, D.J., Jr.; Bull, P.C.; Stins, M.F.; Smith, J.D. A restricted subset of var genes mediates adherence of plasmodium falciparum-infected erythrocytes to brain endothelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, E1782–E1790. [Google Scholar] [CrossRef] [PubMed]
- Heddini, A.; Chen, Q.; Obiero, J.; Kai, O.; Fernandez, V.; Marsh, K.; Muller, W.A.; Wahlgren, M. Binding of plasmodium falciparum-infected erythrocytes to soluble platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31): Frequent recognition by clinical isolates. Am. J. Trop. Med. Hyg. 2001, 65, 47–51. [Google Scholar] [PubMed]
- Claessens, A.; Adams, Y.; Ghumra, A.; Lindergard, G.; Buchan, C.C.; Andisi, C.; Bull, P.C.; Mok, S.; Gupta, A.P.; Wang, C.W.; et al. A subset of group A-like var genes encodes the malaria parasite ligands for binding to human brain endothelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, E1772–E1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, T.E.; Fu, W.J.; Carr, R.A.; Whitten, R.O.; Mueller, J.S.; Fosiko, N.G.; Lewallen, S.; Liomba, N.G.; Molyneux, M.E. Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nat. Med. 2004, 10, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Dorovini-Zis, K.; Schmidt, K.; Huynh, H.; Fu, W.; Whitten, R.O.; Milner, D.; Kamiza, S.; Molyneux, M.; Taylor, T.E. The neuropathology of fatal cerebral malaria in Malawian children. Am. J. Pathol. 2011, 178, 2146–2158. [Google Scholar] [CrossRef] [PubMed]
- Moxon, C.A.; Wassmer, S.C.; Milner, D.A., Jr.; Chisala, N.V.; Taylor, T.E.; Seydel, K.B.; Molyneux, M.E.; Faragher, B.; Esmon, C.T.; Downey, C.; et al. Loss of endothelial protein C receptors links coagulation and inflammation to parasite sequestration in cerebral malaria in African children. Blood 2013, 122, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V.; Griffin, J.H. Cytoprotective protein C pathways and implications for stroke and neurological disorders. Trends Neurosci. 2011, 34, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. The protein c pathway. Chest 2003, 124, 26S–32S. [Google Scholar] [CrossRef] [PubMed]
- Mosnier, L.O.; Zlokovic, B.V.; Griffin, J.H. The cytoprotective protein C pathway. Blood 2007, 109, 3161–3172. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.L.; Hofman, F.M.; Ishii, H.; Fisher, M. Regional distribution of thrombomodulin in human brain. Brain Res. 1991, 556, 1–5. [Google Scholar] [CrossRef]
- Laszik, Z.; Mitro, A.; Taylor, F.B., Jr.; Ferrell, G.; Esmon, C.T. Human protein c receptor is present primarily on endothelium of large blood vessels: Implications for the control of the protein C pathway. Circulation 1997, 96, 3633–3640. [Google Scholar] [CrossRef] [PubMed]
- Van der Poll, T. The endothelial protein c receptor and malaria. Blood 2013, 122, 624–625. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. Protein c anticoagulant system—Anti-inflammatory effects. Semin. Immunopathol. 2012, 34, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Lavstsen, T.; Turner, L.; Saguti, F.; Magistrado, P.; Rask, T.S.; Jespersen, J.S.; Wang, C.W.; Berger, S.S.; Baraka, V.; Marquard, A.M.; et al. Plasmodium falciparum erythrocyte membrane protein 1 domain cassettes 8 and 13 are associated with severe malaria in children. Proc. Natl. Acad. Sci. USA 2012, 109, E1791–E1800. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, P.; Kurniawan, H.; Byrne, M.E.; Wower, J. Therapeutic RNA aptamers in clinical trials. Eur. J. Pharm. Sci. 2013, 48, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, J.; Nilsen-Hamilton, M. Aptamers: Multifunctional molecules for biomedical research. J. Mol. Med. 2013, 91, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Wilson, G.; Hebbard, L.; Duan, W.; Liddle, C.; George, J.; Qiao, L. Aptamers: A promising chemical antibody for cancer therapy. Oncotarget 2016, 7, 13446–13463. [Google Scholar] [PubMed]
- Sun, H.; Zhu, X.; Lu, P.Y.; Rosato, R.R.; Tan, W.; Zu, Y. Oligonucleotide aptamers: New tools for targeted cancer therapy. Mol. Ther. Nucleic Acids 2014, 3, e182. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.Y.; Byun, J. Nucleic acid aptamers: New methods for selection, stabilization, and application in biomedical science. Biomol. Ther. 2013, 21, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Nomura, Y.; Sugiyama, S.; Sakamoto, T.; Miyakawa, S.; Adachi, H.; Takano, K.; Murakami, S.; Inoue, T.; Mori, Y.; Nakamura, Y.; et al. Conformational plasticity of RNA for target recognition as revealed by the 2.15 a crystal structure of a human IgG-aptamer complex. Nucleic Acids Res. 2010, 38, 7822–7829. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, D.K.; Brenner, S.E.; Holbrook, S.R. RNA structural motifs: Building blocks of a modular biomolecule. Q. Rev. Biophys. 2005, 38, 221–243. [Google Scholar] [CrossRef] [PubMed]
- Butcher, S.E.; Pyle, A.M. The molecular interactions that stabilize RNA tertiary structure: RNA motifs, patterns, and networks. Acc. Chem. Res. 2011, 44, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.; Magdesian, M.H.; Alves, M.J.; Colli, W. In vitro selection of RNA aptamers that bind to cell adhesion receptors of trypanosoma cruzi and inhibit cell invasion. J. Biol. Chem. 2002, 277, 20756–20762. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Dis. 2010, 9, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Germer, K.; Leonard, M.; Zhang, X. RNA aptamers and their therapeutic and diagnostic applications. Int. J. Biochem. Mol. Biol. 2013, 4, 27–40. [Google Scholar] [PubMed]
- Citartan, M.; Thean-Hock, T.; Soo-Choon, T.; Chee-Hock, H.; Saini, R.; Tominaga, J.; Gopinath, S.C.B. Asymmetric PCR for good quality ssDNA generation towards DNA aptamer production. Songklanakarin J. Sci. Technol. 2012, 34, 125–131. [Google Scholar]
- Marimuthu, C.; Tang, T.-H.; Tominaga, J.; Tan, S.-C.; Gopinath, S.C.B. Single-stranded DNA (ssDNA) production in DNA aptamer generation. Analyst 2012, 137, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-W.; Lee, S.J.; Ren, S.; Lee, S.; Kim, S.; Laurell, T. Acousto-microfluidics for screening of ssDNA aptamer. Sci. Rep. 2016, 6, 27121. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Scheper, T.; Walter, J.-G. Aptamers: Versatile probes for flow cytometry. Appl. Microbiol. Biotechnol. 2013, 97, 7097–7109. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-A.; Ahn, J.-Y.; Lee, S.-H.; Singh Sekhon, S.; Kim, D.-G.; Min, J.; Kim, Y.-H. Aptamer-based sandwich assay and its clinical outlooks for detecting lipocalin-2 in hepatocellular carcinoma (HCC). Sci. Rep. 2015, 5, 10897. [Google Scholar] [CrossRef] [PubMed]
- Mosing, R.K.; Bowser, M.T. Isolating aptamers using capillary electrophoresis–SELEX (CE–SELEX). In Nucleic Acid and Peptide Aptamers: Methods and Protocols; Mayer, G., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 33–43. [Google Scholar]
- Reinholt, S.J.; Ozer, A.; Lis, J.T.; Craighead, H.G. Highly multiplexed RNA aptamer selection using a microplate-based microcolumn device. Sci. Rep. 2016, 6, 29771. [Google Scholar] [CrossRef] [PubMed]
- Blind, M.; Blank, M. Aptamer selection technology and recent advances. Mol. Ther. Nucleic Acids 2015, 4, e223. [Google Scholar] [CrossRef]
- Long, Y.; Qin, Z.; Duan, M.; Li, S.; Wu, X.; Lin, W.; Li, J.; Zhao, Z.; Liu, J.; Xiong, D.; et al. Screening and identification of DNA aptamers toward Schistosoma japonicum eggs via SELEX. Sci. Rep. 2016, 6, 24986. [Google Scholar] [CrossRef] [PubMed]
- Szeto, K.; Latulippe, D.R.; Ozer, A.; Pagano, J.M.; White, B.S.; Shalloway, D.; Lis, J.T.; Craighead, H.G. Rapid-SELEX for RNA aptamers. PLoS ONE 2013, 8, e82667. [Google Scholar] [CrossRef] [PubMed]
- Song, K.M.; Lee, S.; Ban, C. Aptamers and their biological applications. Sensors 2012, 12, 612–631. [Google Scholar] [CrossRef] [PubMed]
- Mascini, M. Aptamers and their applications. Anal. Bioanal. Chem. 2008, 390, 987–988. [Google Scholar] [CrossRef] [PubMed]
- Ruscito, A.; DeRosa, M.C. Small-molecule binding aptamers: Selection strategies, characterization, and applications. Front. Chem. 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.E.; Medley, C.D.; Tang, Z.; Shangguan, D.; Lofton, C.; Tan, W. Aptamer-conjugated nanoparticles for the collection and detection of multiple cancer cells. Anal. Chem. 2007, 79, 3075–3082. [Google Scholar] [CrossRef] [PubMed]
- Toh, S.Y.; Citartan, M.; Gopinath, S.C.B.; Tang, T.-H. Aptamers as a replacement for antibodies in enzyme-linked immunosorbent assay. Biosens. Bioelectron. 2015, 64, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Thiel, K. Oligo oligarchy[mdash]the surprisingly small world of aptamers. Nat. Biotechnol. 2004, 22, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Kedzierski, S.U.; Khoshnejad, M.A.; Caltagirone, G.T. Synthetic antibodies: The emerging field of aptamers. Bioprocess. J. 2012, 11, 46–49. [Google Scholar] [CrossRef]
- Eyetech Study Group. Preclinical and phase 1A clinical evaluation of an anti-VEGF pegylated aptamer (eye001) for the treatment of exudative age-related macular degeneration. Retina 2002, 22, 143–152. [Google Scholar]
- Ireson, C.R.; Kelland, L.R. Discovery and development of anticancer aptamers. Mol. Cancer Ther. 2006, 5, 2957–2962. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Gu, M.B. Advances in aptamer screening and small molecule aptasensors. Adv. Biochem. Eng. Biotechnol. 2014, 140, 29–67. [Google Scholar] [PubMed]
- Wilson, C.; Keefe, A.D. Building oligonucleotide therapeutics using non-natural chemistries. Curr. Opin. Chem. Biol. 2006, 10, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, S.C. Methods developed for selex. Anal. Bioanal. Chem. 2007, 387, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, A.; Deakin, A.; Spaull, J.; Coomber, B.; Aitken, A.; Life, P.; Rees, S. The generation and characterization of antagonist RNA aptamers to human oncostatin m. J. Biol. Chem. 2000, 275, 28555–28561. [Google Scholar] [CrossRef] [PubMed]
- Tokmakov, A.A.; Kurotani, A.; Takagi, T.; Toyama, M.; Shirouzu, M.; Fukami, Y.; Yokoyama, S. Multiple post-translational modifications affect heterologous protein synthesis. J. Biol. Chem. 2012, 287, 27106–27116. [Google Scholar] [CrossRef] [PubMed]
- Sefah, K.; Shangguan, D.; Xiong, X.; O’Donoghue, M.B.; Tan, W. Development of DNA aptamers using cell-selex. Nat. Protoc. 2010, 5, 1169–1185. [Google Scholar] [CrossRef] [PubMed]
- Parekh, P.; Kamble, S.; Zhao, N.; Zeng, Z.; Portier, B.P.; Zu, Y. Immunotherapy of CD30-expressing lymphoma using a highly stable ssDNA aptamer. Biomaterials 2013, 34, 8909–8917. [Google Scholar] [CrossRef] [PubMed]
- Hicke, B.J.; Marion, C.; Chang, Y.-F.; Gould, T.; Lynott, C.K.; Parma, D.; Schmidt, P.G.; Warren, S. Tenascin-c aptamers are generated using tumor cells and purified protein. J. Biol. Chem. 2001, 276, 48644–48654. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Anand, S.; Schollaardt, T.; Patel, K.D.; Looareesuwan, S.; Ho, M. Synergism of multiple adhesion molecules in mediating cytoadherence of plasmodium falciparum-infected erythrocytes to microvascular endothelial cells under flow. Blood 2000, 96, 2292–2298. [Google Scholar] [PubMed]
- Gray, C.; McCormick, C.; Turner, G.; Craig, A. ICAM-1 can play a major role in mediating P. Falciparum adhesion to endothelium under flow. Mol. Biochem. Parasitol. 2003, 128, 187–193. [Google Scholar] [CrossRef]
- John, C.C.; Kutamba, E.; Mugarura, K.; Opoka, R.O. Adjunctive therapy for cerebral malaria and other severe forms of plasmodium falciparum malaria. Expert Rev. Antiinfect. Ther. 2010, 8, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Barfod, A.; Persson, T.; Lindh, J. In vitro selection of RNA aptamers against a conserved region of the plasmodium falciparum erythrocyte membrane protein 1. Parasitol. Res. 2009, 105, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.W.; Kwok, J.; Law, A.W.; Watt, R.M.; Kotaka, M.; Tanner, J.A. Structural basis for discriminatory recognition of plasmodium lactate dehydrogenase by a DNA aptamer. Proc. Natl. Acad. Sci. USA 2013, 110, 15967–15972. [Google Scholar] [CrossRef] [PubMed]
- Weissbuch, I.; Leiserowitz, L. Interplay between malaria, crystalline hemozoin formation, and antimalarial drug action and design. Chem. Rev. 2008, 108, 4899–4914. [Google Scholar] [CrossRef] [PubMed]
- Okazawa, A.; Maeda, H.; Fukusaki, E.; Katakura, Y.; Kobayashi, A. In vitro selection of hematoporphyrin binding DNA aptamers. Bioorg. Med. Chem. Lett. 2000, 10, 2653–2656. [Google Scholar] [CrossRef]
- Li, Y.; Geyer, C.R.; Sen, D. Recognition of anionic porphyrins by DNA aptamers. Biochemistry 1996, 35, 6911–6922. [Google Scholar] [CrossRef] [PubMed]
- Niles, J.C.; DeRisi, J.L.; Marletta, M.A. Inhibiting plasmodium falciparum growth and heme detoxification pathway using heme-binding DNA aptamers. Proc. Natl. Acad. Sci. USA 2009, 106, 13266–13271. [Google Scholar] [CrossRef] [PubMed]
- Dondorp, A.; Nosten, F.; Stepniewska, K.; Day, N.; White, N. Artesunate versus quinine for treatment of severe falciparum malaria: A randomised trial. Lancet 2005, 366, 717–725. [Google Scholar] [PubMed]
- Panton, L.J.; Leech, J.H.; Miller, L.; Howard, R.J. Cytoadherence of plasmodium falciparum-infected erythrocytes to human melanoma cell lines correlates with surface OKM5 antigen. Infect. Immun. 1987, 55, 2754–2758. [Google Scholar] [PubMed]
- Craig, A.G.; Pinches, R.; Khan, S.; Roberts, D.J.; Turner, G.D.; Newbold, C.I.; Berendt, A.R. Failure to block adhesion of plasmodium falciparum-infected erythrocytes to ICAM-1 with soluble ICAM-1. Infect. Immun. 1997, 65, 4580–4585. [Google Scholar] [PubMed]
- Ruigrok, V.J.; Levisson, M.; Eppink, M.H.; Smidt, H.; van Der Oost, J. Alternative affinity tools: More attractive than antibodies? Biochem. J. 2011, 436, 1–13. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aptamer | Target | Function | Result | Reference |
|---|---|---|---|---|
| RNA | Surface protein of PfEMP1—DBLα domain | To disrupt the rosette formation between infected erythrocyte with normal erythrocyte. | Isolated RNA aptamer reduced rosette formation by 35% at 33 nM concentration and 100% reduction at 387 nM concentration during in vitro study using high rosette-forming strain, P. falciparum FCR3S1.2 | [99] |
| DNA | Heme group | To interfere with heme-detoxification and growth of P. falciparum in infected erythrocyte. | Parasite growth was significantly reduced after 72 h in a culture when compared with the control | [102] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nik Kamarudin, N.A.A.; Mohammed, N.A.; Mustaffa, K.M.F. Aptamer Technology: Adjunct Therapy for Malaria. Biomedicines 2017, 5, 1. https://doi.org/10.3390/biomedicines5010001
Nik Kamarudin NAA, Mohammed NA, Mustaffa KMF. Aptamer Technology: Adjunct Therapy for Malaria. Biomedicines. 2017; 5(1):1. https://doi.org/10.3390/biomedicines5010001
Chicago/Turabian StyleNik Kamarudin, Nik Abdul Aziz, Nurul Adila Mohammed, and Khairul Mohd Fadzli Mustaffa. 2017. "Aptamer Technology: Adjunct Therapy for Malaria" Biomedicines 5, no. 1: 1. https://doi.org/10.3390/biomedicines5010001
APA StyleNik Kamarudin, N. A. A., Mohammed, N. A., & Mustaffa, K. M. F. (2017). Aptamer Technology: Adjunct Therapy for Malaria. Biomedicines, 5(1), 1. https://doi.org/10.3390/biomedicines5010001