Toll-Like Receptor 9 Agonists for Cancer Therapy

 ,
,
Abstract
:
1. Introduction
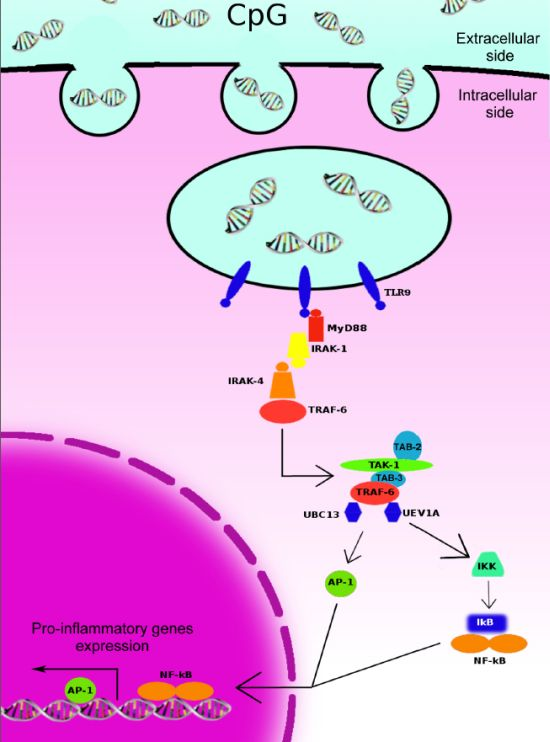
2. Toll-Like Receptors (TLR)-9-Initiated Intracellular Signaling Pathways and Immune Responses
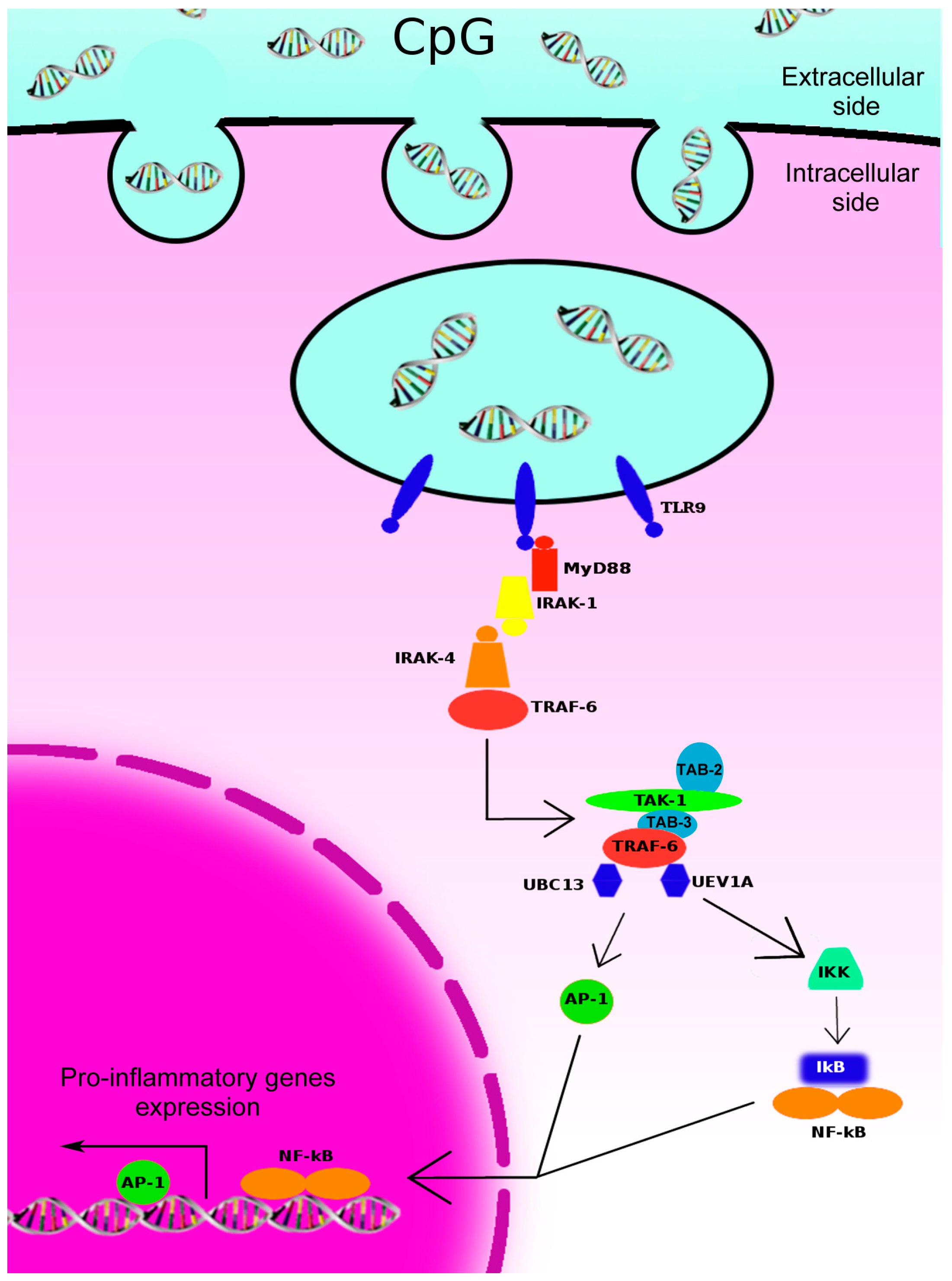
3. TLR-9 Agonists for Cancer Therapy
4. Clinical Evidences for TLR-9 Agonists Antitumor Activity

{kind=link}
{kind=link}
| Agent | Treatment Arms | Study Phase | Cancer Type | No. Patients | Results | References | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PF-3512676 | PF-3512676 8 mg vs. saline | Phase II randomized | Early stage melanoma | 24 | In the experimental arm: larger sentinel lymph nodes (SLN), higher SLN leucocytes, higher maturation markers of DC, lower T-reg, increased cytokines | Molenkamp et al. [61,62] | |||||||
| PF-3512676 | PF-3512676 0.01–5/10 mg | Phase I | BCC and advanced melanoma | 10 | Local tumor regression, post-treatment cytokines levels reduction, dense intra- and peri-tumoral lymphocytic infiltrates | Hofmann et al. [63] | |||||||
| PF-3512676 | PF-3512676 6 mg | Phase II | Advanced melanoma | 20 | PR = 10%, CR = 5%, SD = 15% (DCR = 30%) | Pashenkov et al. [64] | |||||||
| PF-3512676 | PF-3512676 0.08, 0.12, 0.16, 0.36, 0.54, 0.81 mg/kg | Phase I/II | Metastatic RCC | 39 | PR = 5%, DCR = 30% | Thompson et al. [65] | |||||||
| PF-3512676 | PF-3512676 10 mg vs. PF-3512676 40 mg vs. PF-3512676 40 mg + DTIC 850 mg/m2 vs. DTIC 850 mg/m2 | Phase II randomized | Untreated advanced melanoma | 184 | Higher ORR (16%) for PF-3512676 40 mg + DTIC 850 mg/m2 no differences in mTTP and mOS | Weber et al. [66] | |||||||
| PF-3512676 | PF-3512676 0.2 mg/kg + taxane/platinum vs. taxane/platinum | Phase II randomized | Untreated advanced NSCLC | 117 | Higher ORR for PF-3512676 0.2 mg/kg + taxane/platinum (38% vs. 19%) Longer mOS PF-3512676 0.2 mg/kg + taxane/platinum (12.3 vs. 6.8 ms) | Manegold et al. [67] | |||||||
| PF-3512676 | PF-3512676 0.2 mg/kg + CBDCA/TXL vs. CBDCA/TXL | Phase III | Untreated advanced NSCLC | 828 | No significant differences in mOS neither mPFS | Hirsh et al. [68] | |||||||
| PF-3512676 | PF-3512676 0.2 mg/kg + CDDP/GEM vs. CDDP/GEM | Phase III | Untreated advanced NSCLC | 839 | No significant differences in mOS neither mPFS | Manegold et al. [67] | |||||||
| PF-3512676 | PF-3512676 0.2 mg/kg + erlotinib vs. erlotinib | Phase II randomized | EGFR mutated advanced pre-treated NSCLC | 39 | No differences in PFS | Belani et al. [69] | |||||||
| IMO-2055 (EMD1201081) | IMO-2055 0.16, 0.32, 0.48 mg/kg + CDDP/5-FU/Cetuximab | Phase Ib | Recurrent/metastatic SCCHN | 13 | Prematurely stopped for unacceptable toxicity no MTD determined | Machiels et al. [70] | |||||||
| IMO-2055 (EMD1201081) | IMO-2055 0.32 mg/kg + cetuximab vs. cetuximab | Phase II randomized | Recurrent/metastatic SCCHN never treated with anti-EGFR | Ongoing; recruitment terminated | [71] | ||||||||
| IMO-2055 (EMD1201081) | IMO-2055 0.32 mg/kg + FOLFIRI/cetuximab | Phase I | Second-line Kras wt CRC | Ongoing; recruitment terminated | [72] | ||||||||
| AS15 | MAGE-A3 + AS15 vs. MAGE-A3 + AS02b | Phase II randomized | MAGE-A3 positive advanced melanoma | 75 | Higher ORR (5%) for MAGE-A3 + AS15 Longer 6 ms-PFS (25%) and mOS (33 ms) for MAGE-A3 + AS15 | Kruit et al. [73] | |||||||
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Topalian, S.L.; Weiner, G.J.; Pardoll, D.M. Cancer immunotherapy comes of age. J. Clin. Oncol. 2011, 29, 4828–4836. [Google Scholar] [CrossRef]
- Sharma, P.; Wagner, K.; Wolchok, J.D.; Allison, J.P. Novel cancer immunotherapy agents with survival benefit: Recent successes and next steps. Nat. Rev. Cancer 2011, 11, 805–812. [Google Scholar] [CrossRef]
- Hennessy, E.J.; Parker, A.E.; OʼNeill, L.A. Targeting toll-like receptors: Emerging therapeutics? Nat. Rev. Drug. Discov. 2010, 9, 293–307. [Google Scholar] [CrossRef]
- Lotze, M.T.; Zeh, H.J.; Rubartelli, A.; Sparvero, L.J.; Amoscato, A.A.; Washburn, N.R.; Devera, M.E.; Liang, X.; Tor, M.; Billiar, T. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol. Rev. 2007, 220, 60–81. [Google Scholar]
- Krieg, A.M. Therapeutic potential of Toll-like receptor 9 activation. Nat. Rev. Drug. Discov. 2006, 5, 471–484. [Google Scholar] [CrossRef]
- Agrawal, S.; Kandimalla, E.R. Synthetic agonists of toll-like receptors 7, 8 and 9. Biochem. Soc. Trans. 2007, 35, 1461–1467. [Google Scholar] [CrossRef]
- Bauer, S.; Kirschning, C.J.; Hacker, H.; Redecke, V.; Hausmann, S.; Akira, S.; Wagner, H.; Lipford, G.B. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. USA 2001, 98, 9237–9242. [Google Scholar] [CrossRef]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef]
- Barton, G.M.; Kagan, J.C.; Medzhitov, R. Intracellular localization of toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat. Immunol. 2006, 7, 49–56. [Google Scholar] [CrossRef]
- Krieg, A.M. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 2008, 27, 161–167. [Google Scholar] [CrossRef]
- Hayashi, F.; Means, T.K.; Luster, A.D. Toll-like receptors stimulate human neutrophil function. Blood 2003, 102, 2660–2669. [Google Scholar] [CrossRef]
- Saikh, K.U.; Kissner, T.L.; Sultana, A.; Ruthel, G.; Ulrich, R.G. Human monocytes infected with Yersinia pestis express cell surface TLR9 and differentiate into dendritic cells. J. Immunol. 2004, 173, 7426–7434. [Google Scholar] [CrossRef]
- Siren, J.; Pirhonen, J.; Julkunen, I.; Matikainen, S. IFN-α regulates TLR-dependent gene expression of IFN-α, IFN-β, IL-28, and IL-29. J. Immunol. 2005, 174, 1932–1937. [Google Scholar] [CrossRef]
- Gelman, A.E.; Zhang, J.; Choi, Y.; Turka, L.A. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J. Immunol. 2004, 172, 6065–6073. [Google Scholar] [CrossRef]
- Platz, J.; Beisswenger, C.; Dalpke, A.; Koczulla, R.; Pinkenburg, O.; Vogelmeier, C.; Bals, R. Microbial DNA induces a host defense reaction of human respiratory epithelial cells. J. Immunol. 2004, 173, 1219–1223. [Google Scholar] [CrossRef]
- Lee, J.; Mo, J.H.; Katakura, K.; Alkalay, I.; Rucker, A.N.; Liu, Y.T.; Lee, H.K.; Shen, C.; Cojocaru, G.; Shenouda, S.; et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat. Cell Biol. 2006, 8, 1327–1336. [Google Scholar] [CrossRef]
- Ilvesaro, J.M.; Merrell, M.A.; Li, L.; Wakchoure, S.; Graves, D.; Brooks, S.; Rahko, E.; Jukkola-Vuorinen, A.; Vuopala, K.S.; Harris, K.W.; et al. Toll-like receptor 9 mediates CpG oligonucleotide-induced cellular invasion. Mol. Cancer Res. 2008, 6, 1534–1543. [Google Scholar] [CrossRef]
- Lee, J.W.; Choi, J.J.; Seo, E.S.; Kim, M.J.; Kim, W.Y.; Choi, C.H.; Kim, T.J.; Kim, B.G.; Song, S.Y.; Bae, D.S. Increased toll-like receptor 9 expression in cervical neoplasia. Mol. Carcinog. 2007, 46, 941–947. [Google Scholar] [CrossRef]
- Klinman, D.M.; Yi, A.K.; Beaucage, S.L.; Conover, J.; Krieg, A.M. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc. Natl. Acad. Sci. USA 1996, 93, 2879–2883. [Google Scholar] [CrossRef]
- Brignole, C.; Marimpietri, D.; di Paolo, D.; Perri, P.; Morandi, F.; Pastorino, F.; Zorzoli, A.; Pagnan, G.; Loi, M.; Caffa, I.; et al. Therapeutic targeting of TLR9 inhibits cell growth and induces apoptosis in neuroblastoma. Cancer Res. 2010, 70, 9816–9826. [Google Scholar] [CrossRef]
- Damiano, V.; Caputo, R.; Garofalo, S.; Bianco, R.; Rosa, R.; Merola, G.; Gelardi, T.; Racioppi, L.; Fontanini, G.; de Placido, S.; et al. TLR9 agonist acts by different mechanisms synergizing with bevacizumab in sensitive and cetuximab-resistant colon cancer xenografts. Proc. Natl. Acad. Sci. USA 2007, 104, 12468–12473. [Google Scholar] [CrossRef]
- Pradere, J.P.; Dapito, D.H.; Schwabe, R.F. The Yin and Yang of toll-like receptors in cancer. Oncogene 2014, 33, 3485–3495. [Google Scholar] [CrossRef]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukoc. Biol. 2013, 93, 847–863. [Google Scholar]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef]
- Janssens, S.; Beyaert, R. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol. Cell 2003, 11, 293–302. [Google Scholar] [CrossRef]
- Li, S.; Strelow, A.; Fontana, E.J.; Wesche, H. IRAK-4: A novel member of the IRAK family with the properties of an IRAK-kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 5567–5572. [Google Scholar] [CrossRef]
- Melisi, D.; Xia, Q.; Paradiso, G.; Ling, J.; Moccia, T.; Carbone, C.; Budillon, A.; Abbruzzese, J.L.; Chiao, P.J. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J. Natl. Cancer Inst. 2011, 103, 1190–1204. [Google Scholar] [CrossRef]
- Ajibade, A.A.; Wang, H.Y.; Wang, R.F. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013, 34, 307–316. [Google Scholar] [CrossRef]
- Dai, L.; Aye Thu, C.; Liu, X.Y.; Xi, J.; Cheung, P.C. TAK1, more than just innate immunity. IUBMB Life 2012, 64, 825–834. [Google Scholar] [CrossRef]
- Singhirunnusorn, P.; Suzuki, S.; Kawasaki, N.; Saiki, I.; Sakurai, H. Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-β-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J. Biol. Chem. 2005, 280, 7359–7368. [Google Scholar] [CrossRef]
- Yu, Y.; Ge, N.; Xie, M.; Sun, W.; Burlingame, S.; Pass, A.K.; Nuchtern, J.G.; Zhang, D.; Fu, S.; Schneider, M.D.; et al. Phosphorylation of THR-178 and THR-184 in the TAK1 T-loop is required for interleukin (IL)-1-mediated optimal NFκB and AP-1 activation as well as IL-6 gene expression. J. Biol. Chem. 2008, 283, 24497–24505. [Google Scholar] [CrossRef]
- Kanzler, H.; Barrat, F.J.; Hessel, E.M.; Coffman, R.L. Therapeutic targeting of innate immunity with toll-like receptor agonists and antagonists. Nat. Med. 2007, 13, 552–559. [Google Scholar] [CrossRef]
- Takauji, R.; Iho, S.; Takatsuka, H.; Yamamoto, S.; Takahashi, T.; Kitagawa, H.; Iwasaki, H.; Iida, R.; Yokochi, T.; Matsuki, T. CpG-DNA-induced IFN-α production involves p38 MAPK-dependent STAT1 phosphorylation in human plasmacytoid dendritic cell precursors. J. Leukoc. Biol. 2002, 72, 1011–1019. [Google Scholar]
- Krug, A.; Rothenfusser, S.; Hornung, V.; Jahrsdorfer, B.; Blackwell, S.; Ballas, Z.K.; Endres, S.; Krieg, A.M.; Hartmann, G. Identification of CpG oligonucleotide sequences with high induction of IFN-α/β in plasmacytoid dendritic cells. Eur. J. Immunol. 2001, 31, 2154–2163. [Google Scholar] [CrossRef]
- Iparraguirre, A.; Tobias, J.W.; Hensley, S.E.; Masek, K.S.; Cavanagh, L.L.; Rendl, M.; Hunter, C.A.; Ertl, H.C.; von Andrian, U.H.; Weninger, W. Two distinct activation states of plasmacytoid dendritic cells induced by influenza virus and CpG 1826 oligonucleotide. J. Leukoc. Biol. 2008, 83, 610–620. [Google Scholar]
- Sansonetti, P.J. The innate signaling of dangers and the dangers of innate signaling. Nat. Immunol. 2006, 7, 1237–1242. [Google Scholar] [CrossRef]
- Dieu, M.C.; Vanbervliet, B.; Vicari, A.; Bridon, J.M.; Oldham, E.; Ait-Yahia, S.; Briere, F.; Zlotnik, A.; Lebecque, S.; Caux, C. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med. 1998, 188, 373–386. [Google Scholar] [CrossRef]
- Ma, L.; Zhao, G.; Hua, C.; Li, X.; Zhao, X.; Sun, L.; Hou, Y. Down-regulation of TLR9 expression affects the maturation and function of murine bone marrow-derived dendritic cells induced by CpG. Cell. Mol. Immunol. 2009, 6, 199–205. [Google Scholar] [CrossRef]
- MartIn-Fontecha, A.; Sebastiani, S.; Hopken, U.E.; Uguccioni, M.; Lipp, M.; Lanzavecchia, A.; Sallusto, F. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J. Exp. Med. 2003, 198, 615–621. [Google Scholar] [CrossRef]
- Gray, R.C.; Kuchtey, J.; Harding, C.V. CpG-B ODNs potently induce low levels of IFN-αβ and induce IFN-αβ-dependent MHC-I cross-presentation in DCs as effectively as CpG-A and CpG-C ODNs. J. Leukoc. Biol. 2007, 81, 1075–1085. [Google Scholar] [CrossRef]
- Krieg, A.M. Development of TLR9 agonists for cancer therapy. J. Clin. Investig. 2007, 117, 1184–1194. [Google Scholar] [CrossRef]
- So, E.Y.; Ouchi, T. The application of toll like receptors for cancer therapy. Int. J. Biol. Sci. 2010, 6, 675–681. [Google Scholar]
- Tanaka, J.; Sugimoto, K.; Shiraki, K.; Tameda, M.; Kusagawa, S.; Nojiri, K.; Beppu, T.; Yoneda, K.; Yamamoto, N.; Uchida, K.; et al. Functional cell surface expression of toll-like receptor 9 promotescell proliferation and survival in human hepatocellular carcinomas. Int. J. Oncol. 2010, 37, 805–814. [Google Scholar]
- Zhang, Y.; Wang, Q.; Ma, A.; Li, Y.; Li, R.; Wang, Y. Functional expression of TLR9 in esophageal cancer. Oncol. Rep. 2014, 31, 2298–2304. [Google Scholar]
- Vollmer, J.; Weeratna, R.; Payette, P.; Jurk, M.; Schetter, C.; Laucht, M.; Wader, T.; Tluk, S.; Liu, M.; Davis, H.L.; et al. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur. J. Immunol. 2004, 34, 251–262. [Google Scholar] [CrossRef]
- Kerkmann, M.; Costa, L.T.; Richter, C.; Rothenfusser, S.; Battiany, J.; Hornung, V.; Johnson, J.; Englert, S.; Ketterer, T.; Heckl, W.; et al. Spontaneous formation of nucleic acid-based nanoparticles is responsible for high interferon-α induction by CpG-A in plasmacytoid dendritic cells. J. Biol. Chem. 2005, 280, 8086–8093. [Google Scholar] [CrossRef]
- Chinnathambi, S.; Chen, S.; Ganesan, S.; Hanagata, N. Binding mode of CpG oligodeoxynucleotides to nanoparticles regulates bifurcated cytokine induction via toll-like receptor 9. Sci. Rep. 2012, 2, 534. [Google Scholar]
- Wang, H.; Rayburn, E.; Zhang, R. Synthetic oligodeoxynucleotides containing deoxycytidyl-deoxyguanosine dinucleotides (CpG ODNs) and modified analogs as novel anticancer therapeutics. Curr. Pharm. Des. 2005, 11, 2889–2907. [Google Scholar] [CrossRef]
- Kandimalla, E.R.; Bhagat, L.; Li, Y.; Yu, D.; Wang, D.; Cong, Y.P.; Song, S.S.; Tang, J.X.; Sullivan, T.; Agrawal, S. Immunomodulatory oligonucleotides containing a cytosine-phosphate-2'-deoxy-7-deazaguanosine motif as potent toll-like receptor 9 agonists. Proc. Natl. Acad. Sci. USA 2005, 102, 6925–6930. [Google Scholar] [CrossRef]
- Rosa, R.; Melisi, D.; Damiano, V.; Bianco, R.; Garofalo, S.; Gelardi, T.; Agrawal, S.; di Nicolantonio, F.; Scarpa, A.; Bardelli, A.; et al. Toll-like receptor 9 agonist IMO cooperates with cetuximab in K-Ras mutant colorectal and pancreatic cancers. Clin. Cancer Res. 2011, 17, 6531–6541. [Google Scholar] [CrossRef]
- Van Ojik, H.H.; Bevaart, L.; Dahle, C.E.; Bakker, A.; Jansen, M.J.; van Vugt, M.J.; van de Winkel, J.G.; Weiner, G.J. CpG-A and B oligodeoxynucleotides enhance the efficacy of antibody therapy by activating different effector cell populations. Cancer Res. 2003, 63, 5595–5600. [Google Scholar]
- Zoglmeier, C.; Bauer, H.; Norenberg, D.; Wedekind, G.; Bittner, P.; Sandholzer, N.; Rapp, M.; Anz, D.; Endres, S.; Bourquin, C. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin. Cancer Res. 2011, 17, 1765–1775. [Google Scholar] [CrossRef]
- Shirota, Y.; Shirota, H.; Klinman, D.M. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J. Immunol. 2012, 188, 1592–1599. [Google Scholar] [CrossRef]
- Cerullo, V.; Diaconu, I.; Romano, V.; Hirvinen, M.; Ugolini, M.; Escutenaire, S.; Holm, S.L.; Kipar, A.; Kanerva, A.; Hemminki, A. An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol. Ther. 2012, 20, 2076–2086. [Google Scholar] [CrossRef]
- Damiano, V.; Caputo, R.; Bianco, R.; DʼArmiento, F.P.; Leonardi, A.; de Placido, S.; Bianco, A.R.; Agrawal, S.; Ciardiello, F.; Tortora, G. Novel toll-like receptor 9 agonist induces epidermal growth factor receptor (EGFR) inhibition and synergistic antitumor activity with EGFR inhibitors. Clin. Cancer Res. 2006, 12, 577–583. [Google Scholar] [CrossRef]
- Damiano, V.; Garofalo, S.; Rosa, R.; Bianco, R.; Caputo, R.; Gelardi, T.; Merola, G.; Racioppi, L.; Garbi, C.; Kandimalla, E.R.; et al. A novel toll-like receptor 9 agonist cooperates with trastuzumab in trastuzumab-resistant breast tumors through multiple mechanisms of action. Clin. Cancer Res. 2009, 15, 6921–6930. [Google Scholar] [CrossRef]
- Xu, L.; Wen, Z.; Zhou, Y.; Liu, Z.; Li, Q.; Fei, G.; Luo, J.; Ren, T. MicroRNA-7-regulated TLR9 signaling-enhanced growth and metastatic potential of human lung cancer cells by altering the phosphoinositide-3-kinase, regulatory subunit 3/Akt pathway. Mol. Biol. Cell 2013, 24, 42–55. [Google Scholar] [CrossRef]
- Sommariva, M.; de Cecco, L.; de Cesare, M.; Sfondrini, L.; Menard, S.; Melani, C.; Delia, D.; Zaffaroni, N.; Pratesi, G.; Uva, V.; et al. TLR9 agonists oppositely modulate DNA repair genes in tumor versus immune cells and enhance chemotherapy effects. Cancer Res. 2011, 71, 6382–6390. [Google Scholar] [CrossRef]
- Molenkamp, B.G.; van Leeuwen, P.A.; Meijer, S.; Sluijter, B.J.; Wijnands, P.G.; Baars, A.; van den Eertwegh, A.J.; Scheper, R.J.; de Gruijl, T.D. Intradermal CpG-B activates both plasmacytoid and myeloid dendritic cells in the sentinel lymph node of melanoma patients. Clin. Cancer Res. 2007, 13, 2961–2969. [Google Scholar] [CrossRef]
- Molenkamp, B.G.; Sluijter, B.J.; van Leeuwen, P.A.; Santegoets, S.J.; Meijer, S.; Wijnands, P.G.; Haanen, J.B.; van den Eertwegh, A.J.; Scheper, R.J.; de Gruijl, T.D. Local administration of PF-3512676 CpG-B instigates tumor-specific CD8+ T-cell reactivity in melanoma patients. Clin. Cancer Res. 2008, 14, 4532–4542. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Kors, C.; Audring, H.; Walden, P.; Sterry, W.; Trefzer, U. Phase 1 evaluation of intralesionally injected TLR9-agonist PF-3512676 in patients with basal cell carcinoma or metastatic melanoma. J. Immunother. 2008, 31, 520–527. [Google Scholar] [CrossRef]
- Pashenkov, M.; Goess, G.; Wagner, C.; Hormann, M.; Jandl, T.; Moser, A.; Britten, C.M.; Smolle, J.; Koller, S.; Mauch, C.; et al. Phase II trial of a toll-like receptor 9-activating oligonucleotide in patients with metastatic melanoma. J. Clin. Oncol. 2006, 24, 5716–5724. [Google Scholar] [CrossRef]
- Thompson, J.A.; Kuzel, T.; Drucker, B.J.; Urba, W.J.; Bukowski, R.M. Safety and efficacy of PF-3512676 for the treatment of stage IV renal cell carcinoma: An open-label, multicenter phase I/II study. Clin. Genitourin. Cancer 2009, 7, E58–E65. [Google Scholar] [CrossRef]
- Weber, J.S.; Zarour, H.; Redman, B.; Trefzer, U.; OʼDay, S.; van den Eertwegh, A.J.; Marshall, E.; Wagner, S. Randomized phase 2/3 trial of CpG oligodeoxynucleotide PF-3512676 alone or with dacarbazine for patients with unresectable stage III and IV melanoma. Cancer 2009, 115, 3944–3954. [Google Scholar] [CrossRef]
- Manegold, C.; Gravenor, D.; Woytowitz, D.; Mezger, J.; Hirsh, V.; Albert, G.; Al-Adhami, M.; Readett, D.; Krieg, A.M.; Leichman, C.G. Randomized phase II trial of a toll-like receptor 9 agonist oligodeoxynucleotide, PF-3512676, in combination with first-line taxane plus platinum chemotherapy for advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 2008, 26, 3979–3986. [Google Scholar] [CrossRef]
- Hirsh, V.; Paz-Ares, L.; Boyer, M.; Rosell, R.; Middleton, G.; Eberhardt, W.E.; Szczesna, A.; Reiterer, P.; Saleh, M.; Arrieta, O.; et al. Randomized phase III trial of paclitaxel/carboplatin with or without PF-3512676 (toll-like receptor 9 agonist) as first-line treatment for advanced non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 2667–2674. [Google Scholar] [CrossRef]
- Manegold, C.; van Zandwijk, N.; Szczesna, A.; Zatloukal, P.; Au, J.S.; Blasinska-Morawiec, M.; Serwatowski, P.; Krzakowski, M.; Jassem, J.; Tan, E.H.; et al. A phase III randomized study of gemcitabine and cisplatin with or without PF-3512676 (TLR9 agonist) as first-line treatment of advanced non-small-cell lung cancer. Ann. Oncol. 2012, 23, 72–77. [Google Scholar] [CrossRef]
- Machiels, J.P.; Kaminsky, M.C.; Keller, U.; Brummendorf, T.H.; Goddemeier, T.; Forssmann, U.; Delord, J.P. Phase Ib trial of the Toll-like receptor 9 agonist IMO-2055 in combination with 5-fluorouracil, cisplatin, and cetuximab as first-line palliative treatment in patients with recurrent/metastatic squamous cell carcinoma of the head and neck. Investig. New Drugs 2013, 31, 1207–1216. [Google Scholar] [CrossRef]
- A Trial Exploring the Efficacy of EMD 1201081 in Combination With Cetuximab in Second-Line Cetuximab-Naïve Subjects With Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (R/M SCCHN). Available online: https://clinicaltrials.gov/show/NCT01040832 (accessed on 1 July 2014).
- Study of FOLFIRI Plus Cetuximab Plus IMO-2055 in Patients With Colorectal Cancer. Available online: https://clinicaltrials.gov/show/NCT00719199 (accessed on 1 July 2014).
- Kruit, W.H.; Suciu, S.; Dreno, B.; Mortier, L.; Robert, C.; Chiarion-Sileni, V.; Maio, M.; Testori, A.; Dorval, T.; Grob, J.J.; et al. Selection of immunostimulant AS15 for active immunization with MAGE-A3 protein: Results of a randomized phase II study of the European Organisation for Research and Treatment of Cancer Melanoma Group in Metastatic Melanoma. J. Clin. Oncol. 2013, 31, 2413–2420. [Google Scholar] [CrossRef]
- Pinzon-Charry, A.; Maxwell, T.; Lopez, J.A. Dendritic cell dysfunction in cancer: A mechanism for immunosuppression. Immunol. Cell Biol. 2005, 83, 451–461. [Google Scholar] [CrossRef]
- Yang, J.C.; Sherry, R.M.; Steinberg, S.M.; Topalian, S.L.; Schwartzentruber, D.J.; Hwu, P.; Seipp, C.A.; Rogers-Freezer, L.; Morton, K.E.; White, D.E.; et al. Randomized study of high-dose and low-dose interleukin-2 in patients with metastatic renal cancer. J. Clin. Oncol. 2003, 21, 3127–3132. [Google Scholar] [CrossRef]
- Escudier, B.; Pluzanska, A.; Koralewski, P.; Ravaud, A.; Bracarda, S.; Szczylik, C.; Chevreau, C.; Filipek, M.; Melichar, B.; Bajetta, E.; et al. Bevacizumab plus interferon α-2a for treatment of metastatic renal cell carcinoma: A randomised, double-blind phase III trial. Lancet 2007, 370, 2103–2111. [Google Scholar] [CrossRef]
- Belani, C.P.; Nemunaitis, J.J.; Chachoua, A.; Eisenberg, P.D.; Raez, L.E.; Cuevas, J.D.; Mather, C.B.; Benner, R.J.; Meech, S.J. Phase 2 trial of erlotinib with or without PF-3512676 (CPG 7909, a toll-like receptor 9 agonist) in patients with advanced recurrent EGFR-positive non-small cell lung cancer. Cancer Biol. Ther. 2013, 14, 557–563. [Google Scholar] [CrossRef]
- Speiser, D.E.; Lienard, D.; Rufer, N.; Rubio-Godoy, V.; Rimoldi, D.; Lejeune, F.; Krieg, A.M.; Cerottini, J.C.; Romero, P. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J. Clin. Investig. 2005, 115, 739–746. [Google Scholar] [CrossRef]
- Appay, V.; Jandus, C.; Voelter, V.; Reynard, S.; Coupland, S.E.; Rimoldi, D.; Lienard, D.; Guillaume, P.; Krieg, A.M.; Cerottini, J.C.; et al. New generation vaccine induces effective melanoma-specific CD8+ T cells in the circulation but not in the tumor site. J. Immunol. 2006, 177, 1670–1678. [Google Scholar] [CrossRef]
- Haining, W.N.; Davies, J.; Kanzler, H.; Drury, L.; Brenn, T.; Evans, J.; Angelosanto, J.; Rivoli, S.; Russell, K.; George, S.; et al. CpG oligodeoxynucleotides alter lymphocyte and dendritic cell trafficking in humans. Clin. Cancer Res. 2008, 14, 5626–5634. [Google Scholar] [CrossRef]
- Valmori, D.; Souleimanian, N.E.; Tosello, V.; Bhardwaj, N.; Adams, S.; OʼNeill, D.; Pavlick, A.; Escalon, J.B.; Cruz, C.M.; Angiulli, A.; et al. Vaccination with NY-ESO-1 protein and CpG in Montanide induces integrated antibody/Th1 responses and CD8 T cells through cross-priming. Proc. Natl. Acad. Sci. USA 2007, 104, 8947–8952. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Melisi, D.; Frizziero, M.; Tamburrino, A.; Zanotto, M.; Carbone, C.; Piro, G.; Tortora, G. Toll-Like Receptor 9 Agonists for Cancer Therapy. Biomedicines 2014, 2, 211-228. https://doi.org/10.3390/biomedicines2030211
Melisi D, Frizziero M, Tamburrino A, Zanotto M, Carbone C, Piro G, Tortora G. Toll-Like Receptor 9 Agonists for Cancer Therapy. Biomedicines. 2014; 2(3):211-228. https://doi.org/10.3390/biomedicines2030211
Chicago/Turabian StyleMelisi, Davide, Melissa Frizziero, Anna Tamburrino, Marco Zanotto, Carmine Carbone, Geny Piro, and Giampaolo Tortora. 2014. "Toll-Like Receptor 9 Agonists for Cancer Therapy" Biomedicines 2, no. 3: 211-228. https://doi.org/10.3390/biomedicines2030211
APA StyleMelisi, D., Frizziero, M., Tamburrino, A., Zanotto, M., Carbone, C., Piro, G., & Tortora, G. (2014). Toll-Like Receptor 9 Agonists for Cancer Therapy. Biomedicines, 2(3), 211-228. https://doi.org/10.3390/biomedicines2030211