Tumor Restrictions to Oncolytic Virus

Abstract
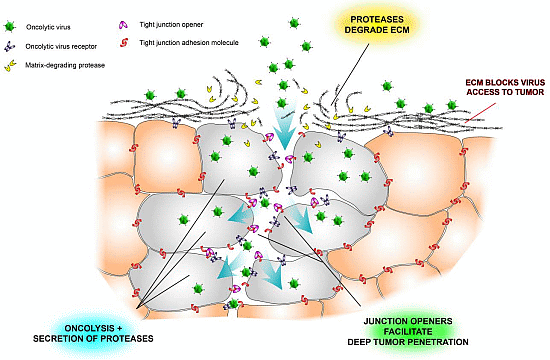
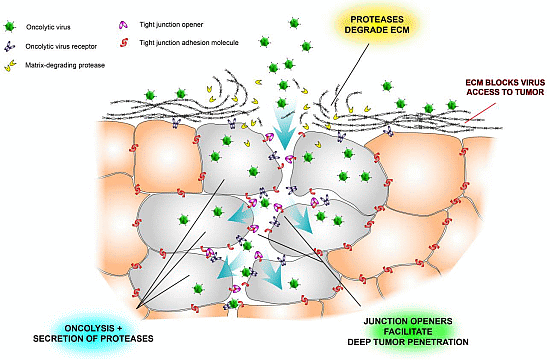
:
1. Introduction
2. Physical Barriers to Oncolytic Viruses
2.1. Interstitial Fluid Pressure
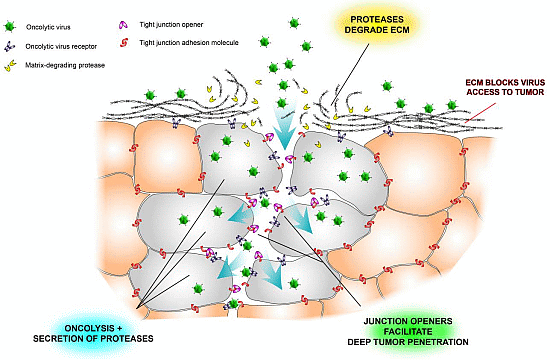
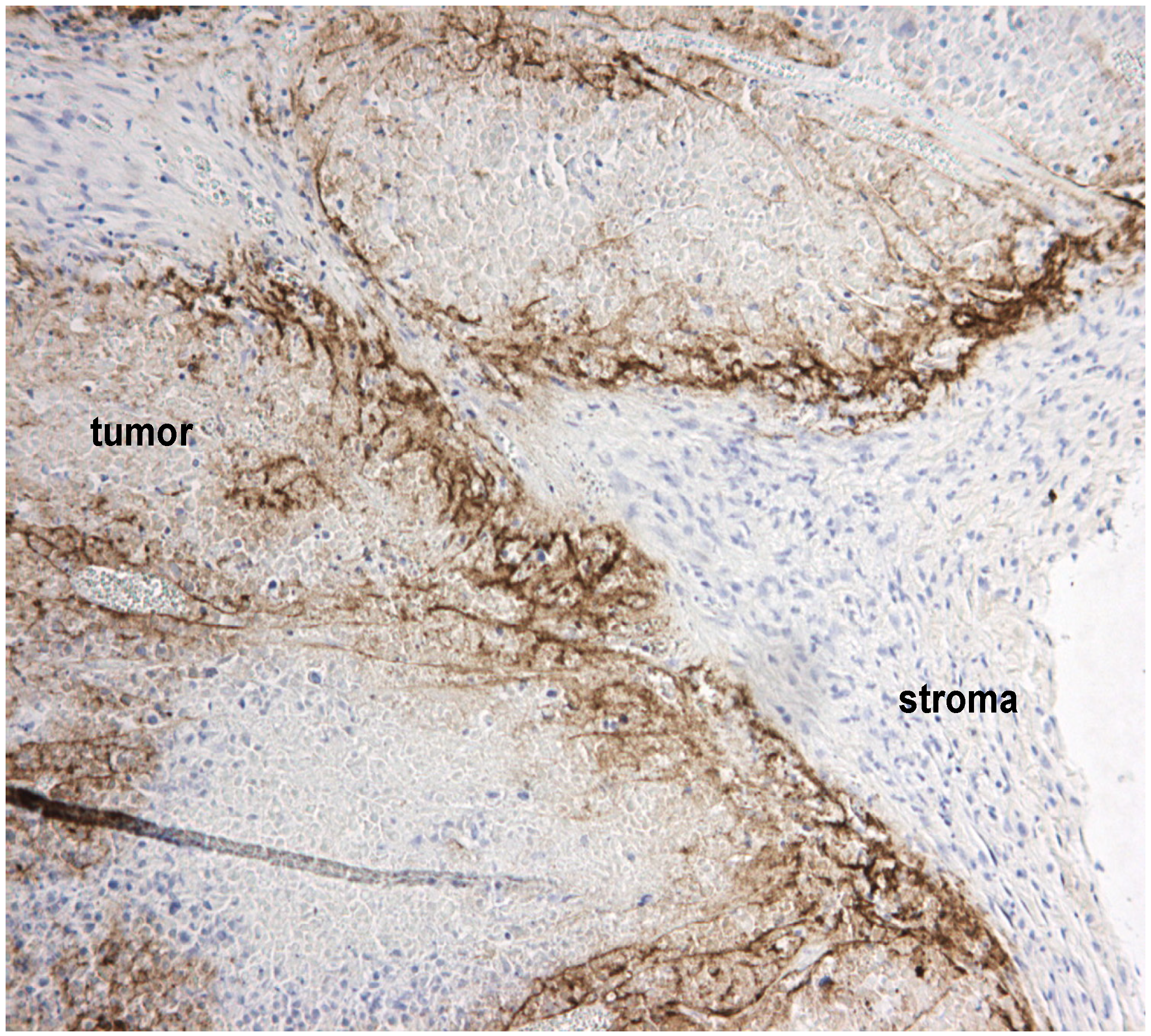
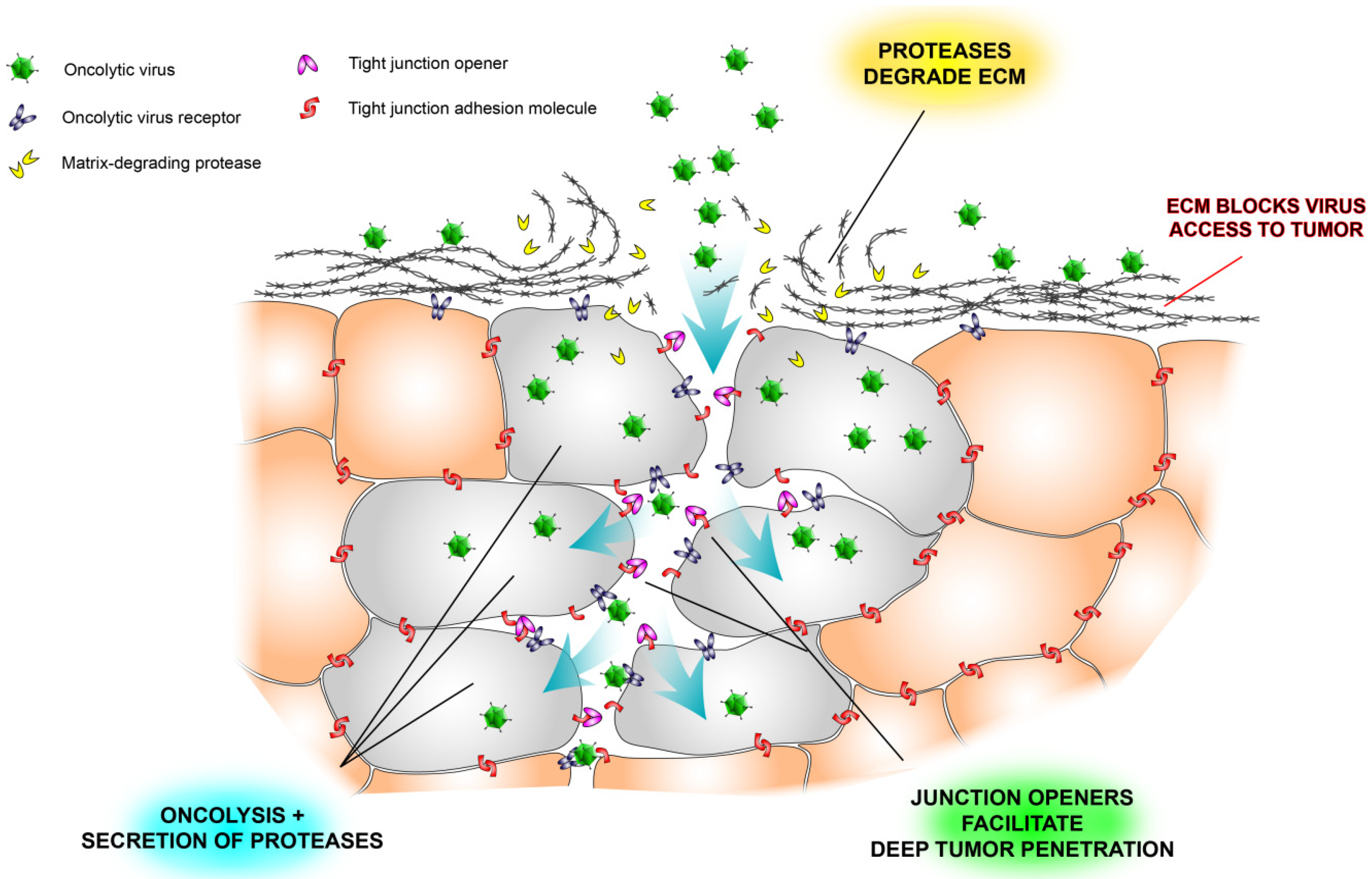
2.2. Extracellular Matrix Deposits
2.3. Tight Junctions Block Virus Penetrance and Hide Virus Receptors
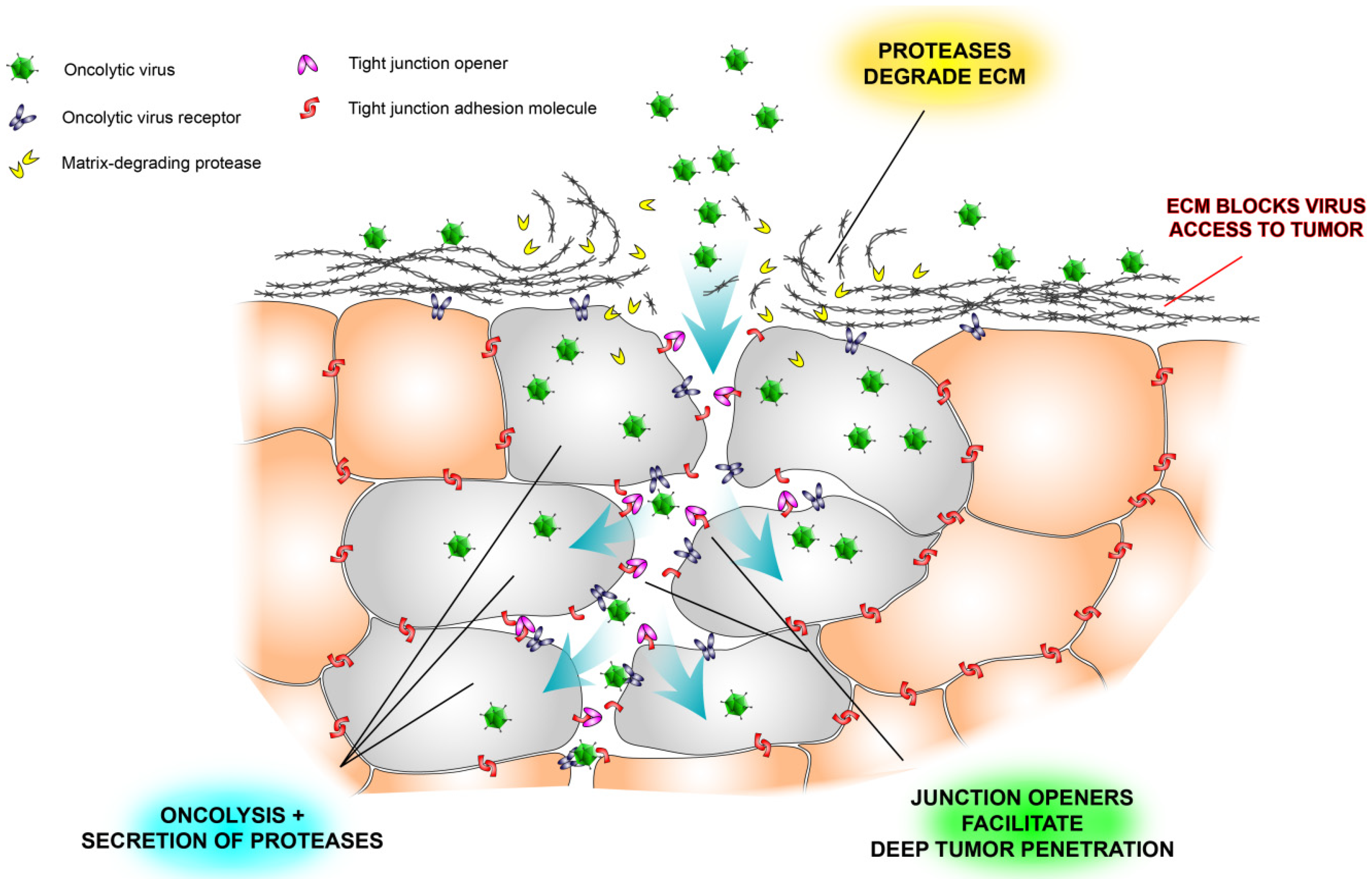
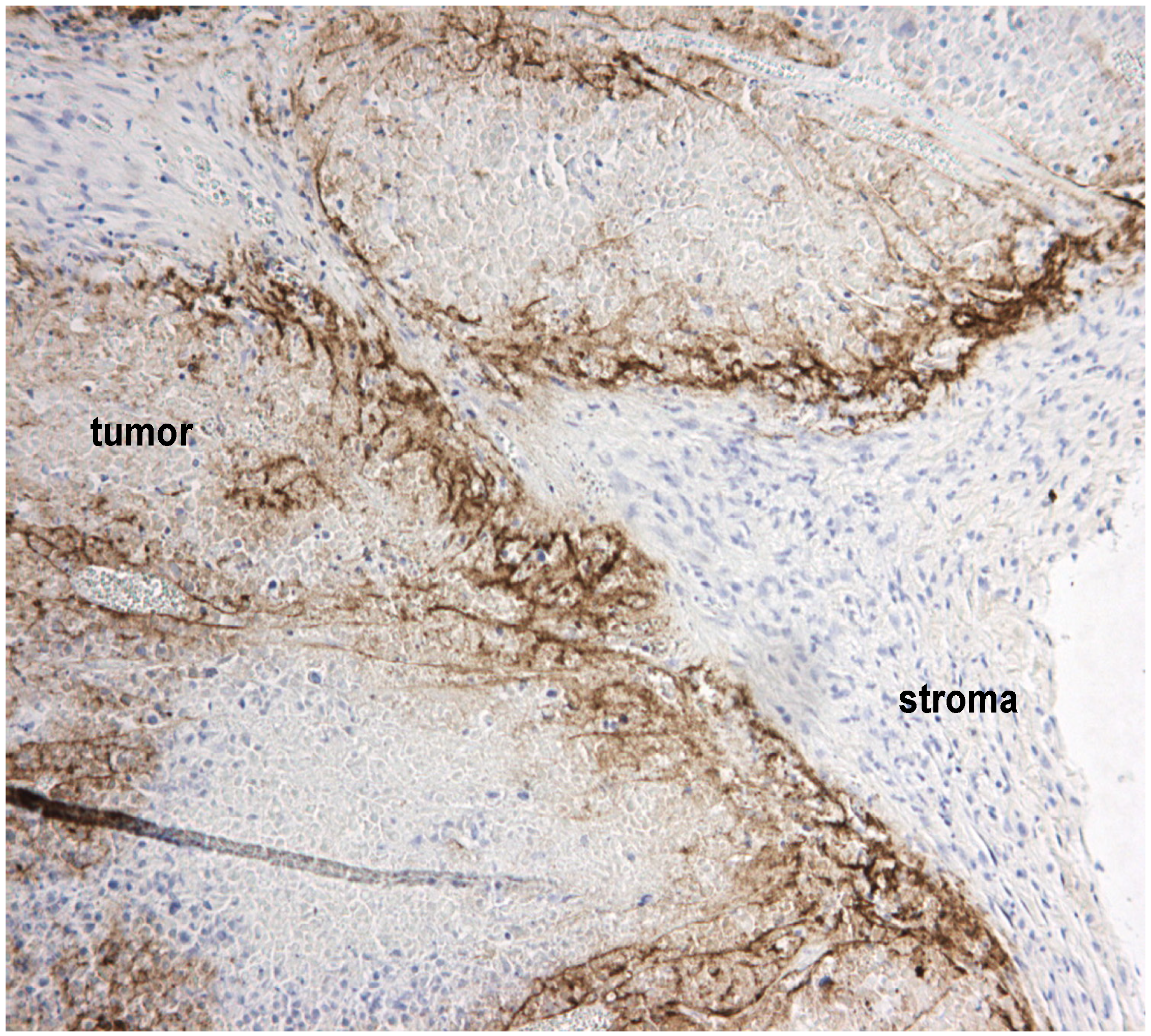
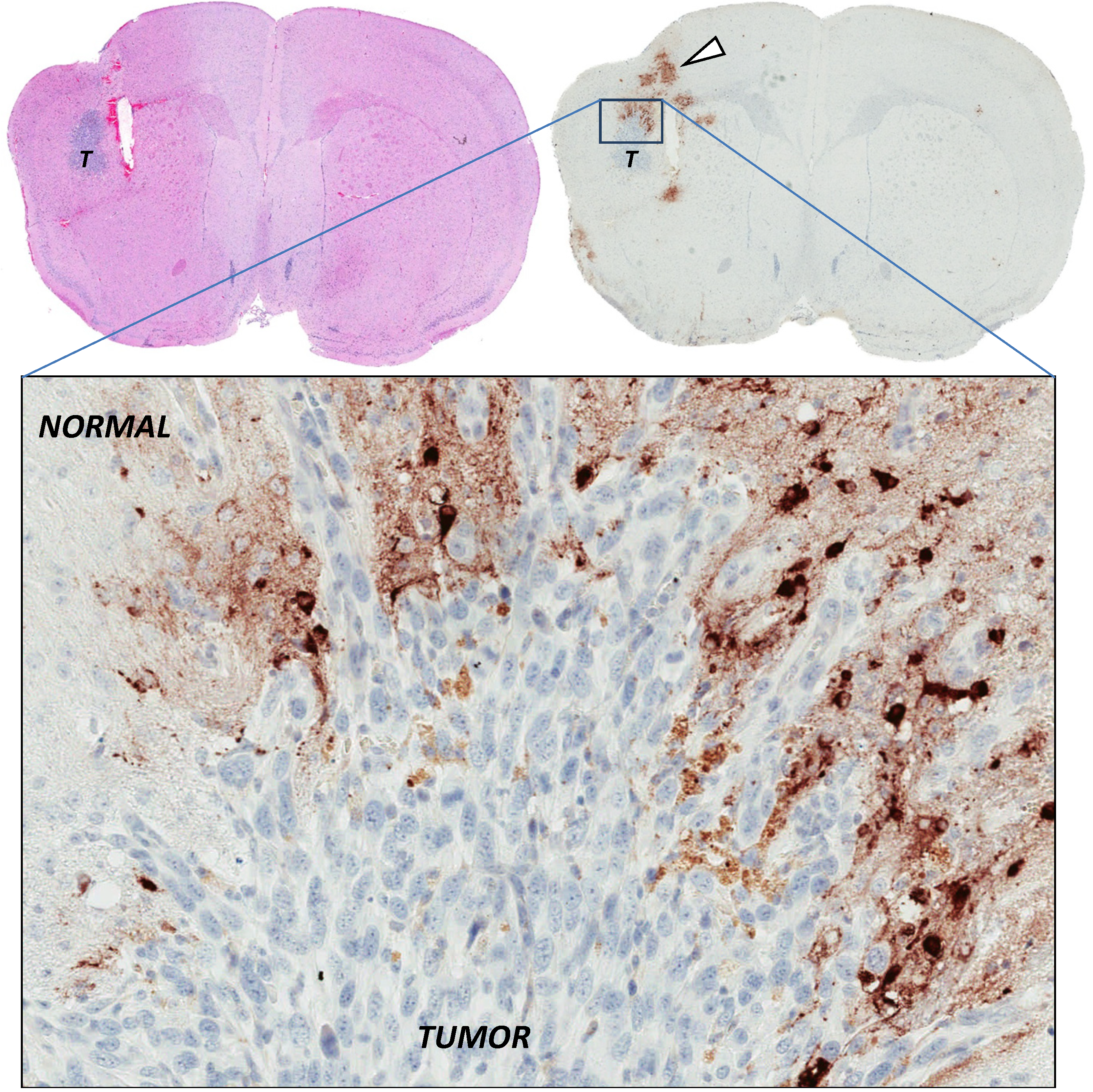
2.4. Stromal Cells Hinder Viruses
3. Tumor Cellular Defenses against Viruses
3.2. Oncolytic Virus Restriction by Innate Defenses

3.3. Exogenous Combination Therapy to Overcome Innate Defenses
3.4. Virus Engineering and Combination to Overcome Innate Defenses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target virus | Target virus modifications | Donor virus | Donor gene(s) | Description | Reference |
|---|---|---|---|---|---|
| measles vaccine strain (Edmonston) | ΔP | measles wild type (IC-B) | P | wild type P gene product V is a stronger inhibitor of MDA5-mediated activation of IRF-3 and IFN-I than V from Edmonston strain | [148] |
| measles vaccine strain (Edmonston) | ΔN, P, L | measles wild type (IC-B) | N, P, L | compared to the construct above, addition of N and L created a chimera with stronger IFN antagonistic capacity | [149] |
| newcastle disease virus F3aa (lentogenic Hitchner B1) | F mutations conferring increased fusogenic capacity | Influenza A/Puerto Rico/8/1934 (PR8), H1N1 | NS1 (between P and M) | chimera showed superior oncolytic efficacy to parental virus | [154] |
| newcastle disease virus (mesogenic Beaudette C) | Influenza H5N1 or H1N1/09 | NS1 (between P and M) | while not tested as oncolytic viruses, these recombinants did display pathogenicity in chickens and increased capacity to replicate in human cells compared to parental virus | [155] | |
| vaccinia virus (Western reserve) | ΔE3L | Influenza | NS1 | This chimera has not yet been evaluated as an oncolytic agent. Vaccinia virus E3L is critical for replication in most cell types and for spread in normal mice by blocking ISG15—influenza NS1 partially restores the capacity to replicate in cells but the resulting chimera is still unable to spread in normal tissues in vivo | [156] |
| herpes simplex type 1 (Synco-2D) | γ34.5−/−, multiple mutations, expressing GALV under UL38 promoter | vaccinia virus | B19R | Vaccinia virus soluble type I IFN scavenger B19R facilitated replication and spread of oncolytic HSV. Oncolytic efficacy in animal models was increased | [157] |
| herpes simplex type 1 | γ34.5−/− | human cytomegalovirus | TRS1 or IRS1 | PKR-antagonists TRS1 and IRS1 conferred increased replication capacity to oncolytic HSV-1, yielding greater therapeutic efficacy in glioma models in mice | [158] |
| vesicular stomatitis virus | ΔΜ51 | vaccinia virus (Western reserve) | B19R | Superior ability to spread due to neutralization of paracrine type I IFN | [159] |
| maraba virus (MG1) | G protein (Q242R) and M protein (L123W) mutations | vaccinia virus (Western reserve) | B19R | Similar to the VSV recombinant but with the enhanced oncolytic capacity of the Maraba backbone. Virus was safe in mice | [159] |
4. Conclusions
Conflicts of Interest
References
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar]
- Polanska, U.M.; Orimo, A. Carcinoma-associated fibroblasts: Non-neoplastic tumour-promoting mesenchymal cells. J. Cell. Physiol. 2013, 228, 1651–1657. [Google Scholar] [CrossRef]
- Gritsenko, P.G.; Ilina, O.; Friedl, P. Interstitial guidance of cancer invasion. J. Pathol. 2012, 226, 185–199. [Google Scholar] [CrossRef]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Inflammatory cytokines in cancer: Tumour necrosis factor and interleukin 6 take the stage. Ann. Rheum. Dis. 2011, 70, i104–i108. [Google Scholar] [CrossRef]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef]
- Lippitz, B.E. Cytokine patterns in patients with cancer: A systematic review. Lancet Oncol. 2013, 14, e218–e228. [Google Scholar]
- Miest, T.S.; Cattaneo, R. New viruses for cancer therapy: Meeting clinical needs. Nat. Rev. Microbiol. 2014, 12, 23–34. [Google Scholar] [CrossRef]
- Nakashima, H.; Kaur, B.; Chiocca, E.A. Directing systemic oncolytic viral delivery to tumors via carrier cells. Cytokine Growth Factor Rev. 2010, 21, 119–126. [Google Scholar] [CrossRef]
- Willmon, C.; Harrington, K.; Kottke, T.; Prestwich, R.; Melcher, A.; Vile, R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol. Ther. 2009, 17, 1667–1676. [Google Scholar] [CrossRef]
- Alemany, R. Viruses in cancer treatment. Clin. Transl. Oncol. 2013, 15, 182–188. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Tanaka, T.; Yamanaka, N.; Oriyama, T.; Furukawa, K.; Okamoto, E. Factors regulating tumor pressure in hepatocellular carcinoma and implications for tumor spread. Hepatology 1997, 26, 283–287. [Google Scholar] [CrossRef]
- Stohrer, M.; Boucher, Y.; Stangassinger, M.; Jain, R.K. Oncotic pressure in solid tumors is elevated. Cancer Res. 2000, 60, 4251–4255. [Google Scholar]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Jain, R.K.; Tong, R.T.; Munn, L.L. Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: Insights from a mathematical model. Cancer Res. 2007, 67, 2729–2735. [Google Scholar] [CrossRef]
- Mok, W.; Boucher, Y.; Jain, R.K. Matrix metalloproteinases-1 and -8 improve the distribution and efficacy of an oncolytic virus. Cancer Res. 2007, 67, 10664–10668. [Google Scholar] [CrossRef]
- McKee, T.D.; Grandi, P.; Mok, W.; Alexandrakis, G.; Insin, N.; Zimmer, J.P.; Bawendi, M.G.; Boucher, Y.; Breakefield, X.O.; Jain, R.K. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006, 66, 2509–2513. [Google Scholar] [CrossRef]
- Bilbao, R.; Bustos, M.; Alzuguren, P.; Pajares, M.J.; Drozdzik, M.; Qian, C.; Prieto, J. A blood-tumor barrier limits gene transfer to experimental liver cancer: The effect of vasoactive compounds. Gene Ther. 2000, 7, 1824–1832. [Google Scholar] [CrossRef]
- Kuriyama, N.; Kuriyama, H.; Julin, C.M.; Lamborn, K.; Israel, M.A. Pretreatment with protease is a useful experimental strategy for enhancing adenovirus-mediated cancer gene therapy. Hum. Gene Ther. 2000, 11, 2219–2230. [Google Scholar] [CrossRef]
- Schäfer, S.; Weibel, S.; Donat, U.; Zhang, Q.; Aguilar, R.J.; Chen, N.G.; Szalay, A.A. Vaccinia virus-mediated intra-tumoral expression of matrix metalloproteinase 9 enhances oncolysis of PC-3 xenograft tumors. BMC Cancer 2012, 12, 366. [Google Scholar]
- Hong, C.S.; Fellows, W.; Niranjan, A.; Alber, S.; Watkins, S.; Cohen, J.B.; Glorioso, J.C.; Grandi, P. Ectopic matrix metalloproteinase-9 expression in human brain tumor cells enhances oncolytic HSV vector infection. Gene Ther. 2010, 17, 1200–1205. [Google Scholar]
- Vähä-Koskela, M.J.; Kallio, J.P.; Jansson, L.C.; Heikkilä, J.E.; Zakhartchenko, V.A.; Kallajoki, M.A.; Kähäri, V.M.; Hinkkanen, A.E. Oncolytic capacity of attenuated replicative semliki forest virus in human melanoma xenografts in severe combined immunodeficient mice. Cancer Res. 2006, 66, 7185–7194. [Google Scholar] [CrossRef]
- Smith, E.; Breznik, J.; Lichty, B.D. Strategies to enhance viral penetration of solid tumors. Human Gene Ther. 2011, 22, 1053–1060. [Google Scholar] [CrossRef]
- Beyer, I.; Li, Z.; Persson, J.; Liu, Y.; van Rensburg, R.; Yumul, R.; Zhang, X.B.; Hung, M.C.; Lieber, A. Controlled extracellular matrix degradation in breast cancer tumors improves therapy by trastuzumab. Mol. Ther. 2011, 19, 479–489. [Google Scholar] [CrossRef]
- Ganesh, S.; Gonzalez Edick, M.; Idamakanti, N.; Abramova, M.; Vanroey, M.; Robinson, M.; Yun, C.O.; Jooss, K. Relaxin-expressing, fiber chimeric oncolytic adenovirus prolongs survival of tumor-bearing mice. Cancer Res. 2007, 67, 4399–4407. [Google Scholar] [CrossRef]
- Choi, I.K.; Strauss, R.; Richter, M.; Yun, C.O.; Lieber, A. Strategies to increase drug penetration in solid tumors. Front. Oncol. 2013, 3, 193. [Google Scholar]
- Eshun, F.K.; Currier, M.A.; Gillespie, R.A.; Fitzpatrick, J.L.; Baird, W.H.; Cripe, T.P. VEGF blockade decreases the tumor uptake of systemic oncolytic herpes virus but enhances therapeutic efficacy when given after virotherapy. Gene Ther. 2010, 17, 922–929. [Google Scholar]
- Thaci, B.; Ulasov, I.V.; Ahmed, A.U.; Ferguson, S.D.; Han, Y.; Lesniak, M.S. Anti-angiogenic therapy increases intratumoral adenovirus distribution by inducing collagen degradation. Gene Ther. 2013, 20, 318–327. [Google Scholar]
- Angarita, F.A.; Acuna, S.A.; Ottolino-Perry, K.; Zerhouni, S.; McCart, J.A. Mounting a strategic offense: Fighting tumor vasculature with oncolytic viruses. Trends Mol. Med. 2013, 19, 378–392. [Google Scholar] [CrossRef]
- Dingwell, K.S.; Brunetti, C.R.; Hendricks, R.L.; Tang, Q.; Tang, M.; Rainbow, A.J.; Johnson, D.C. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 1994, 68, 834–845. [Google Scholar]
- Rottner, K.; Stradal, T.E. Poxviruses taking a ride on actin: New users of known hardware. Cell Host Microbe 2009, 6, 497–499. [Google Scholar] [CrossRef]
- Smith, G.L.; Law, M. The exit of vaccinia virus from infected cells. Virus Res. 2004, 106, 189–197. [Google Scholar] [CrossRef]
- Guedan, S.; Grases, D.; Rojas, J.J.; Gros, A.; Vilardell, F.; Vile, R.; Mercade, E.; Cascallo, M.; Alemany, R. GALV expression enhances the therapeutic efficacy of an oncolytic adenovirus by inducing cell fusion and enhancing virus distribution. Gene Ther. 2012, 19, 1048–1057. [Google Scholar]
- Le Boeuf, F.; Diallo, J.S.; McCart, J.A.; Thorne, S.; Falls, T.; Stanford, M.; Kanji, F.; Auer, R.; Brown, C.W.; Lichty, B.D.; et al. Synergistic interaction between oncolytic viruses augments tumor killing. Mol. Ther. 2010, 18, 888–895. [Google Scholar] [CrossRef]
- Hoffmann, D.; Grunwald, T.; Kuate, S.; Wildner, O. Mechanistic analysis and comparison of viral fusogenic membrane proteins for their synergistic effects on chemotherapy. Cancer Biol. Ther. 2007, 6, 510–518. [Google Scholar] [CrossRef]
- Wang, H.; Liaw, Y.C.; Stone, D.; Kalyuzhniy, O.; Amiraslanov, I.; Tuve, S.; Verlinde, C.L.; Shayakhmetov, D.; Stehle, T.; Roffler, S.; et al. Identification of CD46 binding sites within the adenovirus serotype 35 fiber knob. J. Virol. 2007, 81, 12785–12792. [Google Scholar] [CrossRef]
- Wang, H.; Li, Z.Y.; Liu, Y.; Persson, J.; Beyer, I.; Moller, T.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.B.; et al. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat. Med. 2011, 17, 96–104. [Google Scholar]
- Coyne, C.B.; Bergelson, J.M. CAR: A virus receptor within the tight junction. Adv. Drug Deliv. Rev. 2005, 57, 869–882. [Google Scholar] [CrossRef]
- Cardone, J.; Al-Shouli, S.; Kemper, C. A novel role for CD46 in wound repair. Front. Immunol. 2011, 2, 28. [Google Scholar]
- Thorsteinsson, L.; O’Dowd, G.M.; Harrington, P.M.; Johnson, P.M. The complement regulatory proteins CD46 and CD59, but not CD55, are highly expressed by glandular epithelium of human breast and colorectal tumour tissues. APMIS 1998, 106, 869–878. [Google Scholar] [CrossRef]
- Nandi, S.; Ulasov, I.V.; Rolle, C.E.; Han, Y.; Lesniak, M.S. A chimeric adenovirus with an Ad 3 fiber knob modification augments glioma virotherapy. J. Gene Med. 2009, 11, 1005–1011. [Google Scholar]
- Martin, T.A.; Mason, M.D.; Jiang, W.G. Tight junctions in cancer metastasis. Front. Biosci. 2011, 16, 898–936. [Google Scholar] [CrossRef]
- Ding, L.; Lu, Z.; Lu, Q.; Chen, Y.H. The claudin family of proteins in human malignancy: A clinical perspective. Cancer Manag. Res. 2013, 5, 367–375. [Google Scholar]
- Beyer, I.; van Rensburg, R.; Strauss, R.; Li, Z.; Wang, H.; Persson, J.; Yumul, R.; Feng, Q.; Song, H.; Bartek, J.; et al. Epithelial junction opener JO-1 improves monoclonal antibody therapy of cancer. Cancer Res. 2011, 71, 7080–7090. [Google Scholar] [CrossRef]
- Beyer, I.; Cao, H.; Persson, J.; Song, H.; Richter, M.; Feng, Q.; Yumul, R.; van Rensburg, R.; Li, Z.; Berenson, R.; et al. Coadministration of epithelial junction opener JO-1 improves the efficacy and safety of chemotherapeutic drugs. Clin. Cancer Res. 2012, 18, 3340–3351. [Google Scholar] [CrossRef]
- Nagano, S.; Perentes, J.Y.; Jain, R.K.; Boucher, Y. Cancer cell death enhances the penetration and efficacy of oncolytic herpes simplex virus in tumors. Cancer Res. 2008, 68, 3795–3802. [Google Scholar]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar]
- Binder, C.; Hagemann, T.; Husen, B.; Schulz, M.; Einspanier, A. Relaxin enhances in vitro invasiveness of breast cancer cell lines by up-regulation of matrix metalloproteases. Mol. Hum. Reprod. 2002, 8, 789–796. [Google Scholar]
- Radestock, Y.; Hoang-Vu, C.; Hombach-Klonisch, S. Relaxin reduces xenograft tumour growth of human MDA-MB-231 breast cancer cells. Breast Cancer Res. 2008, 10, R71. [Google Scholar]
- Lavilla-Alonso, S.; Bauer, M.M.; Abo-Ramadan, U.; Ristimaki, A.; Halavaara, J.; Desmond, R.A.; Wang, D.; Escutenaire, S.; Ahtiainen, L.; Saksela, K.; et al. Macrophage metalloelastase (MME) as adjuvant for intra-tumoral injection of oncolytic adenovirus and its influence on metastases development. Cancer Gene Ther. 2012, 19, 126–134. [Google Scholar]
- Toth, K.; Djeha, H.; Ying, B.; Tollefson, A.E.; Kuppuswamy, M.; Doronin, K.; Krajcsi, P.; Lipinski, K.; Wrighton, C.J.; Wold, W.S. An oncolytic adenovirus vector combining enhanced cell-to-cell spreading, mediated by the ADP cytolytic protein, with selective replication in cancer cells with deregulated wnt signaling. Cancer Res. 2004, 64, 3638–3644. [Google Scholar] [CrossRef]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef]
- Fulci, G.; Dmitrieva, N.; Gianni, D.; Fontana, E.J.; Pan, X.; Lu, Y.; Kaufman, C.S.; Kaur, B.; Lawler, S.E.; Lee, R.J.; et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 2007, 67, 9398–9406. [Google Scholar] [CrossRef]
- Lallemand, C.; Lebon, P.; Rizza, P.; Blanchard, B.; Tovey, M.G. Constitutive expression of specific interferon isotypes in peripheral blood leukocytes from normal individuals and in promonocytic U937 cells. J. Leukoc. Biol. 1996, 60, 137–146. [Google Scholar]
- Liu, Y.P.; Suksanpaisan, L.; Steele, M.B.; Russell, S.J.; Peng, K.W. Induction of antiviral genes by the tumor microenvironment confers resistance to virotherapy. Sci. Rep. 2013, 3, 2375. [Google Scholar]
- Ruotsalainen, J.; Martikainen, M.; Niittykoski, M.; Huhtala, T.; Aaltonen, T.; Heikkilä, J.; Bell, J.; Vähä-Koskela, M.; Hinkkanen, A. Interferon-beta sensitivity of tumor cells correlates with poor response to va7 virotherapy in mouse glioma models. Mol. Ther. 2012, 20, 1529–1539. [Google Scholar] [CrossRef]
- Vähä-Koskela, M.J.; Le Boeuf, F.; Lemay, C.; de Silva, N.; Diallo, J.S.; Cox, J.; Becker, M.; Choi, Y.; Ananth, A.; Sellers, C.; et al. Resistance to two heterologous neurotropic oncolytic viruses, Semliki Forest virus and vaccinia virus, in experimental glioma. J. Virol. 2013, 87, 2363–2366. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Becht, E.; Remark, R.; Damotte, D.; Sautes-Fridman, C.; Fridman, W.H. The immune contexture of primary and metastatic human tumours. Curr. Opin. Immunol. 2014, 27C, 8–15. [Google Scholar]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Currier, M.A.; Eshun, F.K.; Sholl, A.; Chernoguz, A.; Crawford, K.; Divanovic, S.; Boon, L.; Goins, W.F.; Frischer, J.S.; Collins, M.H.; et al. VEGF blockade enables oncolytic cancer virotherapy in part by modulating intratumoral myeloid cells. Mol. Ther. 2013, 21, 1014–1023. [Google Scholar] [CrossRef]
- Burger, J.A.; Kipps, T.J. CXCR4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef]
- Gil, M.; Seshadri, M.; Komorowski, M.P.; Abrams, S.I.; Kozbor, D. Targeting CXCL12/CXCR4 signaling with oncolytic virotherapy disrupts tumor vasculature and inhibits breast cancer metastases. Proc. Natl. Acad. Sci. USA 2013, 110, E1291–E1300. [Google Scholar]
- Muthana, M.; Rodrigues, S.; Chen, Y.Y.; Welford, A.; Hughes, R.; Tazzyman, S.; Essand, M.; Morrow, F.; Lewis, C.E. Macrophage delivery of an oncolytic virus abolishes tumor regrowth and metastasis after chemotherapy or irradiation. Cancer Res. 2013, 73, 490–495. [Google Scholar] [CrossRef]
- Verheije, M.H.; Rottier, P.J. Retargeting of viruses to generate oncolytic agents. Adv. Virol. 2012, 2012, 798526. [Google Scholar]
- Power, A.T.; Bell, J.C. Taming the Trojan horse: Optimizing dynamic carrier cell/oncolytic virus systems for cancer biotherapy. Gene Ther. 2008, 15, 772–779. [Google Scholar] [CrossRef]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Ann. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Muller, U.; Steinhoff, U.; Reis, L.F.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.M.; Aguet, M. Functional role of type I and type II interferons in antiviral defense. Science 1994, 264, 1918–1921. [Google Scholar]
- Fragkoudis, R.; Breakwell, L.; McKimmie, C.; Boyd, A.; Barry, G.; Kohl, A.; Merits, A.; Fazakerley, J.K. The type I interferon system protects mice from Semliki Forest virus by preventing widespread virus dissemination in extraneural tissues, but does not mediate the restricted replication of avirulent virus in central nervous system neurons. J. Gen. Virol. 2007, 88, 3373–3384. [Google Scholar] [CrossRef]
- Chapgier, A.; Wynn, R.F.; Jouanguy, E.; Filipe-Santos, O.; Zhang, S.; Feinberg, J.; Hawkins, K.; Casanova, J.L.; Arkwright, P.D. Human complete Stat-1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J. Immunol. 2006, 176, 5078–5083. [Google Scholar]
- Sancho-Shimizu, V.; Perez de Diego, R.; Jouanguy, E.; Zhang, S.Y.; Casanova, J.L. Inborn errors of anti-viral interferon immunity in humans. Curr. Opin. Virol. 2011, 1, 487–496. [Google Scholar] [CrossRef]
- Bax, H.I.; Freeman, A.F.; Anderson, V.L.; Vesterhus, P.; Laerum, D.; Pittaluga, S.; Wilson, W.H.; Holland, S.M. B-cell lymphoma in a patient with complete interferon gamma receptor 1 deficiency. J. Clin. Immunol. 2013, 33, 1062–1066. [Google Scholar]
- Kawamoto, S.; Oritani, K.; Asada, H.; Takahashi, I.; Ishikawa, J.; Yoshida, H.; Yamada, M.; Ishida, N.; Ujiie, H.; Masaie, H.; et al. Antiviral activity of limitin against encephalomyocarditis virus, herpes simplex virus, and mouse hepatitis virus: Diverse requirements by limitin and alpha interferon for interferon regulatory factor 1. J. Virol. 2003, 77, 9622–9631. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Zurney, J.; Kobayashi, T.; Holm, G.H.; Dermody, T.S.; Sherry, B. Reovirus mu2 protein inhibits interferon signaling through a novel mechanism involving nuclear accumulation of interferon regulatory factor 9. J. Virol. 2009, 83, 2178–2187. [Google Scholar] [CrossRef]
- Zurney, J.; Howard, K.E.; Sherry, B. Basal expression levels of IFNAR and Jak-STAT components are determinants of cell-type-specific differences in cardiac antiviral responses. J. Virol. 2007, 81, 13668–13680. [Google Scholar] [CrossRef]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Sheehan, K.C.; Shankaran, V.; Uppaluri, R.; Bui, J.D.; Diamond, M.S.; Koebel, C.M.; Arthur, C.; White, J.M.; et al. A critical function for type I interferons in cancer immunoediting. Nat. Immunol. 2005, 6, 722–729. [Google Scholar]
- Sheehan, K.C.; Lai, K.S.; Dunn, G.P.; Bruce, A.T.; Diamond, M.S.; Heutel, J.D.; Dungo-Arthur, C.; Carrero, J.A.; White, J.M.; Hertzog, P.J.; et al. Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J. Interferon Cytokine Res. 2006, 26, 804–819. [Google Scholar] [CrossRef]
- Saidi, R.F.; Williams, F.; Silberberg, B.; Mittal, V.K.; ReMine, S.G.; Jacobs, M.J. Expression of interferon receptors in pancreatic cancer: Identification of a novel prognostic factor. Surgery 2006, 139, 743–748. [Google Scholar] [CrossRef]
- Kondo, M.; Nagano, H.; Sakon, M.; Yamamoto, H.; Morimoto, O.; Arai, I.; Miyamoto, A.; Eguchi, H.; Dono, K.; Nakamori, S.; et al. Expression of interferon alpha/beta receptor in human hepatocellular carcinoma. Int. J. Oncol. 2000, 17, 83–88. [Google Scholar]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef]
- Khodarev, N.N.; Roach, P.; Pitroda, S.P.; Golden, D.W.; Bhayani, M.; Shao, M.Y.; Darga, T.E.; Beveridge, M.G.; Sood, R.F.; Sutton, H.G.; et al. STAT1 pathway mediates amplification of metastatic potential and resistance to therapy. PLoS One 2009, 4, e5821. [Google Scholar] [CrossRef]
- Khodarev, N.N.; Roizman, B.; Weichselbaum, R.R. Molecular pathways: interferon/stat1 pathway: Role in the tumor resistance to genotoxic stress and aggressive growth. Clin. Cancer Res. 2012, 18, 3015–3021. [Google Scholar] [CrossRef]
- Luszczek, W.; Cheriyath, V.; Mekhail, T.M.; Borden, E.C. Combinations of DNA methyltransferase and histone deacetylase inhibitors induce DNA damage in small cell lung cancer cells: Correlation of resistance with IFN-stimulated gene expression. Mol. Cancer Ther. 2010, 9, 2309–2321. [Google Scholar] [CrossRef]
- Cheon, H.; Yang, J.; Stark, G.R. The functions of signal transducers and activators of transcriptions 1 and 3 as cytokine-inducible proteins. J. Interferon Cytokine Res. 2011, 31, 33–40. [Google Scholar] [CrossRef]
- Wilson, S.S.; Wiens, M.E.; Smith, J.G. Antiviral mechanisms of human defensins. J. Mol. Biol. 2013, 425, 4965–4980. [Google Scholar] [CrossRef]
- Lundy, F.T.; Orr, D.F.; Gallagher, J.R.; Maxwell, P.; Shaw, C.; Napier, S.S.; Gerald Cowan, C.; Lamey, P.J.; Marley, J.J. Identification and overexpression of human neutrophil alpha-defensins (human neutrophil peptides 1, 2 and 3) in squamous cell carcinomas of the human tongue. Oral Oncol. 2004, 40, 139–144. [Google Scholar] [CrossRef]
- Lapteva, N.; Aldrich, M.; Rollins, L.; Ren, W.; Goltsova, T.; Chen, S.Y.; Huang, X.F. Attraction and activation of dendritic cells at the site of tumor elicits potent antitumor immunity. Mol. Ther. 2009, 17, 1626–1636. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Haller, O.; Kochs, G.; Weber, F. The interferon response circuit: Induction and suppression by pathogenic viruses. Virology 2006, 344, 119–130. [Google Scholar] [CrossRef]
- Suzuki, M.; Chiocca, E.A.; Saeki, Y. Early STAT1 activation after systemic delivery of HSV amplicon vectors suppresses transcription of the vector-encoded transgene. Mol. Ther. 2007, 15, 2017–2026. [Google Scholar] [CrossRef]
- Heikkilä, J.E.; Vähä-Koskela, M.J.; Ruotsalainen, J.J.; Martikainen, M.W.; Stanford, M.M.; McCart, J.A.; Bell, J.C.; Hinkkanen, A.E. Intravenously administered alphavirus vector VA7 eradicates orthotopic human glioma xenografts in nude mice. PLoS One 2010, 5, e8603. [Google Scholar]
- Määttä, A.M.; Liimatainen, T.; Wahlfors, T.; Wirth, T.; Vähä-Koskela, M.; Jansson, L.; Valonen, P.; Hakkinen, K.; Rautsi, O.; Pellinen, R.; et al. Evaluation of cancer virotherapy with attenuated replicative Semliki forest virus in different rodent tumor models. Int. J. Cancer 2007, 121, 863–870. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar]
- Paglino, J.C.; van den Pol, A.N. Vesicular stomatitis virus has extensive oncolytic activity against human sarcomas: Rare resistance is overcome by blocking interferon pathways. J. Virol. 2011, 85, 9346–9358. [Google Scholar] [CrossRef]
- Blackham, A.U.; Northrup, S.A.; Willingham, M.; D’Agostino, R.B., Jr.; Lyles, D.S.; Stewart, J.H.T. Variation in susceptibility of human malignant melanomas to oncolytic vesicular stomatitis virus. Surgery 2013, 153, 333–343. [Google Scholar] [CrossRef]
- Berchtold, S.; Lampe, J.; Weiland, T.; Smirnow, I.; Schleicher, S.; Handgretinger, R.; Kopp, H.G.; Reiser, J.; Stubenrauch, F.; Mayer, N.; et al. Innate immune defense defines susceptibility of sarcoma cells to measles vaccine virus-based oncolysis. J. Virol. 2013, 87, 3484–3501. [Google Scholar] [CrossRef]
- Monsurro, V.; Beghelli, S.; Wang, R.; Barbi, S.; Coin, S.; di Pasquale, G.; Bersani, S.; Castellucci, M.; Sorio, C.; Eleuteri, S.; et al. Anti-viral state segregates two molecular phenotypes of pancreatic adenocarcinoma: Potential relevance for adenoviral gene therapy. J. Transl. Med. 2010, 8, 10. [Google Scholar] [CrossRef]
- Liikanen, I.; Monsurro, V.; Ahtiainen, L.; Raki, M.; Hakkarainen, T.; Diaconu, I.; Escutenaire, S.; Hemminki, O.; Dias, J.D.; Cerullo, V.; et al. Induction of interferon pathways mediates in vivo resistance to oncolytic adenovirus. Mol. Ther. 2011, 19, 1858–1866. [Google Scholar] [CrossRef]
- Wollmann, G.; Davis, J.N.; Bosenberg, M.W.; van den Pol, A.N. Vesicular stomatitis virus variants selectively infect and kill human melanomas but not normal melanocytes. J. Virol. 2013, 87, 6644–6659. [Google Scholar] [CrossRef]
- Saloura, V.; Wang, L.C.; Fridlender, Z.G.; Sun, J.; Cheng, G.; Kapoor, V.; Sterman, D.H.; Harty, R.N.; Okumura, A.; Barber, G.N.; et al. Evaluation of an attenuated vesicular stomatitis virus (vsv) vector expressing interferon-beta for use in malignant pleural mesothelioma: heterogeneity in interferon-responsiveness defines potential efficacy. Hum. Gene Ther. 2010, 21, 51–64. [Google Scholar]
- Shmulevitz, M.; Pan, L.Z.; Garant, K.; Pan, D.; Lee, P.W. Oncogenic Ras promotes reovirus spread by suppressing IFN-beta production through negative regulation of RIG-I signaling. Cancer Res. 2010, 70, 4912–4921. [Google Scholar] [CrossRef]
- Battcock, S.M.; Collier, T.W.; Zu, D.; Hirasawa, K. Negative regulation of the alpha interferon-induced antiviral response by the Ras/Raf/MEK pathway. J. Virol. 2006, 80, 4422–4430. [Google Scholar] [CrossRef]
- Christian, S.L.; Zu, D.; Licursi, M.; Komatsu, Y.; Pongnopparat, T.; Codner, D.A.; Hirasawa, K. Suppression of IFN-induced transcription underlies IFN defects generated by activated Ras/MEK in human cancer cells. PLoS One 2012, 7, e44267. [Google Scholar]
- Cascallo, M.; Capella, G.; Mazo, A.; Alemany, R. Ras-dependent oncolysis with an adenovirus VAI mutant. Cancer Res. 2003, 63, 5544–5550. [Google Scholar]
- Schumann, M.; Dobbelstein, M. Activating Ras mutations fail to ensure efficient replication of adenovirus mutants lacking VA-RNA. Cell Cycle 2006, 5, 315–321. [Google Scholar] [CrossRef]
- Kim, M.; Egan, C.; Alain, T.; Urbanski, S.J.; Lee, P.W.; Forsyth, P.A.; Johnston, R.N. Acquired resistance to reoviral oncolysis in Ras-transformed fibrosarcoma cells. Oncogene 2007, 26, 4124–4134. [Google Scholar] [CrossRef]
- Moerdyk-Schauwecker, M.; Shah, N.R.; Murphy, A.M.; Hastie, E.; Mukherjee, P.; Grdzelishvili, V.Z. Resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus: Role of type I interferon signaling. Virology 2013, 436, 221–234. [Google Scholar]
- Evgin, L.; Vähä-Koskela, M.; Rintoul, J.; Falls, T.; Le, B.F.; Barrett, J.W.; Bell, J.C.; Stanford, M.M. Potent oncolytic activity of raccoonpox virus in the absence of natural pathogenicity. Mol. Ther. 2010, 18, 896–902. [Google Scholar] [CrossRef]
- Stanford, M.M.; Breitbach, C.J.; Bell, J.C.; McFadden, G. Innate immunity, tumor microenvironment and oncolytic virus therapy: Friends or foes? Curr. Opin. Mol. Ther. 2008, 10, 32–37. [Google Scholar]
- Zemp, F.J.; McKenzie, B.A.; Lun, X.; Maxwell, L.; Reilly, K.M.; McFadden, G.; Yong, V.W.; Forsyth, P.A. Resistance to oncolytic myxoma virus therapy in nf1−/−/trp53−/− syngeneic mouse glioma models is independent of anti-viral type-I interferon. PLoS One 2013, 8, e65801. [Google Scholar]
- Haseley, A.; Boone, S.; Wojton, J.; Yu, L.; Yoo, J.Y.; Yu, J.; Kurozumi, K.; Glorioso, J.C.; Caligiuri, M.A.; Kaur, B. Extracellular matrix protein CCN1 limits oncolytic efficacy in glioma. Cancer Res. 2012, 72, 1353–1362. [Google Scholar] [CrossRef]
- Raaben, M.; Groot Koerkamp, M.J.; Rottier, P.J.; de Haan, C.A. Type I interferon receptor-independent and -dependent host transcriptional responses to mouse hepatitis coronavirus infection in vivo. BMC Genomics 2009, 10, 350. [Google Scholar] [CrossRef]
- Chairatvit, K.; Wongnoppavich, A.; Choonate, S. Up-regulation of interferon-stimulated gene15 and its conjugates by tumor necrosis factor-alpha via type I interferon-dependent and -independent pathways. Mol. Cell. Biochem. 2012, 368, 195–201. [Google Scholar] [CrossRef]
- Le Boeuf, F.; Niknejad, N.; Wang, J.; Auer, R.; Weberpals, J.I.; Bell, J.C.; Dimitroulakos, J. Sensitivity of cervical carcinoma cells to vesicular stomatitis virus-induced oncolysis: Potential role of human papilloma virus infection. Int. J. Cancer 2012, 131, E204–E215. [Google Scholar] [CrossRef]
- Escobar-Zarate, D.; Liu, Y.P.; Suksanpaisan, L.; Russell, S.J.; Peng, K.W. Overcoming cancer cell resistance to VSV oncolysis with JAK1/2 inhibitors. Cancer Gene Ther. 2013, 20, 582–589. [Google Scholar]
- Heine, A.; Held, S.A.; Daecke, S.N.; Wallner, S.; Yajnanarayana, S.P.; Kurts, C.; Wolf, D.; Brossart, P. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood 2013, 122, 1192–1202. [Google Scholar] [CrossRef]
- Looyenga, B.D.; Hutchings, D.; Cherni, I.; Kingsley, C.; Weiss, G.J.; Mackeigan, J.P. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PLoS One 2012, 7, e30820. [Google Scholar]
- Nguyen, T.L.; Abdelbary, H.; Arguello, M.; Breitbach, C.; Leveille, S.; Diallo, J.S.; Yasmeen, A.; Bismar, T.A.; Kirn, D.; Falls, T.; et al. Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc. Natl. Acad. Sci. USA 2008, 105, 14981–14986. [Google Scholar] [CrossRef]
- Kim, D.R.; Park, M.Y.; Lim, H.J.; Park, J.S.; Cho, Y.J.; Lee, S.W.; Yoon, H.I.; Lee, J.H.; Kim, Y.S.; Lee, C.T. Combination therapy of conditionally replicating adenovirus and histone deacetylase inhibitors. Int. J. Mol. Med. 2012, 29, 218–224. [Google Scholar]
- Otsuki, A.; Patel, A.; Kasai, K.; Suzuki, M.; Kurozumi, K.; Chiocca, E.A.; Saeki, Y. Histone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic viruses. Mol. Ther. 2008, 16, 1546–1555. [Google Scholar] [CrossRef]
- Katsura, T.; Iwai, S.; Ota, Y.; Shimizu, H.; Ikuta, K.; Yura, Y. The effects of trichostatin A on the oncolytic ability of herpes simplex virus for oral squamous cell carcinoma cells. Cancer Gene Ther. 2009, 16, 237–245. [Google Scholar]
- Shulak, L.; Beljanski, V.; Chiang, C.; Dutta, S.M.; van Grevenynghe, J.; Belgnaoui, S.M.; Nguyen, T.L.; di Lenardo, T.; Semmes, O.J.; Lin, R.; et al. Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating nf-kappab-dependent autophagy. J. Virol. 2014, 88, 2927–2940. [Google Scholar] [CrossRef]
- Du, Z.; Whitt, M.A.; Baumann, J.; Garner, J.M.; Morton, C.L.; Davidoff, A.M.; Pfeffer, L.M. Inhibition of type I interferon-mediated antiviral action in human glioma cells by the IKK inhibitors BMS-345541 and TPCA-1. J. Interferon Cytokine Res. 2012, 32, 368–377. [Google Scholar] [CrossRef]
- Forbes, N.E.; Abdelbary, H.; Lupien, M.; Bell, J.C.; Diallo, J.S. Exploiting tumor epigenetics to improve oncolytic virotherapy. Front. Genet. 2013, 4, 184. [Google Scholar]
- Okemoto, K.; Kasai, K.; Wagner, B.; Haseley, A.; Meisen, H.; Bolyard, C.; Mo, X.; Wehr, A.; Lehman, A.; Fernandez, S.; et al. DNA demethylating agents synergize with oncolytic HSV1 against malignant gliomas. Clin. Cancer. 2013, 19, 5952–5959. [Google Scholar] [CrossRef]
- Jha, B.K.; Dong, B.; Nguyen, C.T.; Polyakova, I.; Silverman, R.H. Suppression of antiviral innate immunity by sunitinib enhances oncolytic virotherapy. Mol. Ther. 2013, 21, 1749–1757. [Google Scholar] [CrossRef]
- Randhawa, P.S.; Farasati, N.A.; Huang, Y.; Mapara, M.Y.; Shapiro, R. Viral drug sensitivity testing using quantitative PCR: Effect of tyrosine kinase inhibitors on polyomavirus BK replication. Am. J. Clin. Pathol. 2010, 134, 916–920. [Google Scholar] [CrossRef]
- Wakimoto, H.; Fulci, G.; Tyminski, E.; Chiocca, E.A. Altered expression of antiviral cytokine mRNAs associated with cyclophosphamide’s enhancement of viral oncolysis. Gene Ther. 2004, 11, 214–223. [Google Scholar]
- Ben Yebdri, F.; van Grevenynghe, J.; Tang, V.A.; Goulet, M.L.; Wu, J.H.; Stojdl, D.F.; Hiscott, J.; Lin, R. Triptolide-mediated inhibition of interferon signaling enhances vesicular stomatitis virus-based oncolysis. Mol. Ther. 2013, 21, 2043–2053. [Google Scholar] [CrossRef]
- Alain, T.; Lun, X.; Martineau, Y.; Sean, P.; Pulendran, B.; Petroulakis, E.; Zemp, F.J.; Lemay, C.G.; Roy, D.; Bell, J.C.; et al. Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production. Proc. Natl. Acad. Sci. USA 2010, 107, 1576–1581. [Google Scholar] [CrossRef]
- Diallo, J.S.; Le, B.F.; Lai, F.; Cox, J.; Vaha-Koskela, M.; Abdelbary, H.; MacTavish, H.; Waite, K.; Falls, T.; Wang, J.; et al. A high-throughput pharmacoviral approach identifies novel oncolytic virus sensitizers. Mol. Ther. 2010, 18, 1123–1129. [Google Scholar] [CrossRef]
- Beug, S.T.; Tang, V.A.; Lacasse, E.C.; Cheung, H.H.; Beauregard, C.E.; Brun, J.; Nuyens, J.P.; Earl, N.; St-Jean, M.; Holbrook, J.; et al. Smac mimetics and innate immune stimuli synergize to promote tumor death. Nat. Biotechnol. 2014, 32, 182–190. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Lefebvre, C.; Allan, K.; Brun, J.; Sanaei, C.A.; Baird, S.; Pearce, N.; Gronberg, S.; Wilson, B.; Prakesh, M.; et al. Virus-tumor interactome screen reveals ER stress response can reprogram resistant cancers for oncolytic virus-triggered caspase-2 cell death. Cancer Cell 2011, 20, 443–456. [Google Scholar]
- Passer, B.J.; Cheema, T.; Zhou, B.; Wakimoto, H.; Zaupa, C.; Razmjoo, M.; Sarte, J.; Wu, S.; Wu, C.L.; Noah, J.W.; et al. Identification of the ENT1 antagonists dipyridamole and dilazep as amplifiers of oncolytic herpes simplex virus-1 replication. Cancer Res. 2010, 70, 3890–3895. [Google Scholar] [CrossRef]
- Wennier, S.T.; Liu, J.; McFadden, G. Bugs and drugs: Oncolytic virotherapy in combination with chemotherapy. Curr. Pharm. Biotechnol. 2012, 13, 1817–1833. [Google Scholar] [CrossRef]
- Newman, W.; Southam, C.M. Virus treatment in advanced cancer; a pathological study of fifty-seven cases. Cancer 1954, 7, 106–118. [Google Scholar] [CrossRef]
- Southam, C.M.; Hilleman, M.R.; Werner, J.H. Pathogenicity and oncolytic capacity of RI virus strain RI-67 in man. J. Lab. Clin. Med. 1956, 47, 573–582. [Google Scholar]
- Elankumaran, S.; Chavan, V.; Qiao, D.; Shobana, R.; Moorkanat, G.; Biswas, M.; Samal, S.K. Type I interferon-sensitive recombinant newcastle disease virus for oncolytic virotherapy. J. Virol. 2010, 84, 3835–3844. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.D.; tenOever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 2003, 4, 263–275. [Google Scholar]
- Malilas, W.; Koh, S.S.; Srisuttee, R.; Boonying, W.; Cho, I.R.; Jeong, C.S.; Johnston, R.N.; Chung, Y.H. Cancer upregulated gene 2, a novel oncogene, confers resistance to oncolytic vesicular stomatitis virus through STAT1-OASL2 signaling. Cancer Gene Ther. 2013, 20, 125–132. [Google Scholar]
- Park, E.H.; Park, E.H.; Cho, I.R.; Srisuttee, R.; Min, H.J.; Oh, M.J.; Jeong, Y.J.; Jhun, B.H.; Johnston, R.N.; Lee, S.; et al. CUG2, a novel oncogene confers reoviral replication through Ras and p38 signaling pathway. Cancer Gene Ther. 2010, 17, 307–314. [Google Scholar]
- Haralambieva, I.; Iankov, I.; Hasegawa, K.; Harvey, M.; Russell, S.J.; Peng, K.W. Engineering oncolytic measles virus to circumvent the intracellular innate immune response. Mol. Ther. 2007, 15, 588–597. [Google Scholar] [CrossRef]
- Meng, X.; Nakamura, T.; Okazaki, T.; Inoue, H.; Takahashi, A.; Miyamoto, S.; Sakaguchi, G.; Eto, M.; Naito, S.; Takeda, M.; et al. Enhanced antitumor effects of an engineered measles virus Edmonston strain expressing the wild-type N, P, L genes on human renal cell carcinoma. Mol. Ther. 2010, 18, 544–551. [Google Scholar] [CrossRef]
- Takaki, H.; Watanabe, Y.; Shingai, M.; Oshiumi, H.; Matsumoto, M.; Seya, T. Strain-to-strain difference of V protein of measles virus affects MDA5-mediated IFN-beta-inducing potential. Mol. Immunol. 2011, 48, 497–504. [Google Scholar]
- Shingai, M.; Ebihara, T.; Begum, N.A.; Kato, A.; Honma, T.; Matsumoto, K.; Saito, H.; Ogura, H.; Matsumoto, M.; Seya, T. Differential type I IFN-inducing abilities of wild-type versus vaccine strains of measles virus. J. Immunol. 2007, 179, 6123–6133. [Google Scholar]
- Schuhmann, K.M.; Pfaller, C.K.; Conzelmann, K.K. The measles virus V protein binds to p65 (RelA) to suppress NF-kappaB activity. J. Virol. 2011, 85, 3162–3171. [Google Scholar] [CrossRef]
- Childs, K.; Randall, R.; Goodbourn, S. Paramyxovirus V proteins interact with the RNA Helicase LGP2 to inhibit RIG-I-dependent interferon induction. J. Virol. 2012, 86, 3411–3421. [Google Scholar] [CrossRef]
- Zamarin, D.; Martinez-Sobrido, L.; Kelly, K.; Mansour, M.; Sheng, G.; Vigil, A.; Garcia-Sastre, A.; Palese, P.; Fong, Y. Enhancement of oncolytic properties of recombinant newcastle disease virus through antagonism of cellular innate immune responses. Mol. Ther. 2009, 17, 697–706. [Google Scholar] [CrossRef]
- Kim, S.H.; Samal, S.K. Inhibition of host innate immune responses and pathogenicity of recombinant Newcastle disease viruses expressing NS1 genes of influenza A viruses. J. Gen. Virol. 2010, 91, 1996–2001. [Google Scholar] [CrossRef]
- Guerra, S.; Abaitua, F.; Martinez-Sobrido, L.; Esteban, M.; Garcia-Sastre, A.; Rodriguez, D. Host-range restriction of vaccinia virus E3L deletion mutant can be overcome in vitro, but not in vivo, by expression of the influenza virus NS1 protein. PLoS One 2011, 6, e28677. [Google Scholar]
- Fu, X.; Rivera, A.; Tao, L.; Zhang, X. Incorporation of the B18R gene of vaccinia virus into an oncolytic herpes simplex virus improves antitumor activity. Mol. Ther. 2012, 20, 1871–1881. [Google Scholar] [CrossRef]
- Shah, A.C.; Parker, J.N.; Gillespie, G.Y.; Lakeman, F.D.; Meleth, S.; Markert, J.M.; Cassady, K.A. Enhanced antiglioma activity of chimeric HCMV/HSV-1 oncolytic viruses. Gene Ther. 2007, 14, 1045–1054. [Google Scholar]
- Le Boeuf, F.; Batenchuk, C.; Vähä-Koskela, M.; Breton, S.; Roy, D.; Lemay, C.; Cox, J.; Abdelbary, H.; Falls, T.; Waghray, G.; et al. Model-based rational design of an oncolytic virus with improved therapeutic potential. Nat. Commun. 2013, 4, 1974. [Google Scholar]
- De Vries, W.; Haasnoot, J.; van der, V.; van Montfort, T.; Zorgdrager, F.; Paxton, W.; Cornelissen, M.; van Kuppeveld, F.; de Haan, P.; Berkhout, B. Increased virus replication in mammalian cells by blocking intracellular innate defense responses. Gene Ther. 2008, 15, 545–552. [Google Scholar]
- Shors, S.T.; Beattie, E.; Paoletti, E.; Tartaglia, J.; Jacobs, B.L. Role of the vaccinia virus E3L and K3L gene products in rescue of VSV and EMCV from the effects of IFN-alpha. J. Interferon Cytokine Res. 1998, 18, 721–729. [Google Scholar] [CrossRef]
- Bridle, B.W.; Chen, L.; Lemay, C.G.; Diallo, J.S.; Pol, J.; Nguyen, A.; Capretta, A.; He, R.; Bramson, J.L.; Bell, J.C.; et al. HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol. Ther. 2013, 21, 887–894. [Google Scholar]
- El-Andaloussi, N.; Bonifati, S.; Kaufmann, J.K.; Mailly, L.; Daeffler, L.; Deryckere, F.; Nettelbeck, D.M.; Rommelaere, J.; Marchini, A. Generation of an adenovirus-parvovirus chimera with enhanced oncolytic potential. J. Virol. 2012, 86, 10418–10431. [Google Scholar] [CrossRef]
- Sanchez-Puig, J.M.; Lorenzo, M.M.; Blasco, R. A vaccinia virus recombinant transcribing an alphavirus replicon and expressing alphavirus structural proteins leads to packaging of alphavirus infectious single cycle particles. PLoS One 2013, 8, e75574. [Google Scholar] [CrossRef]
- Yang, Y.; Xiao, F.; Lu, Z.; Li, Z.; Zuo, H.; Zhang, Q.; Li, Q.; Wang, H.; Wang, L.S. Development of a novel adenovirus-alphavirus hybrid vector with RNA replicon features for malignant hematopoietic cell transduction. Cancer Gene Ther. 2013, 20, 429–436. [Google Scholar]
- Guan, M.; Rodriguez-Madoz, J.R.; Alzuguren, P.; Gomar, C.; Kramer, M.G.; Kochanek, S.; Prieto, J.; Smerdou, C.; Qian, C. Increased efficacy and safety in the treatment of experimental liver cancer with a novel adenovirus-alphavirus hybrid vector. Cancer Res. 2006, 66, 1620–1629. [Google Scholar] [CrossRef]
- Kaufmann, J.K.; Nettelbeck, D.M. Virus chimeras for gene therapy, vaccination, and oncolysis: Adenoviruses and beyond. Trends Mol. Med. 2012, 18, 365–376. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vähä-Koskela, M.; Hinkkanen, A. Tumor Restrictions to Oncolytic Virus. Biomedicines 2014, 2, 163-194. https://doi.org/10.3390/biomedicines2020163
Vähä-Koskela M, Hinkkanen A. Tumor Restrictions to Oncolytic Virus. Biomedicines. 2014; 2(2):163-194. https://doi.org/10.3390/biomedicines2020163
Chicago/Turabian StyleVähä-Koskela, Markus, and Ari Hinkkanen. 2014. "Tumor Restrictions to Oncolytic Virus" Biomedicines 2, no. 2: 163-194. https://doi.org/10.3390/biomedicines2020163
APA StyleVähä-Koskela, M., & Hinkkanen, A. (2014). Tumor Restrictions to Oncolytic Virus. Biomedicines, 2(2), 163-194. https://doi.org/10.3390/biomedicines2020163